
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The superior desorption properties of MgCl2-added ammonia borane compared to MgF2-added systems—the unexpected role of MgCl2 interacting with [NH3] units†
Xiaoli Ding,
Jingjing Feng,
Tianlai Xia,
Xiaomin Yuan*,
Dongming Liu,
Yongtao Li * and
Qingan Zhang
* and
Qingan Zhang
School of Materials Science and Engineering, Anhui University of Technology, Maanshan, 243002, China. E-mail: yuan@ahut.edu.cn; toni-li@163.com; Fax: +86-555-2311570; Tel: +86-555-2311570
First published on 25th July 2017
Abstract
An uncommon dehydrogenation mechanism for metal chloride-added ammonia borane (NH3BH3, AB) systems revealed that MgCl2 interacts with the [NH3] units in AB, analogous to the amine complex of Mg(NH3)xCl2, thus not only resulting in a remarkable decrease in the hydrogen desorption temperature but also effectively suppressing undesirable volatile by-products, particularly ammonia gas.
Using hydrogen as an energy carrier is severely fettered by the key fact that there is still no safe and effective method available for storing hydrogen.1,2 To overcome this, significant effort has been focused on the possibility of using hydrides and their complexes as a solid form of hydrogen storage.3–7 Among them, chemical hydrides, particularly ammonia borane (NH3BH3, AB), have been considered as promising hydrogen storage candidates due to its intrinsic high capacity of 19.6 wt%.8,9 More importantly, solid state AB can decompose below 100 °C and can be regenerated via reacting with hydrazine in liquid ammonia at a rather slow rate.10–12 Unfortunately, abundant volatile impurities such as ammonia (NH3), diborane (B2H6) or borazine (N3B3H6) are released along with H2,13 which not only decreases the yield of H2 but also poisons the noble metal catalysts and Nafion membrane used in fuel cells.14,15 Thus, substantial effort has been devoted to suppressing undesirable volatile products to improve the yield of H2 and further promote its practical use as a hydrogen supply to fuel cells.
One effective approach to improve hydrogen release from AB is the chemical modification of its electron donor–acceptor complex structure by adding chloride salts. In an early study, Heldebrant et al.16 reported that the introduction of NH4Cl to AB forms an intermediate [(NH3)2BH2]+Cl−, which results in the rapid release of hydrogen below 90 °C. Later, Chen et al.17 found that approximately 5.8 wt% hydrogen is evolved from CoCl2-doped AB at a temperature as low as 59 °C due to the catalytic effect of partially reduced Co active species. In a subsequent report, Jagirdar et al.18 proposed that by milling AB with CuCl2 we can achieve facile desorption at about 60 °C through an intermediate [NH4]+[BCl4]− phase without borazine emission. Following these observations, our recent study showed that the incorporation of alkaline earth metal chlorides into AB can further decrease the desorption temperature to be as low as 50 °C and suppress the formation of undesirable volatile by-products.19 However, a rival determination of how the modification of alkali-earth metal chlorides into AB decomposition occurs is lacking and the underlying mechanism is not well understood. More recently, Nakagawa et al.20 further found that the Mn+ of MCln with high electronegativity (χp) acts as a Lewis acid and initiates the AB dehydrocoupling reaction by inducing changes in the electronic state of N. Different from all of the above studies, using a comparative mechanism study among pristine, MgCl2- and MgF2-added AB systems, we obtained results presented here that reveal a new dehydrogenation mechanism. In the mechanism, the MgCl2 unexpectedly interacts with the [NH3] units in AB, analogous to the amine complex of Mg(NH3)xCl2, which results in lowering the thermodynamic threshold as well as concomitantly inhibiting undesirable volatile by-products.
