
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetics and electrochemical evolution of binary silicon–polymer systems for lithium ion batteries†
Changling Lia,
Chueh Liub,
Kazi Ahmedb,
Zafer Mutlua,
Yiran Yana,
Ilkeun Leec,
Mihrimah Ozkanb and
Cengiz S. Ozkan *a
*a
aMaterials Science and Engineering Program, Department of Mechanical Engineering, University of California Riverside, CA 92521, USA. E-mail: cozkan@engr.ucr.edu
bDepartment of Electrical and Computer Engineering, Department of Chemistry, University of California, Riverside, CA 92521, USA
cCentral Facility of Advanced Microscopy and Microanalysis, Analytical Chemistry Instrumentation Facility, University of California, Riverside, CA 92521, USA
First published on 24th July 2017
Abstract
Silicon is a promising anode material for lithium-ion batteries owing to its high specific capacity and low discharge potential. To diminish Si structural degradation and its anode-capacity fading due to the vast volume change during alloying and dealloying, effective binders assisted in the encapsulation of Si anode materials and enhanced their integral stability. Herein, two conducting-hydrogel coatings, polyaniline (PANI) and polypyrrole (PPy), are formed to trap the Si surface via a facile and environmentally benign sol–gel polymerization process. Functional groups from polymer hydrogels chemically promote the confinement of conducting shells on the Si surface, rendering the Si-hydrogel frameworks without resistive binders and carbon black. The effects of coating thickness and conductivity of PPy and PANI coatings on the electrochemical properties of Si anodes have been investigated, and compared to insulating polyacrylic acid (PAA)–Si blended electrodes. The kinetics and the physical evolution of the binary Si–polymer systems during electrochemical reactions have been systematically studied via electrochemical impedance spectroscopy (EIS). It has been observed that the degree of improvement of the cycling stability and the rate capability of the three Si–polymer systems decrease in the order of PPy > PANI > PAA.
1. Introduction
Efficient and reliable energy storage platforms with high energy density, high power density and long life span are essential for the evolution of new generations of portable electronics, electrical vehicles (EVs)/plug-in hybrid electric vehicles (PHEVs), as well as renewable energy resources.1–4 Rechargeable lithium ion batteries (LIBs) have become more dominant over other types of batteries due to their high gravimetric energy density (W h kg−1) as well as the balanced large volumetric energy density (W h L−1).5 The development of anode and cathode materials for LIBs with high capacities is essential to achieve the required energy density for EVs/PHEVs. Silicon (Si) is regarded as an anode candidate for the high-performance LIBs and has been applied in several commercial anodes. This is due to the low discharge potential relative to Li/Li+ (0.5 V vs. Li/Li+) and the high theoretical capacity of 3579 mA h g−1 corresponding to the formation of a Li15Si4 phase at ambient environment. In contrast, conventionally used graphite-based anodes for LIBs have a limited theoretical capacity of 372 mA h g−1, owing to the stoichiometric limit of Li-ion intercalation in LiC6.6,7 Moreover, Si has high abundance as a rock-forming element, is non-toxic and environmentally benign. However, Si suffers from poor capacity retention due to its large volume expansion in excess of 300% produced by alloying with large amounts of Li+ during lithiation. The lithiation-induced mechanical stresses during alloying with the subsequent contraction during dealloying can cause Si structure to fracture, eliminate electrical connectivity while deteriorating the solid electrolyte interface (SEI) layer.8 Accordingly, the repeated huge volume changes during Li–Si lithiation and delithiation lead to irreversible capacity loss.To alleviate the aforementioned issues, extensive research has been investigated on optimizing the performance of Si-based anodes. Decreasing the dimension of Si from micro-size to nano-size is an effective route of avoiding pulverization and capacity fading during charging and discharging. Several nanostructured silicon materials such as nano-spheres, double-walled Si nanotubes, and porous silicon structures have been verified to create void spaces necessary for huge volume change during alloying and dealloying while preserving the crucial SEI layer.2,9,10 Another major drawback of Si is its low conductivity. Conductive additives such as carbon black (CB), carbon nanotubes (CNTs), or conformal carbon coating are used to improve the conductivity of the electrodes while offering efficient electric conduction path.11 However, the methods for synthesizing nanostructured silicon involve complex, high cost and high energy-consuming process, which limit their scalability in industrial-level manufacturing. Moreover, the conductive additives potentially increase the cost of the high-throughput fabrication.
The binders, holding the active materials together onto the current collectors, have attracted increased attention to improve the performance of Si anodes. Due to the large volume change of Si during charging and discharging, an effective binder is aimed to have strong interaction with the surface of active materials rather than weak van der Waals forces to accommodate the vast volume change.12 Binders such as PAA, carboxymethyl cellulose (CMC), alginate and carboxymethyl chitosan have been found to enhance the cycling life compared with the traditional polyvinylidene difluoride (PVDF) binder.13–15 The improved binding performance of those binders is primarily attributed to the high polymer modulus values, and the binding between the functional groups on polymer chains and the native oxide on the Si particles. However, the interaction is physically weak bond along the polymer chains while the one-dimensional polymer chains tends to slide upon the repeated volume change of silicon, which lead to the cracking of electrode during lithiation and delithiation.16 To improve the electrochemical performance of Si anodes, three-dimensional (3D) cross-linked binders have been developed to offer a robust network to capture Si with chemical interactions. Hybridized networks and calcium-mediated cross-linked alginate binders have been used as novel gels for Si slurry.17 Self-healing polymers via supramolecular interactions have been proposed for Si binders.18 Wang et al. reported a superabsorbent PAA–PVA gel for Si anodes, which effectively accommodated the volume change due to the adhesively transformable polymer network.19 However, the low conductivities of the polymer gel binders restrict the electron transportation efficiency, which lower the fast charge and discharge rate of the Si electrodes. The incorporation of conductive additives also increase the burden of holding Si particles and those carbonaceous agents, decreasing the capability to accommodate large volume change at the same amount of binders.
Polymers combining excellent electronic conductivity and cross-linked gel-structure to trap Si particles are encouraging binders for Si anodes. An interesting concept was demonstrated by poly(9,9-dioctylfluorene-co-suorenone-comethylbenzoic acid) (PFFOMB) binder, which not only forms continuous framework to capture Si, but also provides conduction path for electron transport.20 In recent, conductive polymers have been investigated as favorable alternative matrices to resistive binders for Si encapsulation. Of notable examples, PANI and PPy have been used to bind SiO2/Si surface adhesively and offer effective electron transport networks, resulting in impressive cycle lifes at high current densities.21,22 Herein we attempt to systematically compare and understand the roles of typical conductive polymer and non-conductive polymer binders on electrochemical performance of Si anodes. PPy- and PANI-coated Si nanoparticles via facile and scalable in situ polymerizations, and Si–PAA blend via mechanical mixing were casted and assembled as anodes. Since commercial Si nanoparticles were used as the common template for PPy, PANI and PAA to form composites, it was a good model to compare the effect of thickness and conductivity of those polymers on the anode performance excluding the factor of various degrees of silicon volume expansion. After 450 cycles at C/2 (1C = 3.6 A g−1), a decreased trend of cycling stability and rate capacity is in the order of PPy > PANI > PAA. For simplicity, the electrodes made from PPy-, PANI-coated and PAA-blended Si are referred as Si–PPy, Si–PANI and Si–PAA.
