
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reaction of sterically encumbered phenols, TEMPO-H, and organocarbonyl insertion reactions with L-AlH2 (L = HC(MeCNDipp)2, Dipp = 2,6-diisopropylphenyl)†
Lauren K. Keyes‡
a,
Angela D. K. Todd‡a,
Nick A. Giffina,
Alex J. Veinot a,
Arthur D. Hendsbeea,
Katherine N. Robertsona,
Stephen J. Geierb and
Jason D. Masuda*a
a,
Arthur D. Hendsbeea,
Katherine N. Robertsona,
Stephen J. Geierb and
Jason D. Masuda*a
aThe Department of Chemistry, Saint Mary's University, Halifax, Nova Scotia, Canada. E-mail: jason.masuda@smu.ca
bDepartment of Chemistry, Mount Alison University, Sackville, New Brunswick, Canada
First published on 26th July 2017
Abstract
The reaction of L-AlH2 (L = HC(MeCNDipp)2, Dipp = 2,6-diisopropylphenyl) with sterically bulky phenols (2,4,6-trimethylphenol, MesOH; 2,6-diisopropylphenol, DippOH) and an N-hydroxylamine (1-hydroxy-2,2,6,6-tetramethyl-piperidine, TEMPO-H) forms an Al–O bond with concomitant loss of hydrogen gas to give L-Al(H)OMes, L-Al(H)ODipp and L-Al(H)TEMPO, respectively. Reaction with 1 or 2 equivalents of benzaldehyde or 1 equivalent of benzophenone results in insertion of carbonyl into the Al–H bond(s) to give the related benzylate and diphenylmethoxide products. Compounds L-Al(H)OMes, L-Al(H)ODipp, L-Al(H)TEMPO, L-Al(H)OBn, L-Al(OBn)2, and L-Al(H)OCHPh2 have been characterized by NMR spectroscopy, elemental analysis, infrared spectroscopy and single crystal X-ray diffraction. The reaction of L-Al(H)OBn with pinacol borane gives a complex mixture of unidentifiable products, providing evidence of the importance of the triflate group in the known aldehyde and ketone hydroboration catalyst L-Al(H)OTf (OTf = CF3SO3−).
Introduction
Main group element-based molecules have been promoted as lower-cost and less toxic alternatives to some transition metals for bond transformations and catalysis. The lighter main group elements (rows 2 and 3) meet these desirable criteria and continue to be an area of intense study. An example is oxidative addition at a low-valent element such as in singlet carbenes,1–3 silylenes,4,5 and Al(I) centers.6–9 The concept of frustrated Lewis pairs (FLPs) has also played a role in the use of lighter main group elements in bond transformation and catalysis reactions, with initial research into boron and phosphorus Lewis pairs10 now expanding to the rest of the periodic table.11Recently, it has been shown that aluminum hydride complexes of β-diketiminato, diamidato, and imidazolin-2-iminato ligands can be used as catalysts for a number of reactions including hydroboration of aldehydes, ketones,12–14 and alkynes,12,15 trimethylcyanide addition to aldehydes and ketones,14 and dehydrocoupling of boranes with amines, phenols, and thiols.15 Catalytic hydroboration of alkynes has also been expanded to more simple aluminum catalysts, such as (iBu2AlH)2 and AlEt3-based molecules.16
We have had a long-standing interest in the reactivity of main group compounds,17–23 including organoaluminum and aluminum-hydride chemistry,17,24,25 and continue to be interested in the reactivity of sterically bulky aluminum hydrides. With the previously mentioned aluminum-based catalysis reactions in mind, we report the reactivity of aluminum β-diketiminato dihydride, L-AlH2 (ref. 26) (L = HC(MeCNDipp)2, Dipp = 2,6-diisopropylphenyl), with two sterically bulky phenols (2,4,6-trimethylphenol, MesOH; 2,6-diisopropylphenol, DippOH), an N-hydroxylamine (1-hydroxy-2,2,6,6-tetramethyl-piperidine, TEMPO-H23), and with the carbonyls of benzaldehyde and benzophenone.