The synchronous MS and TG profiles in Fig. 1 show the hydrogen desorption properties of the pristine AB and ball-milled MgX2 (X = F, Cl)/AB (molar ratio, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) samples. For the detected MS spectrum of H2 (Fig. 1a), two consecutive peaks at ∼110 and ∼154 °C were observed for the pristine AB, being consistent with the reported values.21 Similar peaks somewhat downshifted were also determined in the MgF2/2AB sample. Compared to them, an evident shift to a lower temperature was obtained in H2-MS spectrum for the MgCl2/2AB sample, whereby the release of hydrogen started at approximately 40 °C, with the first peak appearing at approximately 60 °C (similar to the decomposition of a SrH2 + AB mixture22) and was completed below 130 °C (see Fig. S1 (ESI†) for partial enlarged details). More excitingly, considerable amounts of volatile by-products including NH3, B2H6 and N3B3H6 were obtained for both the pristine AB and MgF2/2AB samples, while not detectable or negligible volatile by-products were observed for the MgCl2/2AB sample. These remarkable suppressions of volatile by-products were further demonstrated by the reduced weight loss detected by TG analysis. As shown in Fig. 1b, the weight loss of ∼64 wt% for pristine AB was in two steps and was observed at around 108 and 155 °C, along with the severe expansion that pushed up the lid of the crucible (see photograph in Fig. 1b). This should be attributed to the decomposition of AB into polymeric aminoboranes ((NH2BH2)n, PAB) and polymeric iminoboranes ((NHBH)n, PIB) solid residues.15,23 Similarly, the MgF2/2AB sample also exhibited a two-step decomposition with an inflated feature. Its weight loss of ∼34 wt% was comparable with the weight loss from AB in the mixture (∼32 wt%). These obtained weight loss results were far larger than the expected H-content of 13 wt% in AB, further demonstrating the emission of undesirable volatile by-products, as shown in Fig. 1a. In contrast, the MgCl2/2AB did not exhibit any detectable foaming (see photograph in Fig. 1b), only with a weight loss of 12 wt%, which was close to the theoretical hydrogen capacity of AB (∼13 wt%). These tremendous contrasts presented by both the TG analysis and optical photographs strongly suggest the suppression of volatile by-products via their distinct decomposition routes.
:2) samples. For the detected MS spectrum of H2 (Fig. 1a), two consecutive peaks at ∼110 and ∼154 °C were observed for the pristine AB, being consistent with the reported values.21 Similar peaks somewhat downshifted were also determined in the MgF2/2AB sample. Compared to them, an evident shift to a lower temperature was obtained in H2-MS spectrum for the MgCl2/2AB sample, whereby the release of hydrogen started at approximately 40 °C, with the first peak appearing at approximately 60 °C (similar to the decomposition of a SrH2 + AB mixture22) and was completed below 130 °C (see Fig. S1 (ESI†) for partial enlarged details). More excitingly, considerable amounts of volatile by-products including NH3, B2H6 and N3B3H6 were obtained for both the pristine AB and MgF2/2AB samples, while not detectable or negligible volatile by-products were observed for the MgCl2/2AB sample. These remarkable suppressions of volatile by-products were further demonstrated by the reduced weight loss detected by TG analysis. As shown in Fig. 1b, the weight loss of ∼64 wt% for pristine AB was in two steps and was observed at around 108 and 155 °C, along with the severe expansion that pushed up the lid of the crucible (see photograph in Fig. 1b). This should be attributed to the decomposition of AB into polymeric aminoboranes ((NH2BH2)n, PAB) and polymeric iminoboranes ((NHBH)n, PIB) solid residues.15,23 Similarly, the MgF2/2AB sample also exhibited a two-step decomposition with an inflated feature. Its weight loss of ∼34 wt% was comparable with the weight loss from AB in the mixture (∼32 wt%). These obtained weight loss results were far larger than the expected H-content of 13 wt% in AB, further demonstrating the emission of undesirable volatile by-products, as shown in Fig. 1a. In contrast, the MgCl2/2AB did not exhibit any detectable foaming (see photograph in Fig. 1b), only with a weight loss of 12 wt%, which was close to the theoretical hydrogen capacity of AB (∼13 wt%). These tremendous contrasts presented by both the TG analysis and optical photographs strongly suggest the suppression of volatile by-products via their distinct decomposition routes.