2. Experimental
2.1 Materials synthesis
The Si–PPy composite was prepared via a solution-based sol–gel polymerization process as follows: phytic acid (50 wt% in H2O, Sigma Aldrich) was mixed with pyrrole monomer (98% reagent grade, Sigma Aldrich) in 0.8 ml isopropanol (IPA) to produce a gel in a molar ratio of 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (phytic acid: 66.4 μL, pyrrole: 10.0 μL). 32.9 mg ammonium persulfate (APS) dissolved in 0.3 ml deionized water, acting as the oxidant was then added to the phytic acid/pyrrole solution and vortexed the mixture for 1 min for ample mixing. The molar ratio of phytic acid/pyrrole/APS is 0.5:1:1. After that, the solution was immediately transferred to a vial containing 60 mg Si nanopowder for in situ polymerization. The rich hydroxyl groups (–OH) contained in the six phosphoric acid groups of phytic acid bind with the native SiO2 on Si surfaces via hydrogen bonding, resulting in the continuous polymerization and the further crosslinking of monomers across Si surfaces. These interactions contribute to the formation of a conformal and interconnected network structure to trap Si particles inside. Moreover, the phosphoric acid groups protonate the nitrogen groups on PPy chains to generate a positively charged conductive coating, while electrostatically interacting with the negatively charged surface oxide to form a seamlessly connected Si–PPy nanocomposite.21 After sonicating the mixture for 10 minutes at room temperature, a viscous green solution is produced. Si–PANI composite was prepared in ethanol/water solution, and the synthesis condition is same as that of Si–PPy hydrogel. The Si-to-PPy and Si-to-PANI ratios are both close to 7:3 in weight by TGA measurements. To compare the effects between conductive polymers and resistive polymers on the electrochemical properties of Si anodes, PAA was selected as an insulating binder to mechanically mix with Si in a ratio of 7:3 (Si/PAA, w/w). Lastly, the formed hydrogel- and blended-slurries were used to doctor blade onto Cu foil current collectors and dried in vacuum for overnight at room temperature. The dried Si–PPy and Si–PANI electrodes were generally rinsed with deionized water to remove undoped phytic acid and excess oligomers, then dried in vacuum for the half cells as anodes.
:1 (phytic acid: 66.4 μL, pyrrole: 10.0 μL). 32.9 mg ammonium persulfate (APS) dissolved in 0.3 ml deionized water, acting as the oxidant was then added to the phytic acid/pyrrole solution and vortexed the mixture for 1 min for ample mixing. The molar ratio of phytic acid/pyrrole/APS is 0.5:1:1. After that, the solution was immediately transferred to a vial containing 60 mg Si nanopowder for in situ polymerization. The rich hydroxyl groups (–OH) contained in the six phosphoric acid groups of phytic acid bind with the native SiO2 on Si surfaces via hydrogen bonding, resulting in the continuous polymerization and the further crosslinking of monomers across Si surfaces. These interactions contribute to the formation of a conformal and interconnected network structure to trap Si particles inside. Moreover, the phosphoric acid groups protonate the nitrogen groups on PPy chains to generate a positively charged conductive coating, while electrostatically interacting with the negatively charged surface oxide to form a seamlessly connected Si–PPy nanocomposite.21 After sonicating the mixture for 10 minutes at room temperature, a viscous green solution is produced. Si–PANI composite was prepared in ethanol/water solution, and the synthesis condition is same as that of Si–PPy hydrogel. The Si-to-PPy and Si-to-PANI ratios are both close to 7:3 in weight by TGA measurements. To compare the effects between conductive polymers and resistive polymers on the electrochemical properties of Si anodes, PAA was selected as an insulating binder to mechanically mix with Si in a ratio of 7:3 (Si/PAA, w/w). Lastly, the formed hydrogel- and blended-slurries were used to doctor blade onto Cu foil current collectors and dried in vacuum for overnight at room temperature. The dried Si–PPy and Si–PANI electrodes were generally rinsed with deionized water to remove undoped phytic acid and excess oligomers, then dried in vacuum for the half cells as anodes.
2.2 Physical characterization
The surface morphology is investigated using optical microscopy, scanning microscopy (SEM; leo-supra, 1550). Transmission electron microscopy (TEM, Titan Themis 300) operated at 300 kV is used to further characterize the purity and morphology of Si–PPy and Si–PANI composites. The chemical structures were examined using FT-IR from 4000 to 400 cm−1 at a resolution of 4 cm−1 (Equinox 55 FT-IR, Bruker, Billerica, MA). TGA measurements were performed in air with a flow rate of 50 ml min−1 from 25 °C to 1000 °C at a heating rate of 5 °C min−1. The phase identification was performed by X-ray diffraction (XRD, PANalytical Empyrean) from 10° to 80°. Electrochemical impedance spectroscopy (EIS) analyses were obtained with a Biologic VMPs.2.3 Electrochemical spectroscopy