Results and discussion
Reactions with sterically encumbered phenols
Reaction of L-AlH2 with one equivalent of DippOH or MesOH in pentane at room temperature proceeded with concomitant formation of a gas (H2). After stirring overnight, the reaction mixtures were filtered and stored in the freezer to obtain crystalline materials. The reaction with DippOH gave a high yield (90%) of compound 1, whereas the MesOH reaction had a poor isolated yield due to the high solubility of compound 2. Analysis of the crude reaction mixture of 2 revealed a mixture of 90% 2 and 10% starting material L-AlH2. 1H NMR spectroscopy revealed the expected signals for the ligand framework and the respective phenolates. However, the Al–H signal was not observable, presumably due to broadening and a relatively low intensity. The presence of the hydride was confirmed with IR spectroscopy; the Al–H stretch peaks appeared at 1850 cm−1 (compound 1) and 1865 cm−1 (compound 2) (Scheme 1).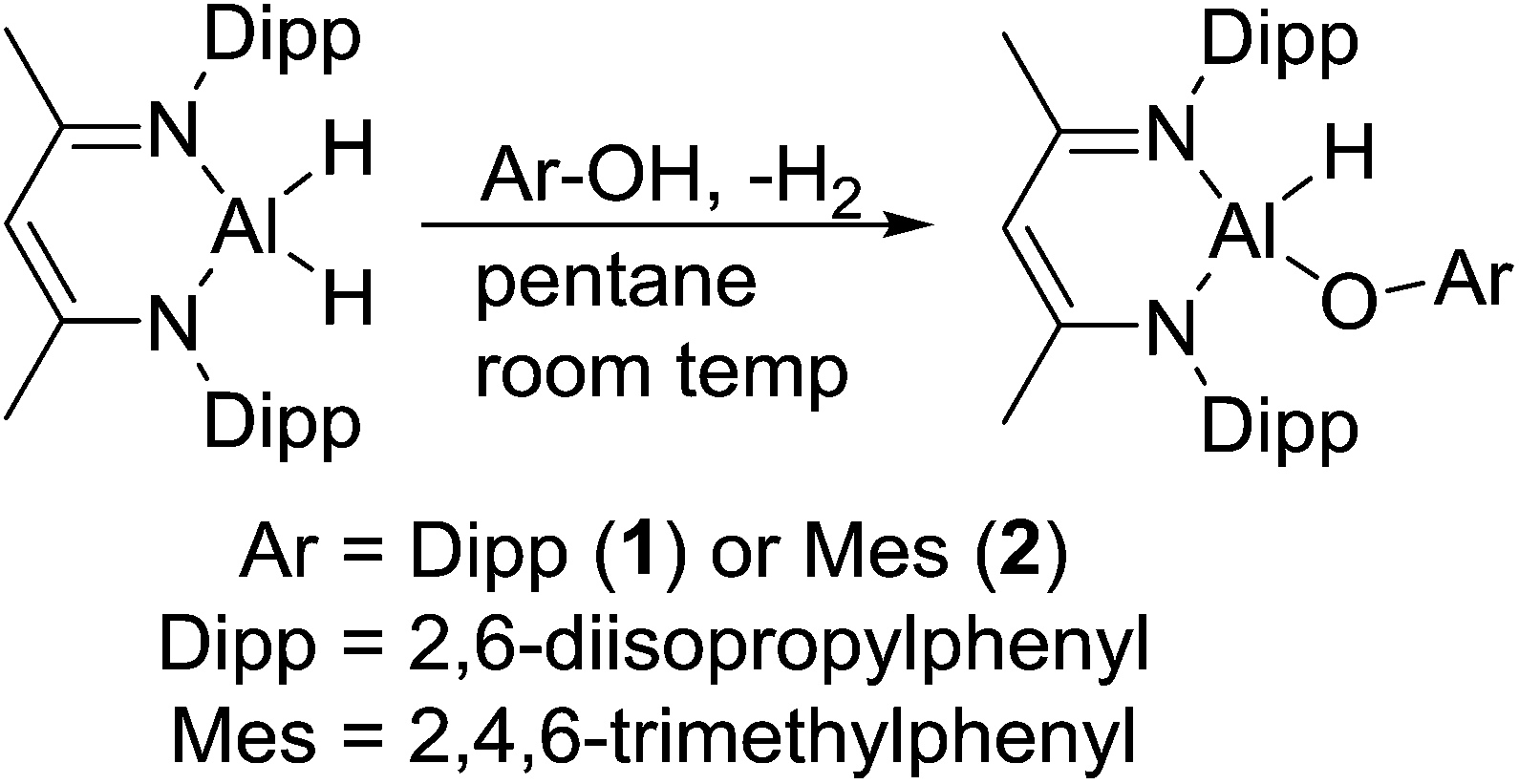 | ||
| Scheme 1 Reaction of L-AlH2 with sterically bulky phenols. | ||
X-ray quality crystals of compounds 1 and 2 were obtained from the pentane solutions after cooling to −35 °C. Compound 1 crystallized in the monoclinic space group P21/c with one equivalent of pentane. Multiple crystals of compound 2 were analysed (triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) but we had difficulty obtaining good data; there were disordered units of co-crystallized pentane that had to be removed using the SQUEEZE routine in PLATON27 to give a reasonable model. Structures are shown in Fig. 1 and 2. The structure of compound 1 reveals the distortions to the six-membered aluminum chelate. The Al atom is 0.549(2) Å out of the plane defined by the N1–C3–C2–C1–N2 β-diketiminate backbone, on the side opposite the phenolate group. This is in contrast to the parent L-AlH2 where the Al atom is in the plane of the ligand backbone.28 This distortion is necessary to accommodate the large Dipp-O group by reducing the interactions with the flanking β-diketiminate Dipp groups. There is also a slight widening of the N–Al–N angle (98.18(6)°) compared to that of L-AlH2 (96.41(5)°). The Al–O–C angle (151.28(11)°) is smaller than that in other aluminum compounds containing bulky phenolates; for example, three- and four-coordinate Al compounds containing the 2,6-tBuC6H3O group have Al–O–C angles ranging from 157.51 Å to 177.71 Å.29 The hydrogen atom attached to aluminum was found in the difference map and refined to give an Al–H distance of 1.519(17) Å, which is the same as that found in L-AlH2 (1.51(2) Å and 1.518(19) Å). For compound 2, compared to 1, the aluminum atom is less distorted (average 0.511 Å) from the mean plane defined by the N–C–C–C–N ligand backbone. The Al–O–C angle (average 161.51°) is larger than that in 1, presumably due to less significant steric interactions between the phenoxide and the ligand N-Dipp groups. In addition, the N–Al–N angle (average 97.26°) is slightly larger than that in 1. Finally, there are no significant intermolecular interactions to report.