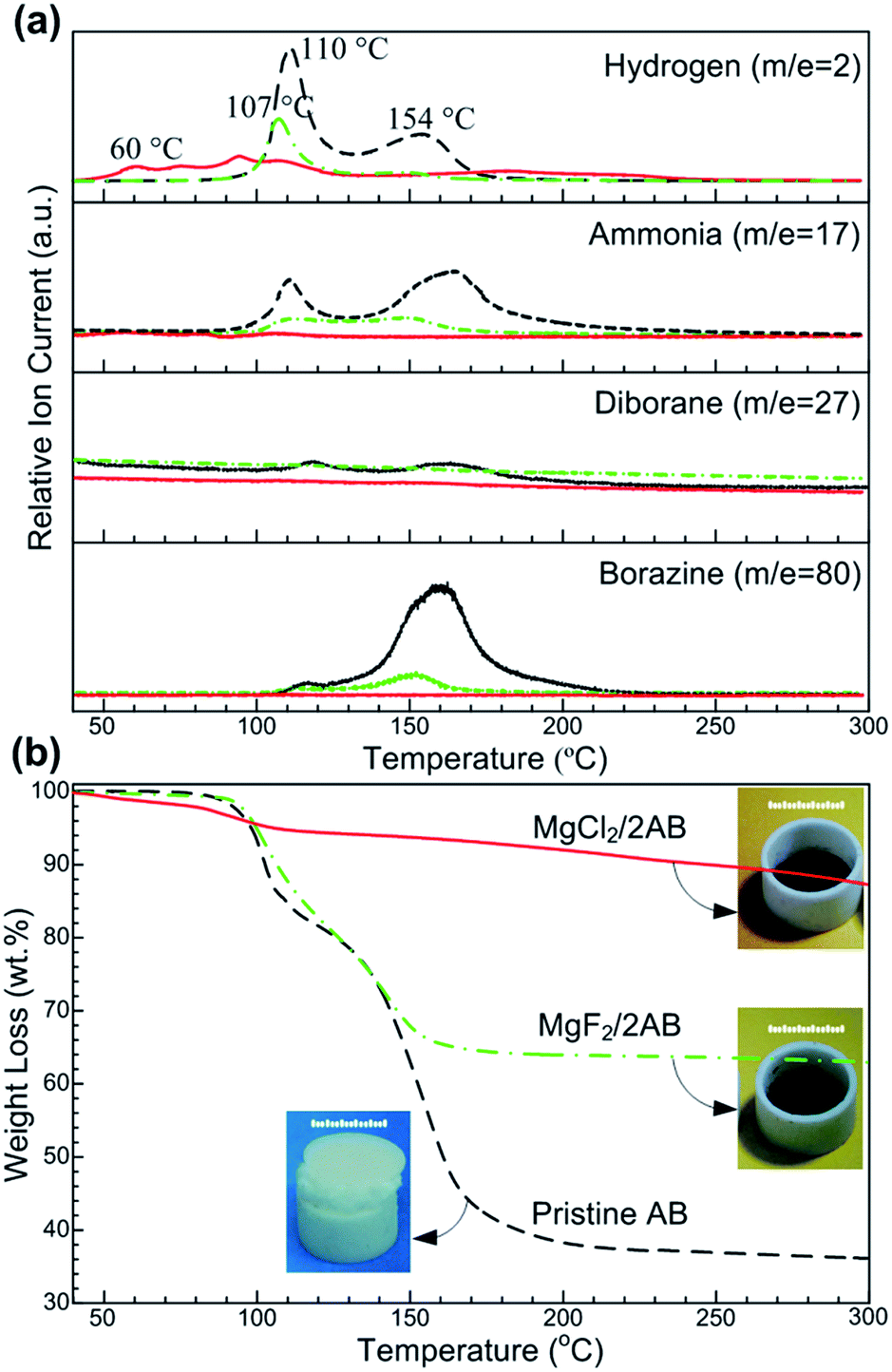 | ||
| Fig. 1 (a) The MS profiles of m/z = 2 (hydrogen, H2), 17 (ammonia, NH3), 27 (diborane, B2H6) and 80 (borazine, c-(NHBH)3) for pristine AB (black dashed line), MgF2/2AB (green dot-dashed line) and MgCl2/2AB (red solid line), and (b) their synchronous TG curves; the ramping rate was 5 °C min−1. The inserts are the optical photographs of these samples after heating in an argon flow. The sample loading was approximately 2.5 mg. The scale bar is 5 mm. | ||
To reveal the intrinsic reason for the superior desorption properties of MgCl2/2AB compared to MgF2/2AB and pristine AB, the structural features of the samples during the milling and heating processes were examined carefully. First, the milled samples were examined by XRD and Raman scattering spectroscopy. It was found that all the peaks can be indexed by the starting phases of AB, MgCl2 and MgF2 (see Fig. 2a) without new peaks being detected, indicating that the solid-state reaction between AB and MgX2 did not occur upon the initial milling step. However, the chemical bonding states of these samples exhibited different results. As shown in Fig. 2b, all the vibrational modes of the typical B–H stretches at ∼2281 and 2380 cm−1, and the N–H stretches at ∼3251 and 3318 cm−1 for the MgX2/2AB samples became significantly broader and weaker relative to those modes for the pristine and post-milled AB (see Fig. S2 (ESI†)), suggesting the reduced bonding stability.24 More interestingly, an evident upfield shift for the B–H stretches and a downfield shift for the N–H stretches were observed for MgCl2/2AB compared with the shifts observed for the pristine AB and MgF2/2AB samples, which strongly indicates that the chemical modification was started during the solid phase ball-milling step upon adding MgCl2. The changed bonding structure caused by MgCl2 but not MgF2 may alter the reaction pathway of AB, thus leading to favourable thermal decomposition behaviour, as shown in Fig. 1.