The anode electrodes were prepared by doctor-blading processing of slurry on pre-cleaned Cu foil with a pre-area Si mass loading for 0.8–0.9 mg cm−2. A button-type (CR 2032) two-electrode half-cell configuration was used for the electrochemical measurements. Cells were assembled in an Argon-filled VAC Omni-lab glovebox with oxygen and H2O level below 0.5 ppm. Pure Li metal was used as the counter electrode. A Celgard 3501 porous PP membrane was used as the separator. In this work, 1 M LiPF6 salt in a mixture of FEC/DMC was used as the electrolyte for half cells. FEC as a co-solvent in carbonate based electrolyte promotes the formation of a conformal and thin SEI with low interface impedance and compatible interface.23 The LiO2, LiF and –CHF–OCO2-type compounds reduced from FEC-containing electrolytes improve the ionic transport capability, and the decomposition of these species lead to the formation of free lithium near Si, thus increasing the yield of lithiation.24,25 Moreover, FEC suppresses the further degradation of LiPF6.26 Consequently, the presence of FEC can substantially enhance the electrochemical performance Si–polymer anodes in terms of capacity, stability and cyclability. Cycling performance and galvanostatic charge–discharge were conducted on Arbin BT300 with a voltage window ranging from 0.01 to 1.0 V (vs. Li+/Li). Capacity and C-rate were determined using 1C = 3.6 A g−1. Cyclic voltammetry scans were tested at a fixed voltage window between 0.01 V and 1.0 V (vs. Li+/Li). Electrochemical impedance spectroscopy measurements were performed to evaluate the charge transfer performance of the anodes on a Biologic VMPs at a range between 0.01 Hz and 1 MHz.3. Results and discussion
In situ polymerization of conductive polymers has garnered significant attention due to its low cost and energy-saving processing at room temperature, compared with other high energy consumption methods operated at elevated temperature.27 The Si–PPy and Si–PANI hydrogel composites are prepared via one-step solution-based polymerization. A detailed schematic of synthesizing conducting hydrogel composites process is shown in Fig. 1. First, pyrrole (or aniline) and phytic acid are dissolved in isopropanol (or ethanol) referred as solution 1. In this light yellow solution, the six phosphoric acid groups from the phytic acid forms hydrogen bonding with nitrogen contained groups from pyrrole (or aniline). Next, ammonium persulfate (APS, oxidizer) in aqueous solution (solution 2) is added to solution 1 to form a sol–gel precursor. The mixed solution is immediately transfer into a vial containing 60 mg Si nanoparticles. The native oxide on the surface of Si binds phytic acid and pyrrole (or aniline) via hydrogen bonding and electrostatic interaction to generate continuous coating hydrogel. Then the mixed solution is sonicated at room temperature to promote polymerizing and crosslinking of monomers. The resulted mixture is a viscously green-gel slurry, which indicates the successful coating of PPy (or PANI) on Si.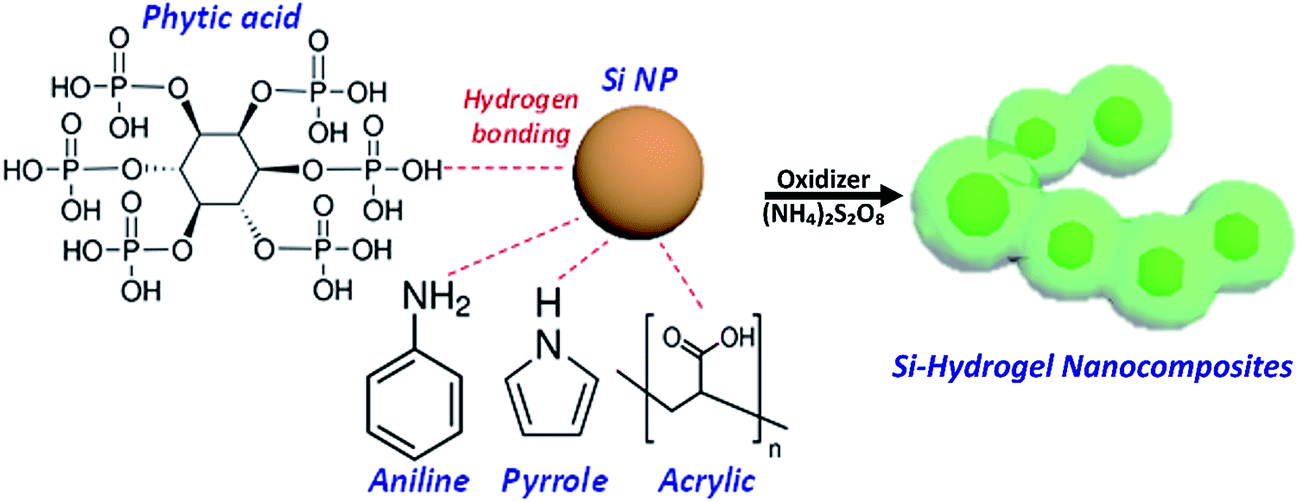 | ||
| Fig. 1 Schematic showing the polymerization route of Si-conductive polymer hydrogels. | ||
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) micrographs are shown to illustrate the structures and morphologies of pure Si nanoparticles, Si–PANI and Si–PPy in Fig. 2a–i. Due to the existence of hydrogen bonding and electrostatic interaction among the ternary components (Si, monomer and phytic acid in precursor), the polymerized PANI (or PPy) hydrogel produces seamless coating to encapsulate Si. Before polymerization, the Si nanoparticles (∼50–70 nm) exhibit a continuously spherical shape with clear edges in Fig. 2a–c. After coating with a layer of PANI (or PPy), the Si nano-clusters are all well-preserved by uniform coverings. A phase separation is observed between the surface of Si and hydrogel coating, and a thickness of 20–30 nm PANI shell is across Si as revealed in Fig. 2e and f. Moreover, the morphology of Si–PPy composite is similar to that of Si–PANI, while the coating thickness of PPy shell is very close to that of PANI coating in Fig. 2h and i.
 | ||
| Fig. 2 (a) SEM and (b, c) TEM images of Si nanoparticles. (d) SEM and (e, f) TEM images of Si–PANI composite. (g) SEM and (h, i) TEM images of Si–PPy composite. | ||
Thermogravimetric analysis (TGA) was performed to estimate the Si content in the Si–PPy and Si–PANI composites (Fig. 3). Upon heating up to 1000 °C under an air atmosphere, Si has an increase of 46.7% in weight due to the partial oxidation of Si particles; pure PPy gel has totally burned at a temperature around 700 °C with a residue of 7.8 wt%. For the TGA behavior of Si–PPy, the weight percentage of this composite drops to the minimum with the value of 83.9 wt% at ∼700 °C, due to the complete burn of the coated PPy hydrogel. Then the Si nanoparticles immediately start oxidation in air verified by the increased weight percentage. Hence, 700 °C is assumed to be the critical temperature for estimation of Si-hydrogel ratios. The Si content in the Si–PPy is calculated to be 70.4 wt% by using Si–PPy retention (83.9 wt%) subtracting the retained weight of PPy (7.7 wt%) and the slightly increased weight of Si (5.8 wt%) at 700 °C. Similarly, the Si content in PANI-coated Si is 66.5 wt%. Moreover, to make a fair comparison between conductive and resistive polymer binders on the electrochemical performance of Si anodes, the blend of Si–PAA is prepared in a weight ratio of 7:3. Therefore, Si–PPy, Si–PANI and Si–PAA composites exhibit the similar Si-to-polymer ratio, and the electrochemical performance of these three electrodes can effectively reveal the order of polymer effects on Si anodes at the same degree of Si volume expansion.