) but we had difficulty obtaining good data; there were disordered units of co-crystallized pentane that had to be removed using the SQUEEZE routine in PLATON27 to give a reasonable model. Structures are shown in Fig. 1 and 2. The structure of compound 1 reveals the distortions to the six-membered aluminum chelate. The Al atom is 0.549(2) Å out of the plane defined by the N1–C3–C2–C1–N2 β-diketiminate backbone, on the side opposite the phenolate group. This is in contrast to the parent L-AlH2 where the Al atom is in the plane of the ligand backbone.28 This distortion is necessary to accommodate the large Dipp-O group by reducing the interactions with the flanking β-diketiminate Dipp groups. There is also a slight widening of the N–Al–N angle (98.18(6)°) compared to that of L-AlH2 (96.41(5)°). The Al–O–C angle (151.28(11)°) is smaller than that in other aluminum compounds containing bulky phenolates; for example, three- and four-coordinate Al compounds containing the 2,6-tBuC6H3O group have Al–O–C angles ranging from 157.51 Å to 177.71 Å.29 The hydrogen atom attached to aluminum was found in the difference map and refined to give an Al–H distance of 1.519(17) Å, which is the same as that found in L-AlH2 (1.51(2) Å and 1.518(19) Å). For compound 2, compared to 1, the aluminum atom is less distorted (average 0.511 Å) from the mean plane defined by the N–C–C–C–N ligand backbone. The Al–O–C angle (average 161.51°) is larger than that in 1, presumably due to less significant steric interactions between the phenoxide and the ligand N-Dipp groups. In addition, the N–Al–N angle (average 97.26°) is slightly larger than that in 1. Finally, there are no significant intermolecular interactions to report.
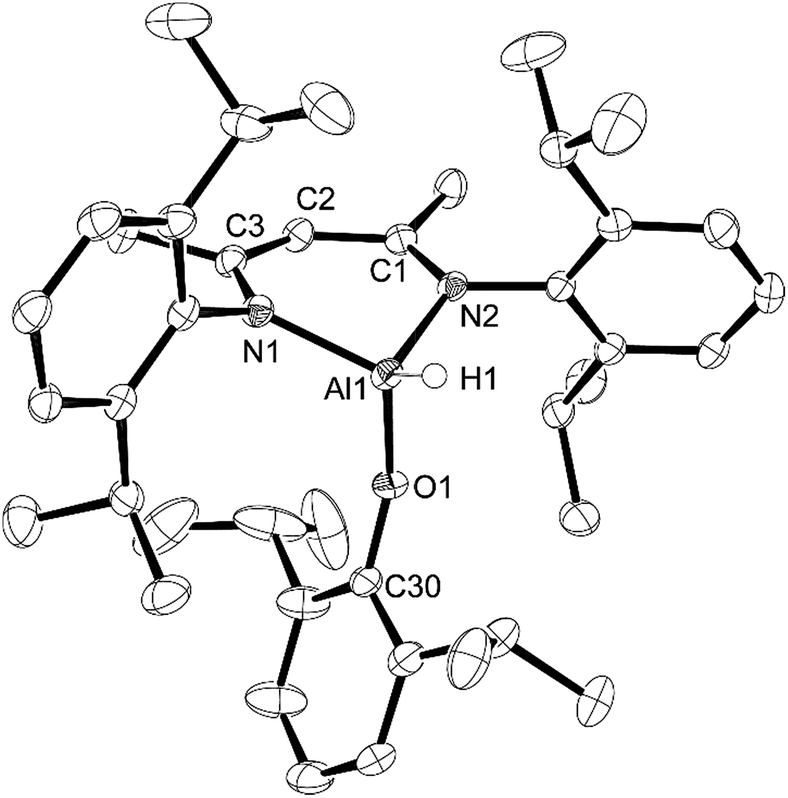 | ||
| Fig. 1 Molecular structure of compound 1, L-Al(H)ODipp, with thermal ellipsoids projected at the 50% probability level. Co-crystallized pentane, hydrogen atoms (except H1) and one component of an isopropyl group disorder have been omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–H1 1.519(17), Al1–O1 1.7115(12), Al1–N1 1.8821(13), Al1–N2 1.8896(14), O1–Al1–H1 117.1(7), O1–Al1–N1 113.36(6), O1–Al1–N2 101.44(6), N1–Al1–H1 111.1(7), N1–Al1–N2 98.18(6), N2–Al1–H1 113.8(7), C30–O1–Al1 151.28(11). | ||
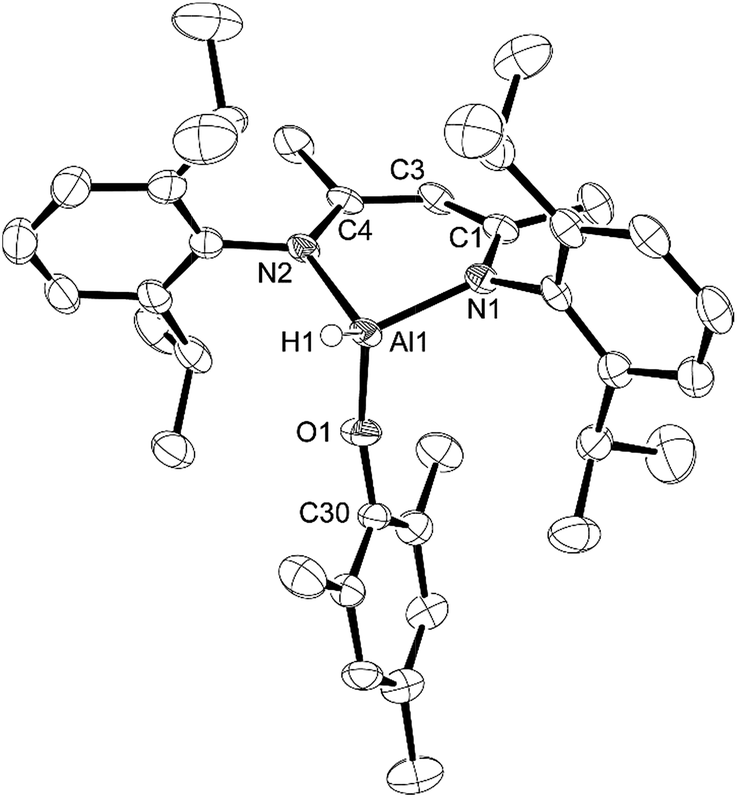 | ||
| Fig. 2 Molecular structure of compound 2, L-Al(H)OMes, with thermal ellipsoids projected at the 50% probability level. One of two in the asymmetric unit. Hydrogen atoms (except H1) have been omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–H1 1.50(2), Al1–O1 1.6955(18), Al1–N1 1.882(2), Al1–N2 1.884(2), O1–Al1–H1 115.1(9), O1–Al1–N1 111.91(9), O1–Al1–N2 107.11(9), N1–Al1–H1 111.6(9), N1–Al1–N2 97.43(8), N2–Al1–H1 112.3(9), C30–O1–Al1 163.28(17). | ||
Reaction of L-AlH2 with 2,4,6-tri-t-butylphenol or BHT resulted in no reaction, even under forcing conditions (110 °C, toluene). Attempts to react a second equivalent of DippOH with 1 under similar conditions resulted in only starting materials when analysed with 1H NMR spectroscopy. Reacting L-ALH2 with two equivalents of MesOH at room temperature gave a crop of crystals from cold pentane. None of the crystals gave a suitable diffraction pattern for crystallographic analysis. Curiously, when the 1H NMR spectrum was measured, it appeared that there was a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 2 and MesOH in the sample. Results from elemental analysis gave the correct values for 2 + MesOH in a 1:1 ratio, implying that this was a co-crystal of the two species. Since single crystal XRD was not possible, we measured the IR spectrum and noted that there were no changes in the Al–H stretching frequency, implying that the co-crystal does not include significant Al–H⋯H–O interactions between 2 and MesOH. We were unable to ascertain any change in the O–H stretch of the co-crystallized MesOH compared to free MesOH as these signals were quite broad.