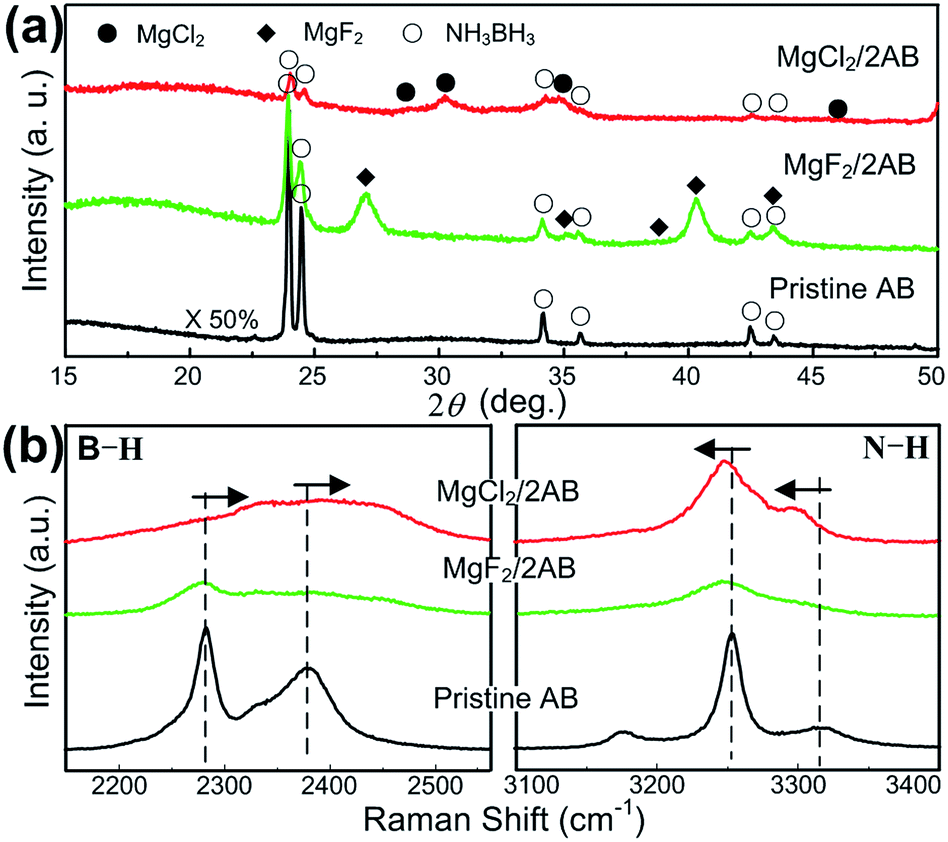 | ||
| Fig. 2 (a) The XRD patterns and (b) Raman spectra recorded for the post-milled AB, MgCl2/2AB and MgF2/2AB samples without heating. | ||
Second, the MgX2/2AB samples after heating at 130 °C were further analysed by XRD and solid-state NMR techniques to determine the difference in their phase and bonding states. As shown in Fig. 3a, only MgF2 peaks were observed in the MgF2-doped samples after heating, indicating the single AB decomposition and its resulting products with an amorphous nature. Different from it, only some unknown phases that did not match both the initial AB and MgCl2 were obtained in the MgCl2-doped sample after heating, strongly suggesting the MgCl2 was involved in the desorption of AB but not for the MgF2-doped sample. Furthermore, except for the similar tridentate 11B NMR spectra that are possibly related to the single or two overlapping quadrupolar resonances of BN3, BN2H or even a mixture of both, an additional peak at approximately −35 ppm was obtained for the MgCl2-doped sample after heating, which can be most likely assigned to the unknown phase, as shown in Fig. 3a, i.e., the complex of MgCl2 with AB after heating at 130 °C.25,26
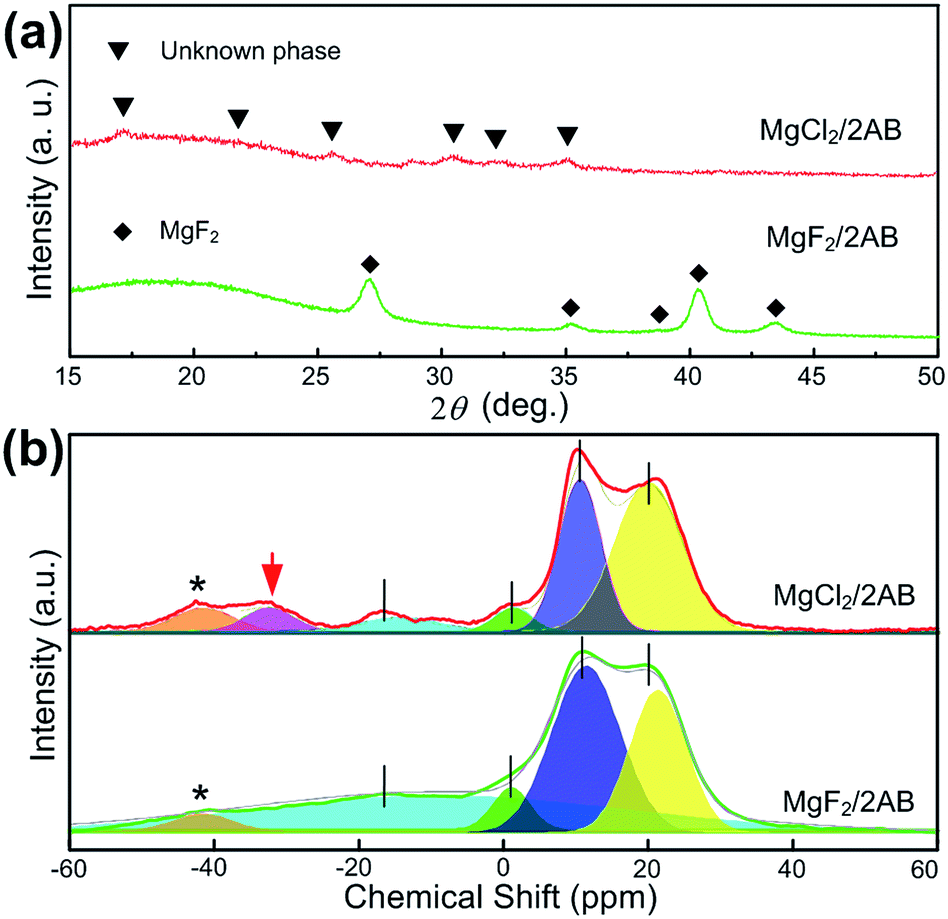 | ||
| Fig. 3 (a) The XRD patterns and (b) 11B NMR spectra for the MgF2/2AB and MgCl2/2AB samples after heating at 130 °C. The spinning sidebands are marked with asterisks (*). | ||