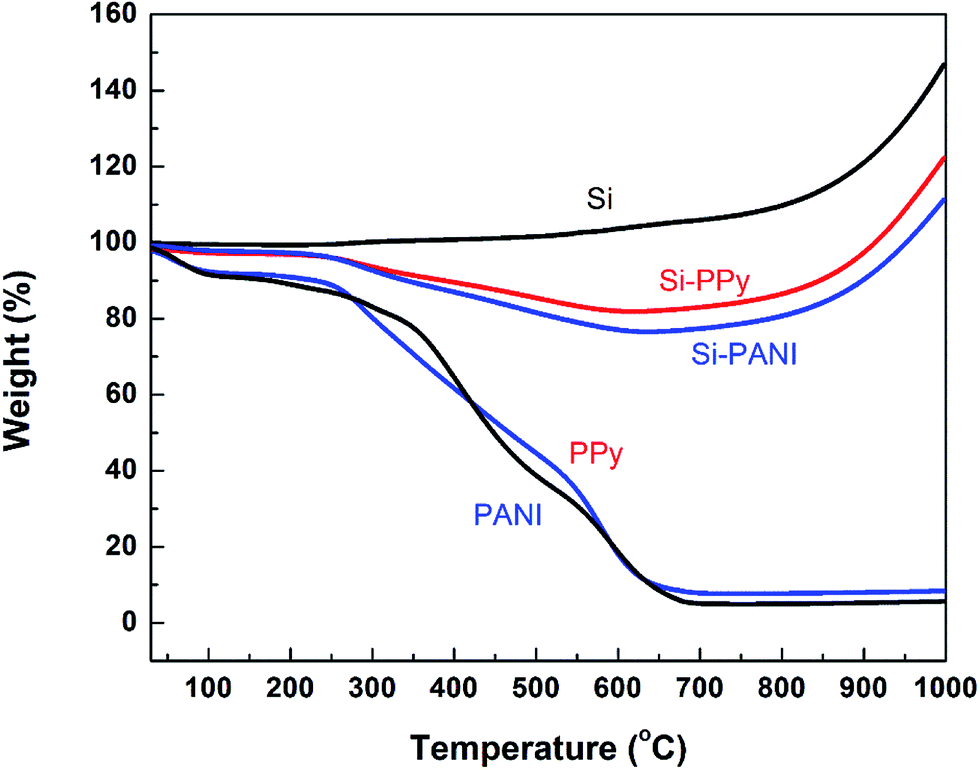 | ||
| Fig. 3 TGA curves of Si nanoparticles, pure PANI and PPy hydrogels, and Si–PANI and Si–PPy composites. | ||
X-ray diffraction (XRD) measurements were carried out to characterize the phase and purity of the as-prepared Si–PPy and Si–PANI composites (Fig. 4a). Both of the sample patterns exhibit narrow and sharp diffraction peaks without obvious amorphous scattering at 2θ of 28.6°, 47.7°, 56.4°, 69.4° and 76.4°, which correspond to the (111), (220), (311), (400) and (331) planes of Si crystals, respectively.11 A relatively weak peak at 23.5° observed in Si–PPy composite spectra, representing the (104) crystal plane of PPy, implies the successful polymerization of PPy coating for Si nanoparticles.28 For Si–PANI pattern, the peaks at 15.6° and 20.3° are associated with the periodicity parallel to the polymer chains, and the intensity peak located at 25.4° is attributed to the periodicity perpendicular to the polymer chains (π–π stacking), indicating the existence of PANI hydrogel.29 Fourier transform infrared spectroscopy (FTIR) is used to further examine the compositions of PPy- and PANI-coated Si composites in Fig. 4b. The transmittance peaks of PPy, the in-ring stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in pyrrole rings at 1548.1 cm−1, the vibrations of C–H at 1315.6 cm−1 and the C–C stretching at 1457.3 cm−1 are consistently observed in the spectrum of Si–PPy composite. Furthermore, the existence of characteristic peak at 1047.3 cm−1 originating from the in-plane deformation of C–H bond and N–H bond reveal the successful polymerization of PPy hydrogel to trap Si.30 The FTIR spectrum of Si–PANI indicates the synthesized hydrogel is emeraldine salt form of PANI with two characteristic peaks at 1480 and 1570 cm−1, corresponding to the stretching vibration of the benzenoid ring and quinoid ring.31–33
C bonds in pyrrole rings at 1548.1 cm−1, the vibrations of C–H at 1315.6 cm−1 and the C–C stretching at 1457.3 cm−1 are consistently observed in the spectrum of Si–PPy composite. Furthermore, the existence of characteristic peak at 1047.3 cm−1 originating from the in-plane deformation of C–H bond and N–H bond reveal the successful polymerization of PPy hydrogel to trap Si.30 The FTIR spectrum of Si–PANI indicates the synthesized hydrogel is emeraldine salt form of PANI with two characteristic peaks at 1480 and 1570 cm−1, corresponding to the stretching vibration of the benzenoid ring and quinoid ring.31–33
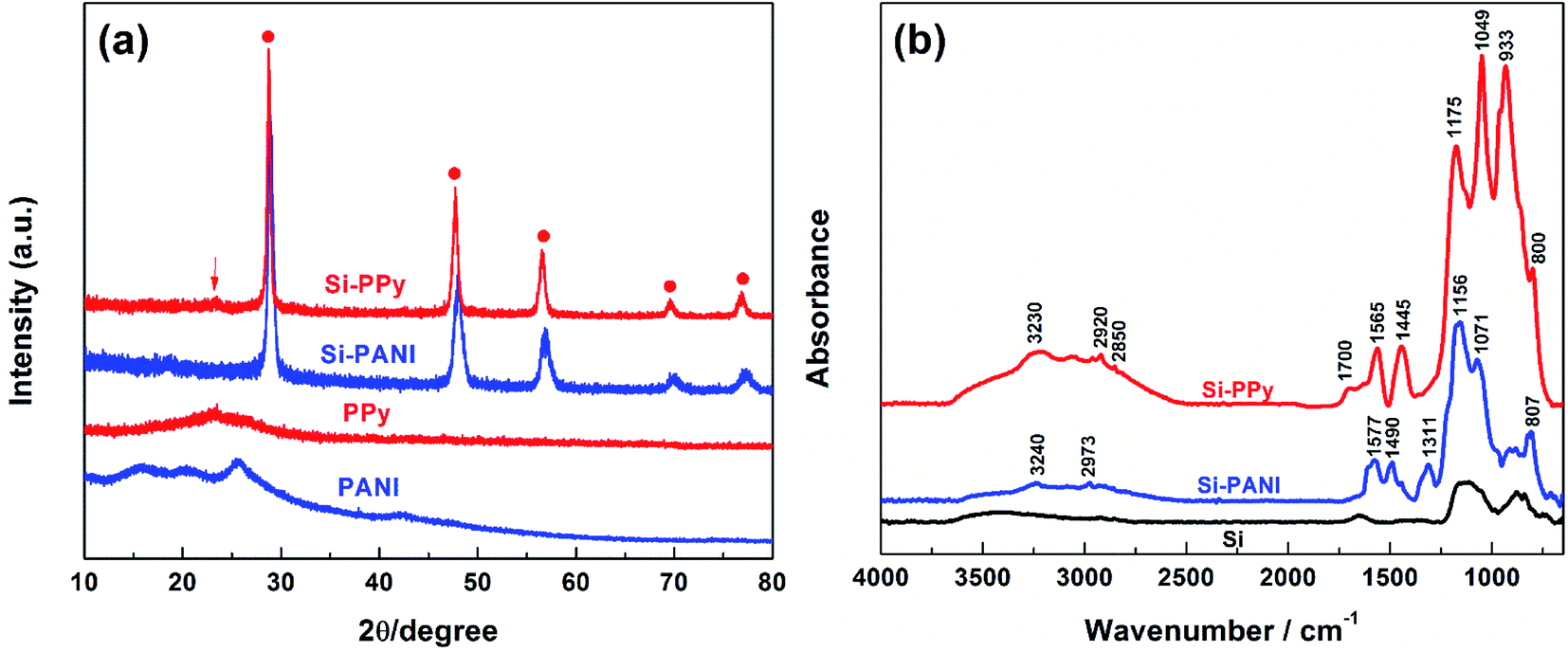 | ||
| Fig. 4 (a) XRD patterns and (b) FTIR spectra of Si–PANI and Si–PPy composites. | ||
LIB 2032-type half-cell batteries were assembled in an Ar-filled glove box (O2 < 0.1. ppm), (H2O < 0.5 ppm) with Si–PPy, Si–PANI and Si–PAA as the anodes, while pure Li metal as the counter electrodes. The electrodes were prepared by doctor-blading the well-mixed slurries onto copper foil as the active material. Conducting hydrogels act as both the binders for Si and the conductive additives to enhance electrical properties. Typical mass loadings of Si of all electrodes are 0.8–0.9 mg cm−2, and the specific capacities are calculated based on the Si mass. A porous polypropylene (PP) membrane