:1 ratio of 2 and MesOH in the sample. Results from elemental analysis gave the correct values for 2 + MesOH in a 1:1 ratio, implying that this was a co-crystal of the two species. Since single crystal XRD was not possible, we measured the IR spectrum and noted that there were no changes in the Al–H stretching frequency, implying that the co-crystal does not include significant Al–H⋯H–O interactions between 2 and MesOH. We were unable to ascertain any change in the O–H stretch of the co-crystallized MesOH compared to free MesOH as these signals were quite broad.
Reaction with TEMPO-H
Originally, we attempted to react L-AlH2 with TEMPO-H produced using methods from the literature, i.e. via reduction of TEMPO, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl, with aqueous ascorbic acid.30 TEMPO-H is known to sublime in a 3:1 ratio with water31 and upon reaction of this material with L-AlH2, we found that the resulting L-Al(H)TEMPO 3 (needle-like crystals, Fig. 3) was contaminated with L-Al(OH)TEMPO 4 (block-like crystals, Fig. 4) when grown from a cooled hexane solutions (Scheme 2). This prompted us to prepare TEMPO-H using anhydrous methods, and we have reported this elsewhere.23 The reaction of L-AlH2 with anhydrous TEMPO-H proceeds smoothly at room temperature in hexanes with rapid evolution of gas. After normal workup, L-Al(H)TEMPO 3 was isolated as colorless, needle-like crystals in moderate isolated yield (59%, due to high solubility of 3). The 1H NMR spectrum has features containing both the β-diketiminate ligand and the TEMPO fragment, including a broad singlet at 1.20 ppm related to the four methyl groups on the piperidine ring. The aluminum hydride signal was not clearly assignable in the 1H NMR spectrum; however, IR spectroscopy revealed the Al–H stretch at 1831 cm−1. This Al–H stretch was absent in the IR spectrum of 4, and was replaced by a new signal at 3710 cm−1, which is in line with the unexpected Al–OH moiety.
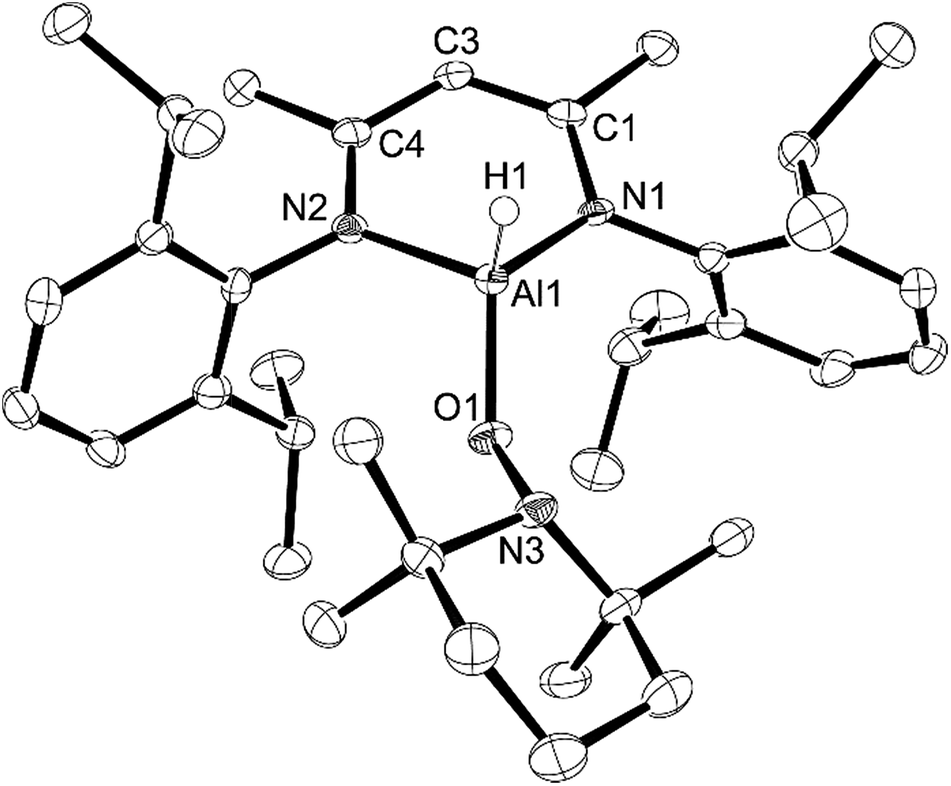 | ||
| Fig. 3 Molecular structure of compound 3, L-Al(H)TEMPO, with thermal ellipsoids projected at the 50% probability level. Hydrogen atoms (except H1) have been omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–H1 1.51(3), Al1–O1 1.745(2), Al1–N1 1.901(3), Al1–N2 1.925(3), O1–N3 1.465(3), O1–Al1–N1 114.70(11), O1–Al1–N2 107.54(11), N1–Al1–H1 109.7(12), N1–Al1–N2 94.60(11), N3–O1–Al1 120.31(17). | ||
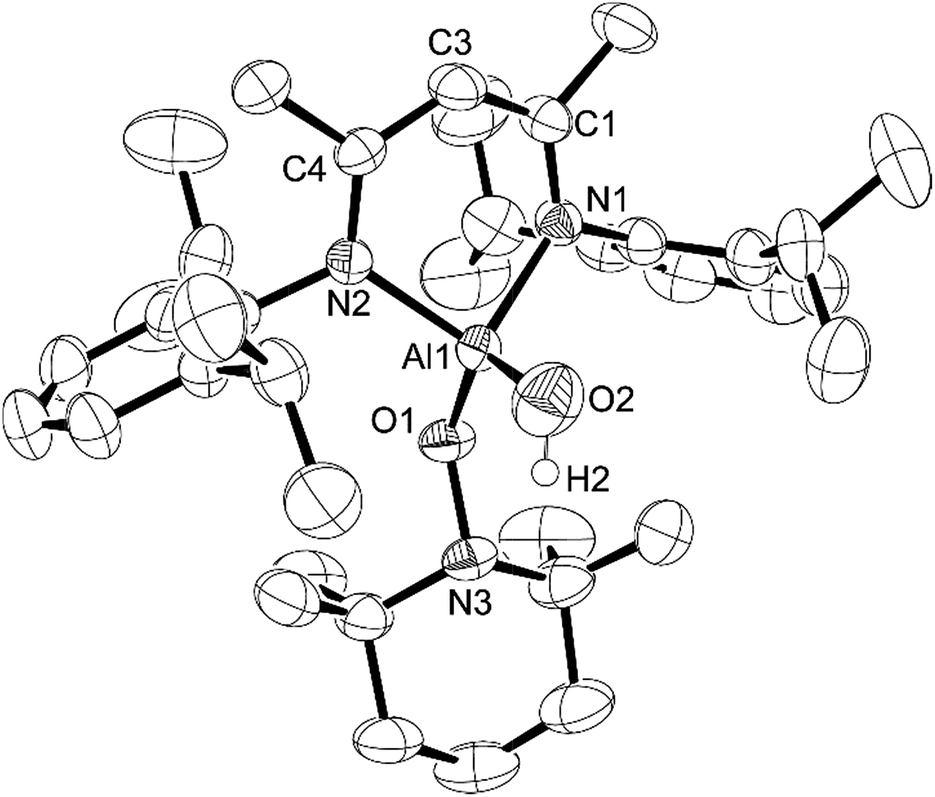 | ||
| Fig. 4 Molecular structure of compound 4, L-Al(OH)TEMPO, with thermal ellipsoids projected at the 50% probability level. Hydrogen atoms (except H2) have been omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–O1 1.7388(19), Al1–O2 1.694(2), Al1–N1 1.929(2), Al1–N2 1.921(2), O1–N3 1.469(3), O1–Al1–N1 111.18(9), O1–Al1–N2 113.23(9), O2–Al1–O1 117.92(11), O2–Al1–N1 109.14(11), O2–Al1–N2 108.02(10), N2–Al1–N1 94.90(9), N3–O1–Al1 120.91(13). | ||
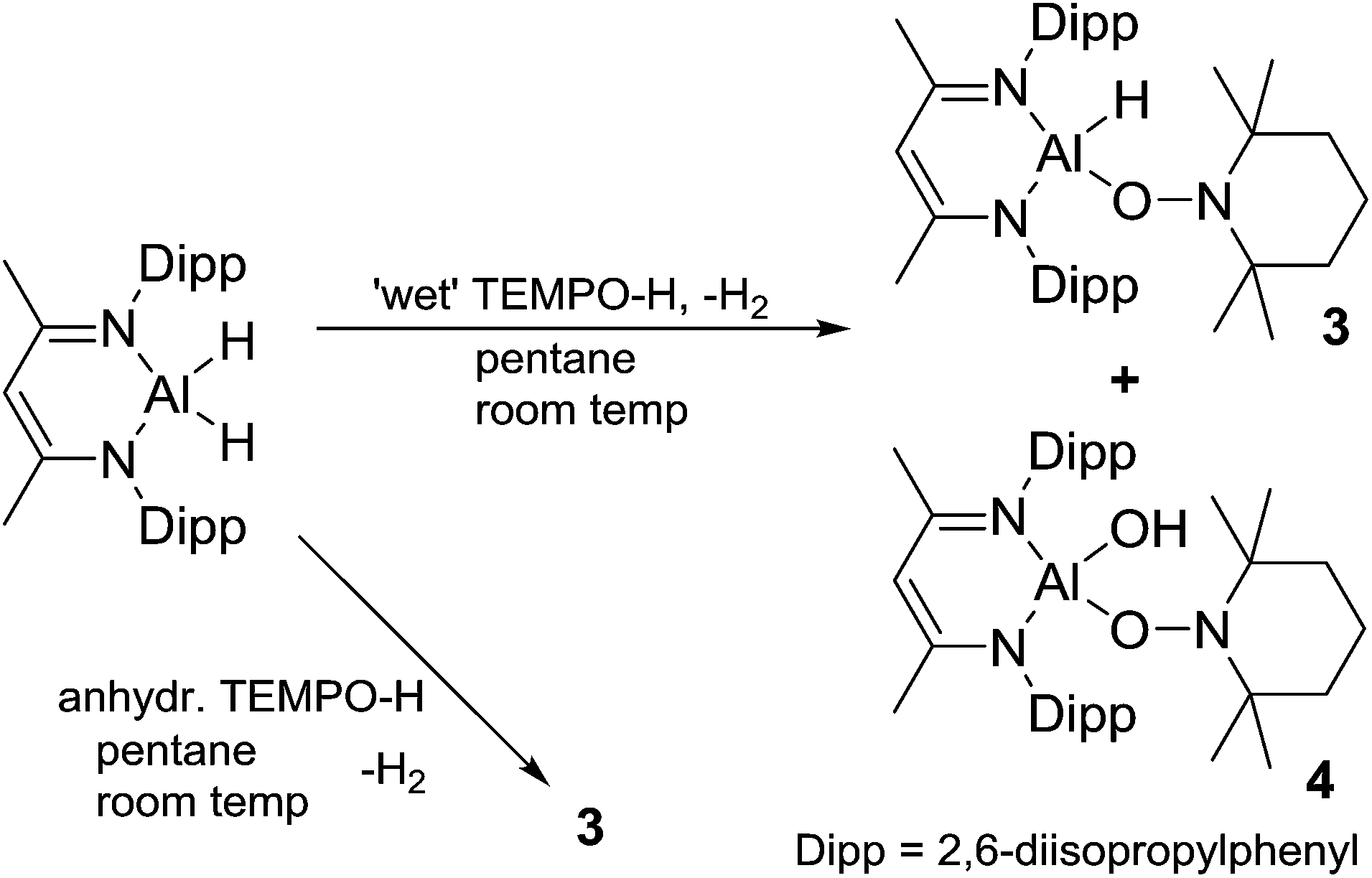 | ||
| Scheme 2 Reaction of L-AlH2 with ‘wet’ TEMPO-H to give compounds 3 and 4. Reaction with anhydrous TEMPO-H to give 3 as the only non-gaseous product. | ||
Compound 3 crystallizes as the monoclinic space group P21/n. The hydrogen atom bound to aluminum was found in the difference map and refined to give a typical distance of 1.51(3) Å. Similar to that in compounds 1 and 2, the aluminum atom in 3 is distorted out of the mean plane (N1–C1–C3–C4–N2) by 0.623(3) Å and the Al–O–N angle is 114.70(11)° to accommodate the bulky TEMPO ligand between the ligand-based N-Dipp groups. Finally, the N–Al–N angle of 94.60(11)° is more acute than that in 1, 2 and L-AlH2, and the Al–O distance (1.745(2) Å) is considerably longer than that of 1 (1.7115(12) Å) and 2 (1.6955(18) Å).