Third, we further employed in situ Raman spectroscopy to examine the entire process of thermal decomposition. Fig. 4 exhibits the in situ Raman spectra for pristine AB and MgX2/2AB during the heating process from room temperature to 250 °C. For the pristine AB sample, the symmetric stretching bands of the B–N, B–H and N–H bonds were gradually weakened at temperatures over 100 °C and completely disappeared at about 125 °C, which corresponded to the first emission of volatile by-products (Fig. 1a). The second emission of volatile by-products between 125 °C and 200 °C became illegible because of their initial remarkable relative intensity. Moreover, MgF2/2AB exhibited a similar curve except for two perpetual peaks at approximately 1250 and 3100 cm−1, which were most probably due to the fluorescent effect of MgF2. This explains why they showed similar thermal decomposition behavior (Fig. 1). In contrast, the B–N, N–H and B–H stretching bands weakened at around 40 °C for the MgCl2/2AB sample and were in good agreement with the TG/MS results shown in Fig. 1. The intensity of these bands rapidly decreased and the bands further disappeared as the temperature increased to 130 °C. More remarkably, with the disappearance of the B–N, N–H and B–H bands, two fresh bands emerged at 505 cm−1 and 916 cm−1, which could not be identified as any known B-, N- and Mg-containing compounds.27–29 With increasing temperature, the intensity of these fresh bands was observed to increase slightly. Combining the TG/MS (Fig. 1) and XRD/NMR (Fig. 2 and 3) results, the resulting product could be preliminarily deduced to be a Mg–N–B–Cl–H complex. In this regard, a complete overview by employing componential determination/theoretical calculations is currently underway in our laboratory. More interestingly, upon further heating to 600 °C (see Fig. S3 (ESI†)), the MgCl2 peaks were recovered in the XRD pattern. This strongly suggests that an uncommon mechanism for the MgCl2 was involved in the AB decomposition. This mechanism was different from the existing desorption mechanism of the other metal chloride doped AB, such as catalytic effect of the in situ formed nano-metals in CuCl2, CoCl2 and NiCl2 doped AB systems,17,18 as well as B–H bonds activated by partially substituting the H of the [BH3] group with Cl anions for the NH4Cl added AB system.16