was used as the separator. In our work, the electrolyte comprised 1 M LiPF6 dissolved in a 1:1 (v/v) mixture of FEC/DMC. The activation process of Si–PPy electrodes were performed by cyclic voltammetry (CV) with a voltage range between 0.01 and 1 V (vs. Li+/Li) at a scan rate of 0.2 mV s−1 in Fig. 5b. The peak in the potential of 0.45 V during the 1st discharge suggests the formation of SEI and is non-existent in the following cycles. The reduction peaks associated with the lithiation (0.21 V and 0.10 V) and the oxidation peaks owing to delithiation (0.38 V and 0.50 V) grow in intensity as the cycles increased, which indicate a slow motion of Li-ion between electrode and electrolyte before fully activating the active materials. The CV of 16th and 17th cycles closely coincides with each other, demonstrating the completion of kinetic enhancement. The existence of activation process is associated with the interaction of PPy hydrogel shell with electrolyte and the gradual alloying of the SiO2 surface and interior Si.7 The CV curves of Si–PANI and Si–PAA exhibit the similar behaviors as that of Si–PPy in Fig. S1a and b.†
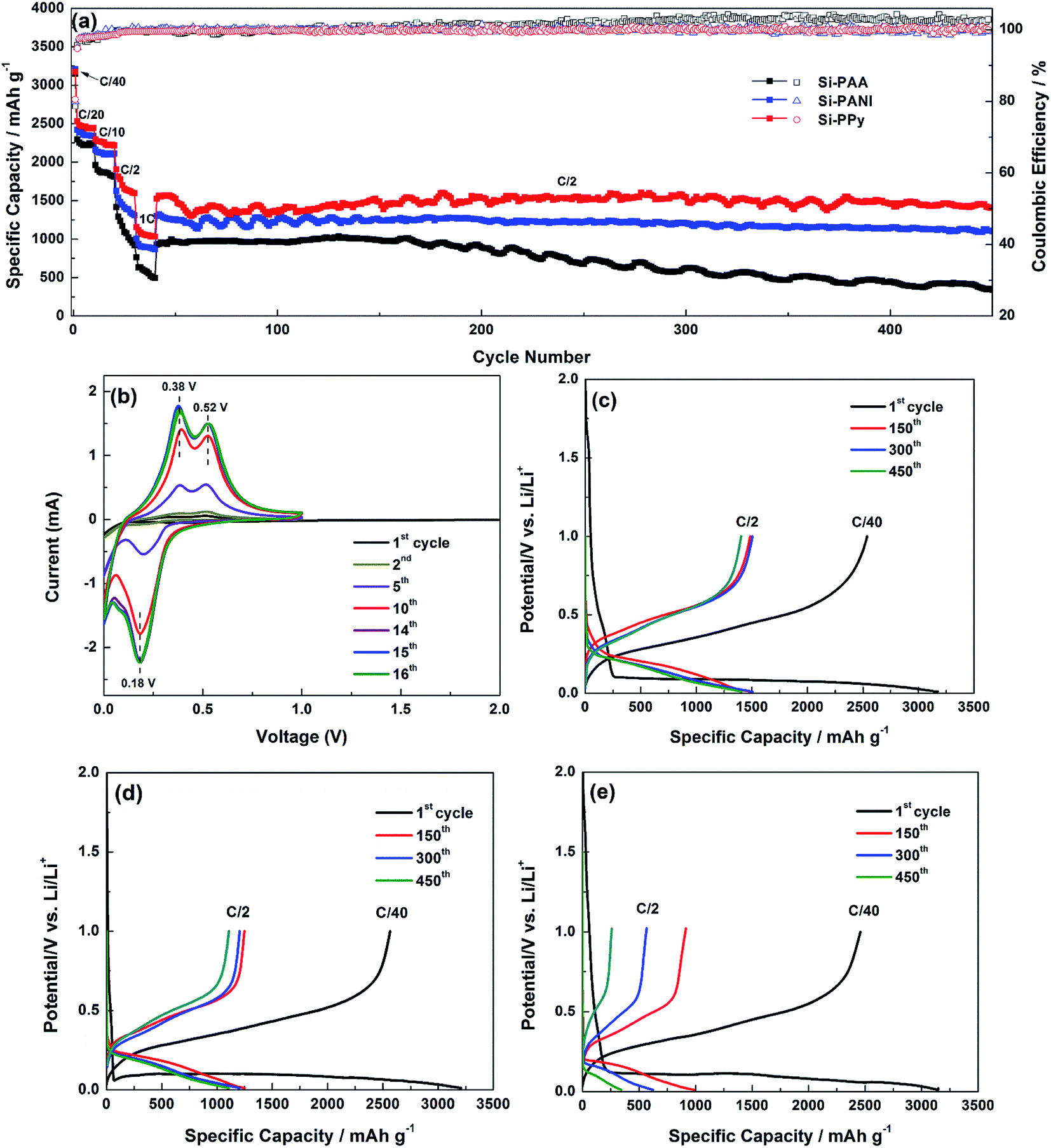 | ||
| Fig. 5 (a) Rate capability, cycling performance and coulombic efficiency of Si–PPy, Si–PANI and Si–PAA composite electrodes. (b) Cyclic voltammetry characteristic of Si–PPy electrodes. (c–e) Galvanostatic charge–discharge profiles of Si–PPy, Si–PANI and Si–PAA at selected cycles, respectively. | ||
Galvanostatic charge–discharge and cycling performance were measured in a voltage ranging from 0.01 to 1 V (vs. Li+/Li). Fig. 5a demonstrates the rate capability of the Si–PPy, Si–PANI and Si–PAA composite electrodes up to the 1C, with additional long-term cycling up to 450 cycles at C/2 (1C = 3.6 A g−1). The 1st cycle is performed at C/40, which is necessary for proper activation of the Si while fostering the formation of a stable SEI confirmed by the CV.34 The 1st discharge capacities of Si–PPy, Si–PANI and Si–PAA electrodes are ∼3170, 3200 and 3150 mA h g−1, corresponding to coulombic efficiencies of 80.6, 79.9 and 78.6%, respectively. The applied potential range (1–0.01 V) for alloying lithium with silicon is beyond the lowest unoccupied molecular orbital (LUMO) of the carbonate based LiPF6/FEC electrolyte (≈1 V). Therefore, an amorphous SEI forms due to the reductive decomposition of the electrolyte during the lithiation of Si under the applied voltage. In the initial lithiation, the native oxide covering the surface of Si is destroyed and generates an inner SEI primarily composed of LixSiOy and lithium ethylene dicarbonate (LEDC). An outer SEI mainly composed of LEDC and LiF forms as discharge continues. These SEI components containing lithium are stable under the applied potential window. Thus, the consumed Li can't be extracted completely from the established SEI components during delithiation, which leads to the irreversible capacity loss and low coulombic efficiency for the 1st cycle.26 The slightly higher coulombic efficiency in the initial charge–discharge of Si–PPy over those of Si–PANI and Si–PAA reveals the superior electrochemical compatibility of PPy hydrogel with Si and LiPF6/FEC electrolyte.