Compound 4 crystallizes in the triclinic space group P. Visually, compound 4 is similar to 3 except that the aluminum hydride has been replaced by an OH group. The Al–O1 distance of 1.7388(19) Å is similar to that in 3, whereas the N–Al–N angle of 94.90(9)° is slightly larger than that in 3. The Al atom is 0.596(3) Å out of the N1–C1–C3–C4–N2 mean plane, which is less than that in 3 and coincides with the larger Al–O–N angle (120.91(13)°). The Al–O2 distance of 1.694(2) Å is considerably shorter than that of the Al–O1 distance; however, it is similar to the Al–O distances in NacNacAl(OH)2 (1.6947(15) Å and 1.7107(16) Å).32 It should be noted that there are no intermolecular hydrogen bonding interactions of the O–H group with other atoms. This is in line with other β-diketiminate-based terminal aluminum monohydroxides in the literature.33–42
Reactions with benzaldehyde and benzophenone
Aluminum hydrides are common stoichiometric reducing agents for organo-carbonyl groups, and recently they have been reported to act as catalysts in the hydroboration of acetylenes13,15 and organo-carbonyls.12–14 We were curious about the insertion of a carbonyl group into the Al–H bond as these products are postulated to be intermediates in the catalytic hydroboration process.12 Reaction of L-AlH2 with 1 or 2 equivalents of benzaldehyde in pentane at room temperature gave insertion products 5 or 6, respectively (Scheme 3). It should be noted that when only one equivalent of benzaldehyde is added, a mixture of compounds 5, 6, and L-AlH2 results, providing evidence that carbonyl insertion into the Al–H bond is competitive between L-AlH2 and the mono-insertion product 5. In 5, the characteristic benzyl CH2 signal appears at δ 4.58 ppm. The asymmetric nature of the molecule is clear with two sets of septets and two pairs of doublets arising from the isopropyl groups on the Al–H side or on the Al–OCH2Ph side of the molecule. There is a very broad signal between 3.9 and 5.3 ppm that is hidden in the baseline; this has been tentatively assigned to the single aluminum hydride. Correspondingly, IR spectroscopy reveals an Al–H stretch at 1818 cm−1. In 6, the benzyl CH2 signal appears at δ 4.82 ppm and the symmetrical nature of the molecule is evident with only one septet and two doublets from the isopropyl groups.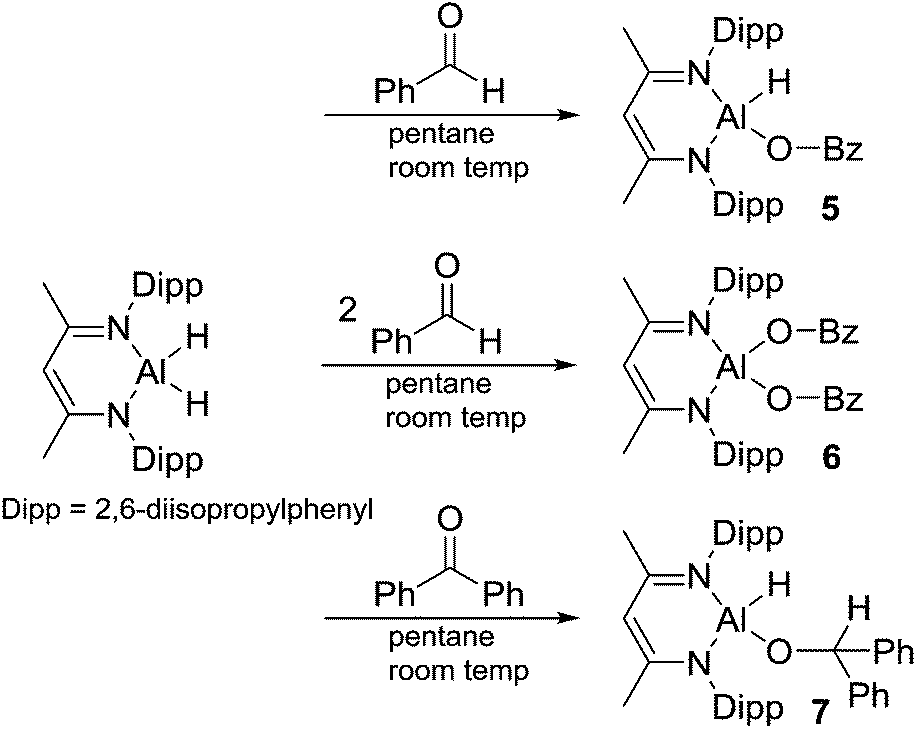 | ||
| Scheme 3 Reaction of L-AlH2 with benzaldehyde and benzophenone. | ||
In the solid state, mono-insertion product 5 crystallizes in the orthorhombic space group P212121, with one molecule of 5 in the asymmetric unit (Fig. 5). In order to minimize the steric interactions between the OCH2Ph group and the flanking Dipp groups, the aluminum atom is pushed 0.544(2) Å out of the plane defined by the N1–C1–C3–C4–N2 ligand backbone. Again, to minimize steric interactions, the Ph ring of the OCH2Ph group is twisted so that it is nearly coplanar with the N–C–C–C–N ligand backbone at a dihedral angle of 8.68(12)° between the two planes. The N–Al–N angle 97.04° is slightly more obtuse than that observed in 3 or 4, and is more in line with the angles observed in compounds 1 and 2. In the solid state, double-insertion product 6 crystallizes in the P21/c space group with one molecule in the asymmetric unit (Fig. 6). One of the two OCH2Ph groups exhibits a two component disorder in a 56:44 ratio. Due to the steric constraints of two OCH2Ph groups on aluminum, the Al atom is only 0.388(2) Å out of the ligand N1–C1–C3–C4–N2 plane. In order to accommodate the two OCH2Ph groups, one OCH2Ph is twisted in a manner similar to that seen in compound 5, while the other is significantly twisted with the CH2Ph moiety sandwiched between the isopropyl groups of the two Dipp groups. The N–Al–N angle (97.32(7)°) is similar to that in mono-insertion compound 5. The O–Al–O angle (115.3(5)°) is slightly more acute than that observed in compound 4.