 | ||
| Fig. 4 The temperature-dependent Raman spectra recorded for the thermal decomposition of (a) pristine AB, (b) MgF2/2AB and (c) MgCl2/2AB upon heating from room temperature to 250 °C. | ||
In terms of the abovementioned properties and structural analyses, it is reasonable to deduce that in the MgCl2/AB system, MgCl2 first interacted with the [NH3] units of the AB molecule due to the strong electron donor–acceptor coupling observed under the milling effect, as shown in Scheme 1. This interaction is similar to the ligand effect of MgCl2 with NH3 in the amine complex of Mg(NH3)xCl2,30 which not only results in suppressing NH3 release (Fig. 1a), but also changes the bonding state of N–H and B–H (Fig. 2b). Along with heating, the routine combination of H+/H− will release H2 gas. Moreover, the electronegative Cl− is able to coordinate with the electropositive B atom via its lone electronic pairs, thus resulting in the occurrence of those unknown intermediates as confirmed by the XRD and Raman patterns (Fig. 3 and 4). When heated to 600 °C (Fig. S3 (ESI†)), these intermediates further decompose into conventional amorphous B–N polymerides and unexpected MgCl2 crystals. This magical disappearance/reappearance of MgCl2 strongly confirms that an unusual desorption mechanism exists in the MgCl2-doped AB system, i.e., the initial ligand interaction of MgCl2 and NH3BH3 only disturbs the B–N, B–H and N–H bonding, but does not combine with them to form any stable compounds, thus leading to its superior properties compared to the pristine and MgF2-doped AB samples.