Subsequent cycling at C/20, C/10, C/2 and 1C demonstrate that the rate capabilities of the anodes are in the order: Si–PPy > Si–PANI > Si–PAA in Fig. 5a. This is also verified by the typical charge–discharge curves of these electrodes at selected rates, while the voltage hysteresis decreases in the order of Si–PAA > Si–PANI > Si–PPy during discharge curve at those rates in Fig. S2a–c.† At C/20, cells made from Si–PPy and Si–PANI display reversible capacities of ∼2450 and 2350 mA h g−1, respectively, which are higher than ∼2220 mA h g−1 exhibited by Si–PAA electrodes. When the rate is further increased to 1C, Si–PPy still achieves a high capacity of ∼1050 mA h g−1, revealing its fast kinetic motion of Li-ions between the electrode and electrolyte or within active materials of the electrode; however, Si–PANI and Si–PAA demonstrate lower capacities of 850 and 550 mA h g−1 at 1C. After comparing the rate capability in the initial cycles, an additional cycling stability test is followed at C/2. Si–PPy electrodes show a higher capacity than those of Si–PANI and Si–PAA anodes when the current is switched from 1C to C/2, indicating the superior robustness and flexibility of PPy as shells to encapsulate Si for accommodating large volume expansion and contraction. The anodes made from Si–PPy and Si–PANI demonstrate capacities of ∼1440 and 1230 mA h g−1 at C/2 after 150 cycles, while Si–PAA shows a lower capacity of ∼980 mA h g−1. Relative to the first cycle starting at C/2 for the long-term test, the capacity retention obtained at the 150 cycles for these anodes declined in the order of Si–PPy (98.7%) > Si–PANI (98.6%) > Si–PAA (98.4%). Even after 450 cycles, reversible capacities of ∼1400, 1100 and 340 mA h g−1 are still retained for Si–PPy, Si–PANI and Si–PAA electrodes in Fig. 5c–e, respectively, corresponding to capacity retentions of 92.4%, 85.3% and 36.7% of their initial capacity for cycling test. It has been reported that PPy and PANI react with Li-ions in the voltage window of 2.5–4 V, which indicates their stable electrochemical properties operated at the voltage window of 0.01–1 V for Si anodes.35 Consequently, the better rate performance of Si-hydrogels over that of nonconductive binder (PAA)–Si blend can be attributed to their faster electron and ion transport capability. Moreover, PPy hydrogel has superior physical confinement on Si compared with PANI confirmed by the more stable cyclability of Si–PPy electrodes, while PPy network have a higher electronic conductivity over that of PANI as conductive binders, which is indicated by the higher specific capacities of Si–PPy electrodes at high current densities.
The electrode kinetics are mainly related to the conductivity of the whole electrode.4 In our system, Si–PPy and Si–PANI composites not only have the same structures, but also the thicknesses of conducting hydrogels on Si are close. Therefore, the variations of rate capability of Si–PPy, Si–PANI and Si–PAA electrodes are primarily affected by the conductivities of those polymer binders. Electrochemical impedance spectroscopy (EIS), a powerful electroanalytical route for studying the kinetics occurring in the electrode of Li-ion batteries, has been carried out to better understand the different rate capability and cyclability of Si–PPy, Si–PANI and Si–PAA composites, and their Nyquist plots of these three electrodes are shown in Fig. 6, respectively. The intercept at high frequency (HF) is generally interpreted as equivalent series resistance (ESR or Rs), which is associated with the ohmic portion of the electrode impedance, the ionic conductivity of electrolyte, the overall electronic conductivity of the electrodes, as well as any electronic contact resistances related to current collectors, electrode materials and cell hardware.36 As shown in Fig. 6b and c, all the Nyquist plots of Si–PPy, Si–PANI and Si–PAA electrodes consist of two depressed semicircles in HF region, one semicircle in the middle frequency region coupled with a slope line in the low frequency region. The generally accepted interpretation of the two HF depressed semicircles is that one semicircle represents the SEI film resistance, while another HF semicircle is related to the resistance of the interphase electronic contacts between the current collector and the active materials/conductive additives/binder (CPEsei+int and Rsei+int). The depressed semicircle in the mid-frequency region is interpreted as the charge-transfer resistance contributing from the reaction at the interface of electrolyte and active material (Rct).37 The linear diffusion drift responsible for circuit element at low-frequency (<200 mHz) is Warburg impedance tail (Wo), which is associated with the diffusion of salt in the electrolyte and the diffusion of Li-ions into active materials on the electrode. All the resistances at different frequencies are useful in understanding the kinetics of various anodes.38
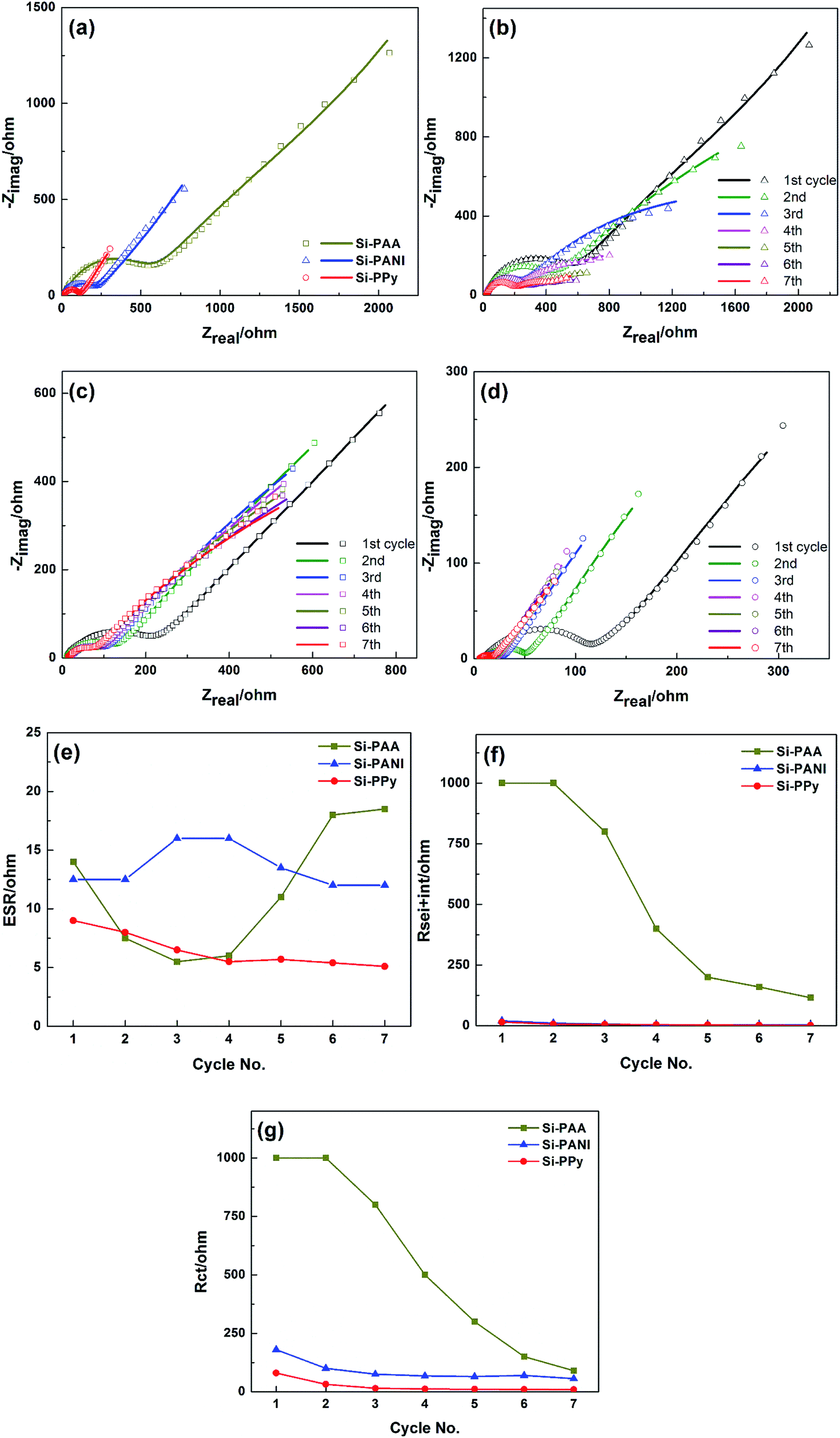 | ||
| Fig. 6 (a) Nyquist plots of Si–PPy, Si–PANI and Si–PAA based LIB cells in the first EIS cycle. (b–d) Nyquist plots of Si–PPy, Si–PANI and Si–PAA over 7 charge–discharge cycles, respectively. (e) Equivalent series resistance. (f) SEI and interphase electronic contact resistance. (g) Charge transfer resistance as a function of cycle number. | ||