 | ||
| Fig. 5 Molecular structure of compound 5, L-Al(H)OCH2Ph, with thermal ellipsoids projected at the 50% probability level. Hydrogen atoms (except H1) have been omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–H1 1.54(3), Al1–O1 1.7175(15), Al1–N1 1.8893(17), Al1–N2 1.8816(18), N2–Al1–N1 97.04(8), O1–Al1–N1 110.50(8), O1–Al1–N2 110.07(8), C30–O1–Al1 123.51(13). | ||
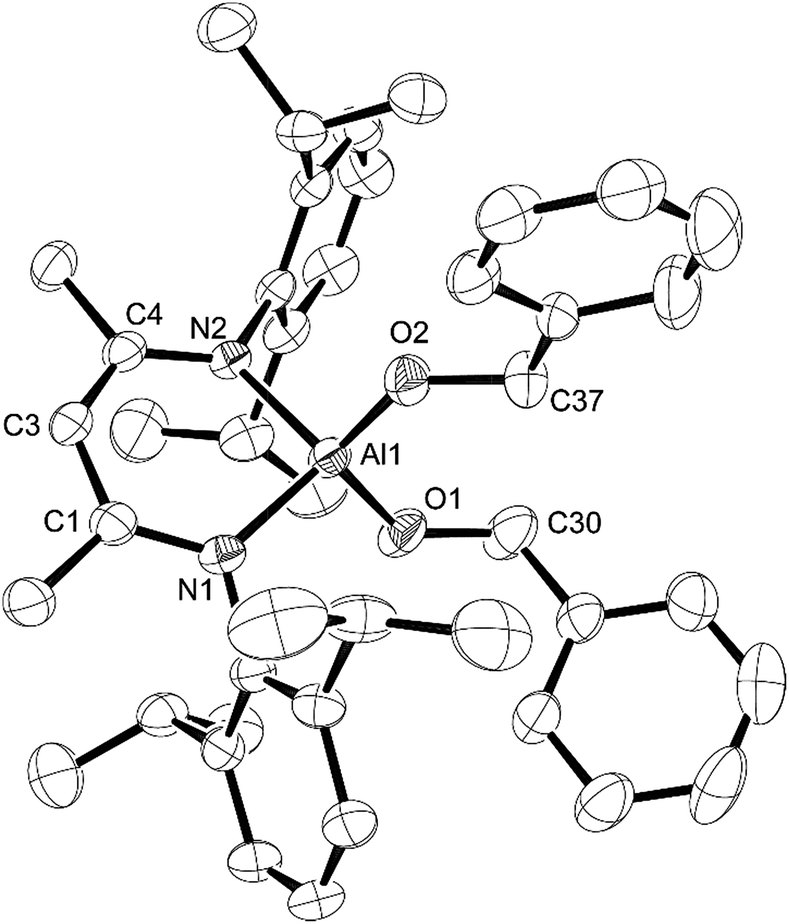 | ||
| Fig. 6 Molecular structure of compound 6, L-Al(OCH2Ph)2, with thermal ellipsoids projected at the 50% probability level. The OCH2Ph group that belongs to O2 exhibits a two-component disorder (56:44) and only one component is shown for clarity. Hydrogen atoms have also been omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–O1 1.7066(14), Al1–N1 1.8803(17), Al1–N2 1.8882(15), Al1–O2 1.680(10), O1–Al1–N1 107.09(7), O1–Al1–N2 113.77(7), N1–Al1–N2 97.32(7), O2–Al1–O1 115.3(5), O2–Al1–N1 115.7(5), O2–Al1–N2 106.4(4), C30–O1–Al1 132.47(15). | ||
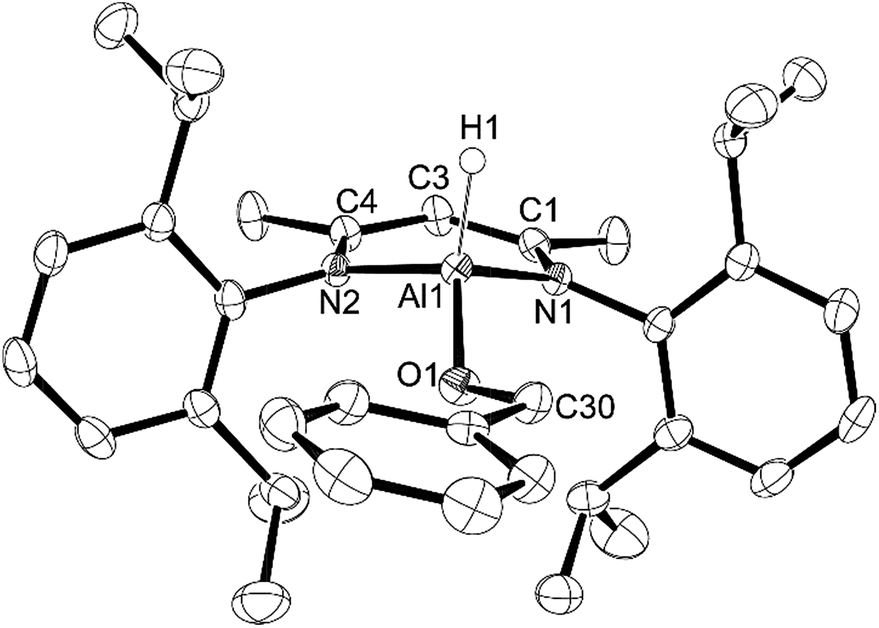
Next, we looked at benzophenone as an example ketone for reactivity. Reaction of L-AlH2 with one equivalent of benzophenone proceeded smoothly at room temperature in pentane. Upon work up, the ketone insertion product, L-Al(H)OCHPh2, was isolated as colorless crystals from cold pentane. Analysis by IR spectroscopy revealed an Al–H stretch at 1814 cm−1, similar to that for 5 (1818 cm−1), but much lower than that in 1 and 2 (1850 and 1865 cm−1, respectively), presumably due to the stronger σ-withdrawing effects of the phenoxide derivatives. The 1H NMR spectrum reveals signals from the ligand that are typical of a complex that is unsymmetrical with respect to the top and bottom halves, as is expected for this complex. Most diagnostic is the methine signal of the alkoxide, which appears at δ 5.79 ppm. Unfortunately, we were unable to observe the remaining Al–H signal in the 1H NMR spectrum, presumably due to the quadrupolar nature of the Al atom. Compound 7 crystallizes in the triclinic space group P with one molecule in the asymmetric unit (Fig. 7). Just as with the other mono-substituted variants in this report, the aluminum atom in 7 is distorted out of the mean plane (N1–C1–C3–C4–N2) by 0.583(4) Å, and the Al–O–C angle is 123.9(3)° to maximize the distance between the OCHPh2 substituent and the Dipp groups. The N–Al–N angle (96.38(14)°) is similar to that in benzaldehyde derivatives 5 and 6. Finally, addition of a second equivalent of benzophenone and heating to 100 °C in toluene showed no reaction when analysed with 1H NMR spectroscopy. Presumably this is due to the extreme steric bulk of the ligands surrounding the aluminum center.
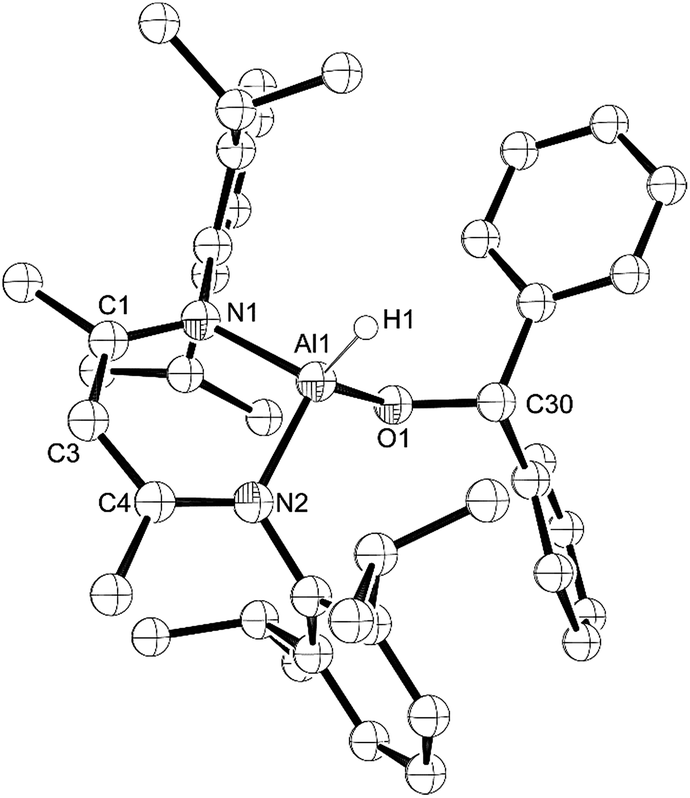 | ||
| Fig. 7 Molecular structure of compound 7, L-Al(H)CHPh2 with thermal ellipsoids projected at the 50% probability level. Hydrogen atoms (except H1) have been omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–H1 1.46(4), Al1–O1 1.729(3), Al1–N1 1.887(3), Al1–N2 1.887(3), O1–Al1–N1 109.06(15), O1–Al1–N2 111.06(15), N2–Al1–N1 96.38(14), C30–O1–Al1 123.9(3). | ||
We were curious to see if mono-benzylate derivative 5 would react with pinacol borate (HBpin) to eliminate PhCH2OBPin and L-AlH2 in a manner similar to that observed in catalytic hydroboration using L-Al(H)(O3SCF3). In an NMR tube, 5 and HBpin were combined in C6D6. Unfortunately, a complex mixture of products was obtained, with none being either L-AlH2 or PhCH2OBPin. This provides evidence that the triflate anion plays a key role in the hydroboration reaction when using L-Al(O3SCF3)H as a catalyst.14
Conclusion
The addition of bulky phenols (MesOH and DippOH) or bulky N-hydroxylamine (TEMPO-H) to L-AlH2 results in the mono-substitution of the aluminum center. All attempts to add a second equivalent of bulky phenols or TEMPO-H were unsuccessful, even at elevated temperatures, and provide evidence of how protected the aluminum hydride fragment is. Aldehyde and ketone insertion into the Al–H bond readily occurred at room temperature. With benzaldehyde, insertion occurs in both 1:1 and 1:2 ratios, giving the corresponding benzylate derivatives. In the case of benzophenone, only one equivalent inserts into the Al–H bond, even at elevated temperatures.