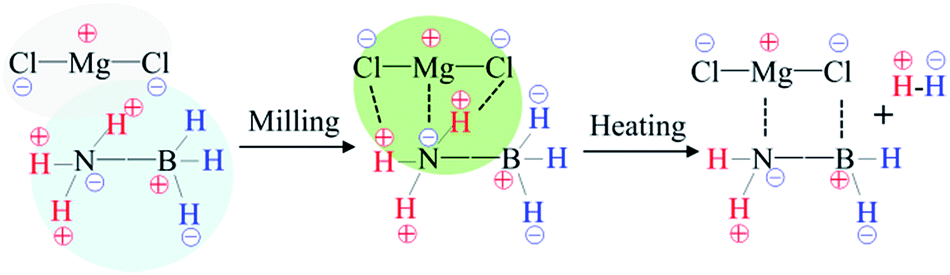 | ||
| Scheme 1 The proposed route for the dehydrogenation of AB activated by MgCl2. | ||
In summary, it has been demonstrated that the hydrogen from AB is released at a temperature as low as 40 °C, and the release of undesirable volatile gases (especially for NH3) is remarkably suppressed upon the addition of the MgCl2. These improvements are attributed to the MgCl2 unexpectedly interacting with the [NH3] units in AB, analogous to the amine complex of Mg(NH3)xCl2. These results not only deepen the understanding of the desorption mechanism of metal chloride–AB systems, but also provides further insight into the promotion of hydrogen release from metal amidoborane and related borohydride amine complexes.
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (No. 51671001, U1503192) and the Natural Science Foundation of Anhui Province (No. 1708085ME99).Notes and references
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353 CrossRef CAS PubMed.
- Y. Kojima and Y. Kawai, Chem. Commun., 2004, 19, 2210 RSC.
- Y. Li, F. Fang, Q. Zhang, L. Ouyang, M. Zhu and D. Sun, Acta Mater., 2011, 59, 1829 CrossRef CAS.
- Q. Zhang, Y. Nakamur, K. Oikaw, T. Kamiyam and E. Akiba, Inorg. Chem., 2002, 41, 6547 CrossRef CAS PubMed.
- Q. Zhang, M. Fang, T. Si, F. Fang, D. Sun, L. Ouyang and M. Zhu, J. Phys. Chem. C, 2010, 114, 11686 CAS.
- G. Xia, D. Li, X. Chen, Z. Tang, Z. Guo and X. Yu, Adv. Mater., 2013, 25, 6238 CrossRef CAS PubMed.
- Y. Li, X. Ding and Q. Zhang, Sci. Rep., 2016, 6, 31144 CrossRef CAS PubMed.
- A. Rossin and M. Peruzzini, Chem. Rev., 2016, 116, 8848 CrossRef CAS PubMed.
- D. W. Himmelberger, C. W. Yoon, M. E. Bluhm, P. J. Carroll and L. G. Sneddo, J. Am. Chem. Soc., 2009, 131, 14101 CrossRef CAS PubMed.