The EIS plots of Si–PPy, Si–PANI and Si–PAA in the first cycle are shown in Fig. 6a. The ESR of those three composite systems are similar. However, it is worth noting that Si–PPy shows a slightly lower ESR value than those of Si–PANI and Si–PAA, while Si–PAA displays much higher interphase electronic contact resistance coupled with SEI resistance (Rsei+int) as those of Si–PPy and Si–PANI electrodes, indicating the conductivity of the binders and the compatibility of formed SEI decreased in the order of PPy > PANI > PAA. To further investigate the EIS evolution and capacity fade mechanism, EIS measurements were performed with 7 continuous cycles for all Si–polymer systems, as shown in Fig. S3a–f.† Gaberscek et al. demonstrated that the magnitude of the HF semicircle of Nyquist plot of negative electrodes reduced as the increase in external load, which is in agreement with the behaviors of our Si–polymer electrodes EIS spectra.39 Dees et al. reported that superior interphase electronic contact decreases the HF impedance semicircle.40 Based on the behavior of our EIS spectra and the previous studies, an equivalent circuit for our Si–polymer systems is proposed, as shown in Fig. S4a–c.† The fitted impedance plots and their parameters including ESR, Rsei+int and Rct using this equivalent circuit are shown in Fig. 6b–d, respectively.
All three Si–polymer composites present low and stable ESR values with a small fluctuation in the first 7 cycles. The ESRs of Si–PPy of Si–PANI slowly decrease and then gradually stabilize afterwards, signifying the formation of stable SEI and the activation of active materials. In contrast, the dramatic increase in ESR of Si–PAA after the 3rd cycle indicates continuous reformation of SEI due to the volume change of non-preserved Si. Fig. 6f shows the SEI film resistance as function of cycle number. The SEI resistances of Si–PPy and Si–PANI start at a similar level and both exhibit fluctuations with a small range in the first 4 cycles and then stabilized at 1–3 Ω in the 7th cycle. The decrease in SEI resistance in the first 7 cycles is primarily owing to the effective encapsulation of Si nanoparticles by conductive coatings and the compatible electrochemical interface between polymer shells and SEI. Although the SEI resistance of Si–PAA decreases to a stable value, the large drop of SEI resistance reveals the formation of SEI consumes large amount of electrolyte. The magnitudes of the SEI resistance for Si–PPy, Si–PANI and Si–PAA are 1.5, 3 and 115 Ω, respectively, which are consistent with their initial irreversible capacity sequence of Si–PPy (19.4%) < Si–PANI (20.1%) < Si–PAA (21.4%). In addition, all three Si–polymer systems exhibit continuous decrease in Rint during the first 7 cycles in Fig. 6f, which is associated with the irreversible electrodes volume increase via the insertion/extraction of Li ions. The gradually increased thickness of active materials improved the pressure on electrodes pressed by a spring in the coin cell, which results in the better electronic contact between current collector and the Si–polymer electrodes. It is noteworthy that Si–PPy composite demonstrates lower Rint than those of Si–PANI and Si–PAA, confirming the superior conductivity of PPy as a binder. As shown in Fig. 6g, the charge transfer resistances of Si–PPy and Si–PANI decrease as the cycle increases, and Si–PPy shows a fast drop in a low resistance range. On the contrary, the Rct of Si–PAA still demonstrates much higher Rct after 7 cycles compared to those of Si–PPy and Si–PANI electrodes. Since the charge transfer reaction primarily takes place on the surface of Si, the electronic contact between Si and polymer has great impact on the charge transfer reaction. Accordingly, compared with insulating PAA as binder for Si, conducting hydrogels not only form uniform coating on Si, but also generate an interconnected conductive network to bridge the whole active materials, which facilitate the charge transfer and minimize the degradation of electronic contact of Si-hydrogels. Furthermore, compared to Si–PPy and Si–PANI anodes, the low frequency tails of Si–PAA, related to diffusion impedance, is shifted off 45° as the cycle increases, indicating the slower and more inefficient diffusion of Li-ions into electrode when using resistive polymers as binders.
4. Conclusion
We have successfully demonstrated the synthesis of conducting hydrogels coated Si composites via a facile and scalable in situ polymerization process. Resistive PAA–Si blend is used as control to investigate the difference of electrochemical performance with Si–PPy and Si–PANI composites. It is found that Si-conducting hydrogel systems showed superior cycling stability and rate capability than that of Si-insulating polymer composite in a decreased order of PPy > PANI > PAA in term of the polymer-binder effects. EIS is employed to verify that the excellent cyclability and rate performance of Si–PPy composite is related to its better conductivity, the formation of a more compatible interface between PPy shell and LiPF6/FEC electrolyte, and its superior kinetic capability that permits the fast motion of Li-ions between the electrode and electrolyte or within active materials.Acknowledgements
Financial support for this work was provided by the Office of the Vice Chancellor for Research at the University of California Riverside.References
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 310–315 CrossRef CAS PubMed.
- W. Wang, S. Guo, M. Penchev, I. Ruiz, K. N. Bozhilov, D. Yan, M. Ozkan and C. S. Ozkan, Nano Energy, 2013, 2, 294–303 CrossRef CAS.
- C. Liu, C. Li, K. Ahmed, W. Wang, I. Lee, F. Zaera, C. S. Ozkan and M. Ozkan, Adv. Mater. Interfaces, 2016, 3, 1500503 CrossRef.