Experimental
General synthetic procedures
All reactions were performed in dry, O2-free conditions under an atmosphere of N2 within an mBraun Labmaster SP inert atmosphere drybox or PTFE sealed reaction vessels using standard Schlenk techniques. L-AlH2 (ref. 26) and anhydrous TEMPO-H23 were prepared using procedures from the literature. All other reagents were purchased from Sigma-Aldrich and used as received, unless otherwise noted. Alumina and molecular sieves were pre-dried in a 150 °C oven before being dried at 300 °C in vacuo. Solvents were purified using an Innovative Technology solvent purification system or purchased as ‘anhydrous’ from Sigma-Aldrich. Solvents were then dried using KH and subsequently filtered through dry alumina and stored over previously dried 4 Å molecular sieves. Glassware was dried at 150 °C overnight prior to experimentation. NMR spectra were recorded on a Bruker Avance 300 MHz or 500 MHz NMR spectrometer. Trace amounts of non- or partially-deuterated solvent were used as internal references for 1H NMR spectra and were referenced relative to tetramethylsilane. The deuterated solvent was used as an internal reference for 13C{1H} NMR spectra and referenced relative to tetramethylsilane. Coupling constants are reported as absolute values. Melting points were recorded on an Electrothermal MEL-Temp 3.0 using glass capillaries sealed under inert conditions. Elemental analysis was performed by the Centre for Environmental Analysis and Remediation (CEAR) facility at Saint Mary's University using a Perkin Elmer 2400 II series Elemental Analyser.In an alternate reaction using similar conditions as mentioned above, but with non-anhydrous TEMPO-H, two types of crystals formed: needle-like crystals, determined to be compound 4 by single crystal X-ray crystallography, and block-like crystals that were determined to be compound 3, L-Al(OH)TEMPO, by single crystal X-ray crystallography. IR of 3 (KBr, cm−1): ν 3710 (s, Al–O–H), 3062 (w), 2964 (s), 1834 (m, Al–H), 1534 (s), 1394 (s), 1317 (s), 1255 (s), 1179 (s), 1135 (w), 1097 (m), 1022 (s), 935 (s), 875 (m), 795 (s), 760 (s), 715 (m), 666 (m).
X-ray crystallography
Crystals of compounds 1–7 were mounted from Paratone-N oil onto an appropriately sized MiTeGen MicroMount. The data were collected on a Bruker APEX II charge-coupled-device (CCD) diffractometer, with an Oxford 700 Cryocool sample cooling device. The instrument was equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å; 30 mA, 50 mV) and MonoCap X-ray source optics. For data collection, typically four ω-scan frame series were collected with 0.5° wide scans, 5–60 second frames and 366 frames per series at varying ϕ angles (ϕ = 0°, 90°, 180°, 270°). Data collection, unit cell refinement, data processing and multi-scan absorption correction were applied using the APEX2 (ref. 43) or APEX3 (ref. 44) software packages. The structures were solved using SHELXT,45 and all non-hydrogen atoms were refined anisotropically with SHELXL46 using shelXle47 or OLEX2 (ref. 48) graphical user interfaces. Unless otherwise noted, all hydrogen atom positions were idealized and rode on the atom to which they were attached. The final refinement included anisotropic temperature factors on all non-hydrogen atoms. Details of crystal data, data collection, and structure refinement are listed in Table 1. All figures were made using ORTEP-3 for Windows.49 For compound 2, one of the iPr groups on the DippO ligand was modelled with a two-site disorder in a 57:43 ratio. For compound 6, one of the Bn groups was modelled with a two-site disorder in an 84:16 ratio. For compound 7, there were two badly disordered pentane molecules that could not be adequately modelled. The SQUEEZE routine as implemented in PLATON27 was used. The program removed 87 electrons from asymmetric unit, which is roughly equivalent to two pentane molecules (42 electrons each). Additional details of the data collection and structure refinement and tables of bond lengths and angles are given in the ESI.† CCDC 1548229–1548235 contain the supplementary crystallographic data for complexes 1–7.†
| Compound reference | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Chemical formula | 2(C41H59AlN2O)·C5H11 | C38H53AlN2O | C38H60AlN3O | C38H60AlN3O2 | C36H49AlN2O | C43H55AlN2O2 | C42H53AlN2O |
| Formula mass | 1316.89 | 580.80 | 601.87 | 617.99 | 552.75 | 658.87 | 630.86 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Triclinic | Orthorhombic | Monoclinic | Triclinic |
| a/Å | 12.5098(16) | 12.120(3) | 9.0846(18) | 9.292(3) | 9.4049(6) | 18.513(5) | 10.589(2) |
| b/Å | 15.1447(19) | 14.739(3) | 20.082(4) | 11.936(3) | 17.1433(11) | 9.348(2) | 11.828(3) |
| c/Å | 22.230(3) | 22.976(4) | 19.763(4) | 17.610(5) | 20.5503(13) | 22.483(6) | 17.768(4) |
| α/° | 90 | 90.781(2) | 90 | 98.473(4) | 90 | 90 | 74.343(2) |
| β/° | 104.731(2) | 94.755(2) | 93.894(3) | 104.305(4) | 90 | 92.259(3) | 78.384(2) |
| γ/° | 90 | 108.004(2) | 90 | 100.237(4) | 90 | 90 | 81.887(2) |
| Unit cell volume/Å3 | 4073.2(9) | 3886.5(13) | 3597.0(12) | 1824.6(9) | 3313.3(4) | 3887.7(17) | 2090.0(8) |
| Temperature/K | 100(2) | 125(2) | 293(2) | 125(2) | 125(2) | 150(2) | 100(2) |
| Space group | P21/c | P |
P21/n | P |
P212121 | P21/c | P |
| Z | 2 | 4 | 4 | 2 | 4 | 4 | 2 |
| Radiation type | MoKα | MoKα | MoKα | MoKα | MoKα | MoKα | MoKα |
| No. of reflections measured | 49947 |
48994 |
37851 |
11683 |
41740 |
37041 |
25497 |
| No. of independent reflections | 10430 |
19018 |
7065 | 7616 | 8415 | 6839 | 10130 |
| Rint | 0.0839 | 0.0766 | 0.1272 | 0.0258 | 0.0774 | 0.0478 | 0.0762 |
| Final R1 values (I > 2σ(I)) | 0.0570 | 0.0711 | 0.0777 | 0.0624 | 0.0413 | 0.0427 | 0.1186 |
| Final wR (F2) values (I > 2σ(I)) | 0.1379 | 0.1753 | 0.1855 | 0.1475 | 0.0972 | 0.0951 | 0.3381 |
| Final R1 values (all data) | 0.0847 | 0.1261 | 0.1021 | 0.1132 | 0.0517 | 0.0676 | 0.1798 |
| Final wR (F2) values (all data) | 0.1537 | 0.2030 | 0.1996 | 0.177 | 0.1031 | 0.1095 | 0.3795 |
| Goodness of fit on F2 | 1.043 | 1.028 | 1.121 | 1.019 | 1.029 | 1.014 | 1.231 |
| Largest diff. peak and hole (e Å−3) | 0.437, −0.466 | 0.878, −0.314 | 0.556, −0.301 | 0.227, −0.354 | 0.197, −0.242 | 0.393, −0.242 | 2.856, −0.510 |
| CCDC number | 1548232 | 1548234 | 1548229 | 1548230 | 1548233 | 1548235 | 1548231 |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
J. D. M. acknowledges support from Saint Mary's University as well as the support of a Discovery Grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada and equipment funding from the Canadian Foundation for Innovation, the Nova Scotia Research and Innovation Trust and NSERC of Canada. J. D. M. also thanks Saint Mary's University for support over the sabbatical in which this manuscript was compiled and written.References
- L. J. Murphy, K. N. Robertson, J. D. Masuda and J. A. C. Clyburne, in N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, ed. S. P. Nolan, Wiley, 2014, p. 427 Search PubMed.