- H. Diyabalanage, R. Shrestha, T. Semelsberger, B. Scott, M. Bowden and B. Davis, Angew. Chem., Int. Ed., 2007, 46, 8995 CrossRef PubMed.
- Q. Zhang, C. Tang, C. Fang, F. Fang, D. Sun, L. Ouyang and M. Zhu, J. Phys. Chem. C, 2010, 114, 1709 CAS.
- Z. Xiong, G. Wu, Y. Chua, H. Hu, T. He, W. Xu and P. Chen, Energy Environ. Sci., 2008, 1, 360 CAS.
- N. C. Smythe and J. C. Gordon, Eur. J. Inorg. Chem., 2010, 4, 509 CrossRef.
- N. J. Hess, M. E. Bowden, V. M. Parvanov, C. Mundy, S. M. Kathman and G. Schenter, J. Chem. Phys., 2008, 128, 034508 CrossRef PubMed.
- V. S. Nguyen, M. H. Matus, D. J. Grant, M. T. Nguyen and D. A. Dixon, J. Phys. Chem. A, 2007, 111, 8844 CrossRef CAS PubMed.
- D. J. Heldebrant, A. Karkamkar, N. J. Hess, M. Bowden, S. Rassat and F. Zheng, Chem. Mater., 2008, 20, 5332 CrossRef CAS.
- T. He, Z. Xiong, G. Wu, H. Chu, C. Wu, T. Zhang and P. Chen, Chem. Mater., 2009, 21, 2315 CrossRef CAS.
- S. Kalidindi, J. Joseph and B. Jagirdar, Energy Environ. Sci., 2009, 2, 1274 CAS.
- Y. Li, F. Fang, Y. Song, Y. Li, Q. Zhang, L. Ouyang, M. Zhu and D. Sun, Int. J. Hydrogen Energy, 2012, 37, 427 Search PubMed.
- Y. Nakagawa, T. Zhang, M. Kitamura, S. Isobe, S. Hino, N. Hashimoto and S. Ohnuki, J. Chem. Eng. Data, 2016, 61, 1924 CrossRef CAS.
- J. Huang, Y. Tan, Q. Gu, L. Ouyang, X. Yu and M. Zhu, J. Mater. Chem. A, 2015, 3, 5299 CAS.
- Q. Zhang, C. Tang, C. Fang, F. Fang, D. Sun, L. Ouyang and M. Zhu, J. Phys. Chem. C, 2010, 114, 1709 CAS.
- C. R. Miranda and G. Ceder, J. Chem. Phys., 2007, 126, 184703 CrossRef PubMed.
- X. Ding, Y. Li, F. Fang, D. Sun and Q. Zhang, J. Mater. Chem. A, 2017, 5, 5067 CAS.
- L. Gao, Y. H. Guo, Q. Li and X. B. Yu, J. Phys. Chem. C, 2010, 114, 9534 CAS.
- M. Mostajeran, D. J. Wolstenholme, C. Frazee, G. S. McGrady and R. T. Baker, Chem. Commun., 2016, 52, 2581 RSC.
- A. M. Heyns and L. C. Prinsloo, J. Solid State Chem., 1998, 137, 33 CrossRef CAS.
- L. Machtoub, Y. Takano and H. Kito, Phys. C, 2006, 445–448, 478 CrossRef CAS.
- R. G. Schlecht and H. K. Bockelmann, Phys. Rev. Lett., 1973, 31, 930 CrossRef CAS.
- C. H. Christensen, R. Z. Sørensen, T. Johannessen, U. J. Quaade, K. Honkala, T. D. Elmøe, R. Køhler and J. K. Nørskov, J. Mater. Chem., 2005, 15, 4106 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials preparing and measurements, Fig. S1–S3. See DOI: 10.1039/c7ra06428d |
| This journal is © The Royal Society of Chemistry 2017 |