- W. Wang, Z. Favors, C. Li, C. Liu, R. Ye, C. Fu, K. Bozhilov, J. Guo, M. Ozkan and C. S. Ozkan, Sci. Rep., 2017, 7, 44838 CrossRef CAS PubMed.
- C. Li, C. Liu, W. Wang, J. Bell, Z. Mutlu, K. Ahmed, R. Ye, M. Ozkan and C. S. Ozkan, Chem. Commun., 2016, 52, 11398–11401 RSC.
- Z. Favors, W. Wang, H. H. Bay, Z. Mutlu, K. Ahmed, C. Liu, M. Ozkan and C. S. Ozkan, Sci. Rep., 2014, 4, 5623 CrossRef CAS PubMed.
- C. Liu, C. Li, W. Wang, M. Ozkan and C. S. Ozkan, Energy Technol., 2017, 5, 422–427 CrossRef CAS.
- W. Wang, Z. Favors, R. Ionescu, R. Ye, H. H. Bay, M. Ozkan and C. S. Ozkan, Sci. Rep., 2015, 5, 8781 CrossRef CAS PubMed.
- J. Cho, J. Mater. Chem., 2010, 20, 4009–4014 RSC.
- C. Li, C. Liu, W. Wang, Z. Mutlu, J. Bell, K. Ahmed, R. Ye, M. Ozkan and C. S. Ozkan, Sci. Rep., 2017, 7, 917 CrossRef PubMed.
- N. Lin, J. Zhou, L. Wang, Y. Zhu and Y. Qian, ACS Appl. Mater. Interfaces, 2015, 7, 409–414 CAS.
- A. Magasinski, B. Zdyrko, I. Kovalenko, B. Hertzberg, R. Burtovyy, C. F. Huebner, T. F. Fuller, I. Luzinov and G. Yushin, ACS Appl. Mater. Interfaces, 2010, 2, 3004–3010 CAS.
- J. Li, R. B. Lewis and J. R. Dahn, Electrochem. Solid-State Lett., 2007, 10, A17–A20 CrossRef CAS.
- J. Liu, Q. Zhang, Z.-Y. Wu, J.-H. Wu, J.-T. Li, L. Huang and S.-G. Sun, Chem. Commun., 2014, 50, 6386–6389 RSC.
- L. Wei, C. Chen, Z. Hou and H. Wei, Sci. Rep., 2016, 6, 19583 CrossRef CAS PubMed.
- X. Yu, H. Yang, H. Meng, Y. Sun, J. Zheng, D. Ma and X. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 15961–15967 CAS.
- C. Wang, H. Wu, Z. Chen, M. T. McDowell, Y. Cui and Z. Bao, Nat. Chem., 2013, 5, 1042–1048 CrossRef CAS PubMed.
- J. Song, M. Zhou, R. Yi, T. Xu, M. L. Gordin, D. Tang, Z. Yu, M. Regula and D. Wang, Adv. Funct. Mater., 2014, 24, 5904–5910 CrossRef CAS.
- G. Liu, S. Xun, N. Vukmirovic, X. Song, P. Olalde-Velasco, H. Zheng, V. S. Battaglia, L. Wang and W. Yang, Adv. Mater., 2011, 23, 4679–4683 CrossRef CAS PubMed.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 Search PubMed.
- B. Liu, P. Soares, C. Checkles, Y. Zhao and G. Yu, Nano Lett., 2013, 13, 3414–3419 CrossRef CAS PubMed.
- V. Etacheri, O. Haik, Y. Goffer, G. A. Roberts, I. C. Stefan, R. Fasching and D. Aurbach, Langmuir, 2011, 28, 965–976 CrossRef PubMed.
- C. Xu, F. Lindgren, B. Philippe, M. Gorgoi, F. Björefors, K. Edström and T. r. Gustafsson, Chem. Mater., 2015, 27, 2591–2599 CrossRef CAS.
- S. Dalavi, P. Guduru and B. L. Lucht, J. Electrochem. Soc., 2012, 159, A642–A646 CrossRef CAS.
- K. Schroder, J. Alvarado, T. A. Yersak, J. Li, N. Dudney, L. J. Webb, Y. S. Meng and K. J. Stevenson, Chem. Mater., 2015, 27, 5531–5542 CrossRef CAS.
- L. Karen, C. Nicha, E. Cristina, L. Changling, B. Wayne, V. M. Nosang and N. Jin, Nanotechnology, 2014, 25, 115501 CrossRef PubMed.
- H. Zhou, T. Ni, X. Qing, X. Yue, G. Li and Y. Lu, RSC Adv., 2014, 4, 4134–4139 RSC.
- K. Wang, X. Zhang, C. Li, H. Zhang, X. Sun, N. Xu and Y. Ma, J. Mater. Chem. A, 2014, 2, 19726–19732 CAS.
- Y. Lu, W. He, T. Cao, H. Guo, Y. Zhang, Q. Li, Z. Shao, Y. Cui and X. Zhang, Sci. Rep., 2014, 4, 5792 CrossRef PubMed.
- L. Pan, G. Yu, D. Zhai, H. R. Lee, W. Zhao, N. Liu, H. Wang, B. C.-K. Tee, Y. Shi, Y. Cui and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9287–9292 CrossRef CAS PubMed.
- C. Li, N. Chartuprayoon, W. Bosze, K. Low, K. H. Lee, J. Nam and N. V. Myung, Electroanalysis, 2014, 26, 711–722 CrossRef CAS.
- K. Low, C. B. Horner, C. Li, G. Ico, W. Bosze, N. V. Myung and J. Nam, Sens. Actuators, B, 2015, 207, 235–242 CrossRef CAS.
- Z. Favors, H. H. Bay, Z. Mutlu, K. Ahmed, R. Ionescu, R. Ye, M. Ozkan and C. S. Ozkan, Sci. Rep., 2015, 5, 8246 CrossRef CAS PubMed.
- W. Li, Q. Zhang, G. Zheng, Z. W. Seh, H. Yao and Y. Cui, Nano Lett., 2013, 13, 5534–5540 CrossRef CAS PubMed.
- C. Liu, C. Li, K. Ahmed, W. Wang, I. Lee, F. Zaera, C. S. Ozkan and M. Ozkan, RSC Adv., 2016, 6, 81712–81718 RSC.
- C. Liu, C. Li, K. Ahmed, Z. Mutlu, C. S. Ozkan and M. Ozkan, Sci. Rep., 2016, 6, 29183 CrossRef CAS PubMed.
- J. Guo, A. Sun, X. Chen, C. Wang and A. Manivannan, Electrochim. Acta, 2011, 56, 3981–3987 CrossRef CAS.
- M. Gaberscek, J. Moskon, B. Erjavec, R. Dominko and J. Jamnik, Electrochem. Solid-State Lett., 2008, 11, A170–A174 CrossRef CAS.
- D. Dees, E. Gunen, D. Abraham, A. Jansen and J. Prakash, J. Electrochem. Soc., 2005, 152, A1409–A1417 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06023h |
| This journal is © The Royal Society of Chemistry 2017 |