- M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266, DOI:10.1021/ar5003494.
- M. Melaimi, R. Jazzar, M. Sioleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017 DOI:10.1002/anie.201702148.
- B. Blom, M. Stoelzel and M. Driess, Chem.–Eur. J., 2013, 19, 40–62, DOI:10.1002/chem.201203072.
- B. Blom, D. Gallego and M. Driess, Inorg. Chem. Front., 2014, 1, 134–148, 10.1039/c3qi00079f.
- C. Ganesamoorthy, S. Loerke, C. Gemel, P. Jerabek, M. Winter, G. Frenking and R. A. Fischer, Chem. Commun., 2013, 49, 2858–2860, 10.1039/c3cc38584a.
- T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195–9202, DOI:10.1021/ja5038337.
- X. Zhang and Z. Cao, Dalton Trans., 2016, 45, 10355–10365, 10.1039/c6dt01154c.
- S. J. Urwin, D. M. Rogers, G. S. Nichol and M. J. Cowley, Dalton Trans., 2016, 45, 13695–13699, 10.1039/c6dt02698b.
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354 DOI:10.1126/science.aaf7229.
- D. Franz, L. Sirtl, A. Pöthig and S. Inoue, Z. Anorg. Allg. Chem., 2016, 642, 1245–1250, DOI:10.1002/zaac.201600313.
- V. K. Jakhar, M. K. Barman and S. Nembenna, Org. Lett., 2016, 18, 4710–4713, DOI:10.1021/acs.orglett.6b02310.
- Z. Yang, M. Zhong, X. Ma, S. De, C. Anusha, P. Parameswaran and H. W. Roesky, Angew. Chem., Int. Ed., 2015, 54, 10225–10229, DOI:10.1002/anie.201503304.
- Z. Yang, M. Zhong, X. Ma, K. Nijesh, S. De, P. Parameswaran and H. W. Roesky, J. Am. Chem. Soc., 2016, 138, 2548–2551, DOI:10.1021/jacs.6b00032.
- A. Bismuto, S. P. Thomas and M. J. Cowley, Angew. Chem., Int. Ed., 2016, 55, 15356–15359, DOI:10.1002/anie.201609690.
- A. D. K. Todd, W. L. McClennan and J. D. Masuda, RSC Adv., 2016, 6, 69270–69276, 10.1039/c6ra15507c.
- N. A. Giffin, A. D. Hendsbee and J. D. Masuda, Dalton Trans., 2016, 45, 12636–12638, 10.1039/c6dt02790c.
- O. Puntigam, D. Förster, N. A. Giffin, S. Burck, J. Bender, F. Ehret, A. D. Hendsbee, M. Nieger, J. D. Masuda and D. Gudat, Eur. J. Inorg. Chem., 2013, 2013, 2041–2050, DOI:10.1002/ejic.201201471.
- A. D. Hendsbee, N. A. Giffin, Y. Zhang, C. C. Pye and J. D. Masuda, Angew. Chem., Int. Ed., 2012, 51, 10836–10840, DOI:10.1002/anie.201206112.
- N. A. Giffin, A. D. Hendsbee, T. L. Roemmele, M. D. Lumsden, C. C. Pye and J. D. Masuda, Inorg. Chem., 2012, 51, 11837–11850, DOI:10.1021/ic301758k.
- N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342–1359 CrossRef CAS.
- N. A. Giffin, M. Makramalla, A. D. Hendsbee, K. N. Robertson, C. Sherren, C. C. Pye, J. D. Masuda and J. A. C. Clyburne, Org. Biomol. Chem., 2011, 9, 3672–3680, 10.1039/c0ob00999g.
- J. D. Masuda and D. W. Stephan, Dalton Trans., 2006, 2089–2097 RSC.
- J. D. Masuda, D. M. Walsh, P. Wei and D. W. Stephan, Organometallics, 2004, 23, 1819–1824 CrossRef CAS.
- C. Cui, H. W. Roesky, H. Hao, H. Schmidt and M. Noltemeyer, Angew. Chem., Int. Ed., 2000, 39, 1815–1817, DOI:10.1002/(SICI)1521-3773(20000515)39:103.0.CO;2-W.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155, DOI:10.1107/S090744490804362X.
- B. Twamley, N. J. Hardman and P. P. Power, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, m227–m228, DOI:10.1107/S1600536801007413.
- H. Nöth, A. Schlegel, J. Knizek, I. Krossing, W. Ponikwar and T. Seifert, Chem.–Eur. J., 1998, 4, 2191–2203, DOI:10.1002/(SICI)1521-3765(19981102)4:113.0.CO;2-Y.
- H. Henry-Riyad and T. T. Tidwell, J. Phys. Org. Chem., 2003, 16, 559–563 CrossRef CAS.
- E. A. Mader, E. R. Davidson and J. M. Mayer, J. Am. Chem. Soc., 2007, 129, 5153–5166, DOI:10.1021/ja0686918.
- G. Bai, Y. Peng, H. W. Roesky, J. Li, H. Schmidt and M. Noltemeyer, Angew. Chem., Int. Ed., 2003, 42, 1132–1135, DOI:10.1002/anie.200390297.
- G. Bai, H. W. Roesky, J. Li, M. Noltemeyer and H. Schmidt, Angew. Chem., Int. Ed., 2003, 42, 5502–5506, DOI:10.1002/anie.200352163.
- Y. Peng, G. Bai, H. Fan, D. Vidovic, H. W. Roesky and J. Magull, Inorg. Chem., 2004, 43, 1217–1219, DOI:10.1021/ic0351803.
- G. Bai, S. Singh, H. W. Roesky, M. Noltemeyer and H. Schmidt, J. Am. Chem. Soc., 2005, 127, 3449–3455, DOI:10.1021/ja043585w.
- V. Jancik and H. W. Roesky, Angew. Chem., Int. Ed., 2005, 44, 6016–6018, DOI:10.1002/anie.200501509.
- X. Li, L. Duan, H. Song, C. Ni and C. Cui, Organometallics, 2006, 25, 5665–5667, DOI:10.1021/om060722k.
- K. Leszczyńska, I. D. Madura, A. R. Kunicki, J. Zachara and M. Łoś, J. Organomet. Chem., 2007, 692, 3907–3913 CrossRef.
- S. González-Gallardo, V. Jancik, R. Cea-Olivares, R. Toscano and M. Moya-Cabrera, Angew. Chem., Int. Ed., 2007, 46, 2895–2898, DOI:10.1002/anie.200605081.
- Y. Yang, P. M. Gurubasavaraj, H. Ye, Z. Zhang, H. W. Roesky and P. G. Jones, J. Organomet. Chem., 2008, 693, 1455–1461 CrossRef CAS.
- Y. Yang, T. Schulz, M. John, Z. Yang, V. M. Jiménez-Pérez, H. W. Roesky, P. M. Gurubasavaraj, D. Stalke and H. Ye, Organometallics, 2008, 27, 769–777, DOI:10.1021/om700969g.
- B. Li, C. Zhang, Y. Yang, H. Zhu and H. W. Roesky, Inorg. Chem., 2015, 54, 6641–6646, DOI:10.1021/acs.inorgchem.5b00990.
- Bruker, APEX2, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- Bruker, APEX3, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8, DOI:10.1107/S2053273314026370.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8, DOI:10.1107/S2053229614024218.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284, DOI:10.1107/S0021889811043202.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341, DOI:10.1107/S0021889808042726.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854, DOI:10.1107/S0021889812029111.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional crystallographic details and tables of bond lengths and angles. CCDC 1548229–1548235. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra06526d |
| ‡ L. K. Keyes and A. D. K. Todd contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |