DOI:
10.1039/C7RA06788G
(Paper)
RSC Adv., 2017,
7, 44954-44960
Amphiphilic acrylamide block copolymer: RAFT block copolymerization and monolayer behaviour†
Received
18th June 2017
, Accepted 13th September 2017
First published on 20th September 2017
Abstract
RAFT polymerization of acrylamide monomers with long alkyl sidechains was studied for application to block copolymer synthesis and nanoscale assembly using the Langmuir–Blodgett technique. Controlled radical polymerization was demonstrated by optimizing the chain transfer agent (CTA) concentration: amphiphilic acrylamide block copolymer, poly(N-dodecylacrylamide)-block-poly(N-tert-pentylacrylamide) (pDDA-b-ptPA) was obtained with high molecular weight (1.3 × 104) and narrow molecular distribution (Mw/Mn = 1.4). The diblock copolymer formed a stable monolayer at the air–water interface under a surface pressure of 30 mN m−1, but the presence of the junction between pDDA and ptPA affected the monolayer mechanical properties at higher surface pressures. Results provide insight into the two-dimensional self-assembly of amphiphilic block copolymers consisting of molecular-level amphiphilic block components.
1. Introduction
Controlling the nanoscale self-assembly of polymer materials is a key issue in various fields such as biomimetic materials1–5 and flexible electronics device science.6–10 To achieve and improve control, both material synthesis and processing methodology have been developing synergistically, thereby providing molecular design and bottom-up approaches for the simple and precise control of nanostructures. The Langmuir–Blodgett (LB) technique is a notable example of nanoscale assembly using an air–water interface.11–13 For the LB technique, amphiphilic materials including amphiphilic polymers are necessary to form a Langmuir monolayer. We have investigated formation and functionalization of polymer LB films consisting of poly(N-alkylacrylamide)s.14–16 Actually, poly(N-dodecylacrylamide) (pDDA) and poly(N-tert-pentylacrylamide) (ptPA) exhibit excellent LB film formation ability: these materials provide highly oriented and densely packed monolayers at the air–water interface. The nanoassembly structure of pDDA and ptPA can be tuned at high surface pressures. They bring two-dimensional (2D) monolayers, which differ greatly from their three-dimensional random conformations at the equilibrium state.11,14–16 Well-defined layer structures are readily obtained by adjusting the compression pressure of the monolayer and the number of stacking layers. Furthermore, various functional groups were incorporated and were distributed uniformly in the 2D film. These acrylamide polymers present the benefit of forming a 2D hydrogen-bonding network among polymer backbones,11,14–16 which engenders high monolayer stability at the air–water interface. Consequently, the quantitative deposition of functional molecules onto solid substrates was realized for the formation of nanostructured monolayer stacks to the out-of-plane direction from the substrate plane. For nanostructure formation in the in-plane direction, block copolymer LB films are expected to provide novel phase-separated structures that are not achieved using homopolymers. Earlier reports of block copolymer LB films have described 2D microphase separation and micro-patterning based on phase separated structures.17–21 Some block copolymers lack monolayer formation ability at higher surface pressures (>15 mN m−1), leading to difficulties from multilayer formation caused by poor mechanical properties. Enhancement of mechanical properties is expected to lead to monolayer formation and multilayer deposition at higher pressures by Langmuir monolayers consisting of amphiphilic block components including acrylamide monomers that provide high monolayer stability at the air–water interface. Block copolymers used in earlier studies were synthesized using controlled polymerization such as atom-transfer radical polymerization (ATRP)22–25 and reversible addition–fragmentation chain transfer (RAFT) polymerization.26,27 Acrylamide polymers with long alkyl side chains (e.g., pDDA) and short branched side chains (e.g., ptPA) have been polymerized using free radical polymerization: controlled polymerizations such as ATRP or RAFT polymerization have been investigated only rarely. These results derive from the fact that acrylamide-based monomers are weakly active in ATRP or RAFT polymerization. Earlier reports describing RAFT polymerization and block copolymer synthesis of acrylamides are limited to monomers with short side chains such as N,N-dimethylacrylamide (DMA) and N-isopropylacrylamide (NIPAM).28–30 Statistical RAFT polymerization of 2-acrylamido-2-methylpropane sulfonic acid (AMPS) and n-dodecyl acrylamide was conducted to investigate micelle formation in water.31,32 Therefore, block copolymer synthesis of pDDA is anticipated as a breakthrough for the investigation and fabrication of self-assembled nanostructures of acrylamide-based amphiphilic block copolymers. This study examined RAFT polymerization ability of pDDA homopolymer with narrow molecular weight distribution and the synthesis of diblock copolymers with the pDDA unit. Further investigations were conducted to examine monolayer properties of the diblock copolymers. The monolayer behaviour was compared with relevant homopolymers and blend monolayers.
2. Experimental section
2.1. Materials
Monomers, DDA and tPA were prepared as reported.4,14–16 Monomers and azoisobutyronitrile (AIBN) were purified respectively using recrystallization from chloroform/hexane mixture and methanol. Anhydrous toluene (Wako Pure Chemical Industries Ltd.), anhydrous DMF (Wako Pure Chemical Industries Ltd.), acetonitrile (Kanto Chemical Co. Inc.), cyanomethyl dodecyl trithiocarbonate (Aldrich), 2-cyano-2-propyl dodecyl trithiocarbonate (Aldrich), and tris(trimethylsilyl)silane were used as received.
2.2. Instruments and analysis
SEC measurements were taken using a GPC system (8020; Tosoh Corp.) with THF as eluent and a mixed gel column (TSK gel SuperHZM-M; Tosoh Corp.). The obtained data were converted into molecular weight using polystyrene standards. UV-vis spectra were measured using a UV-vis spectrometer (V-670; Jasco Corp.). NMR spectra were measured using a Bruker Avance III 400 spectrometer (400 MHz).
2.3. Synthesis of pDDA macro-CTA
A mixed solution of monomer, CTA, and AIBN was degassed using freeze–pump–thaw cycles more than three times before polymerization reaction at 60 °C in an oil bath. The resulting solution was concentrated and poured into acetonitrile for reprecipitation. The obtained polymer was reprecipitated two more times and was then dried in vacuum overnight. Light yellow powder (pDDA macro-CTA) was obtained (yield: 51% for Run 7). 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, –CH3), 1.2–1.7 (br, side chain), 2.1 (br, main chain), 3.2 (br, –NHCH2–), and 3.6 (br, weak, CTA), and 6.5 ppm (br, NH). To trace the reaction, the polymerization reaction was conducted in an Ar-filled glove box (1ADB-3; Miwa Mfg. Co. Ltd.). The solution was kept at 60 °C in an aluminum bead bath. A sample of the reaction solution was extracted and analyzed using NMR and SEC.
2.4. Terminal CTA group removal of pDDA macro-CTA
To a toluene solution of macro-CTA (ca. 150 mg mL−1) were added 30 μL of tris(trimethylsilyl)silane and ca. 4 mg of AIBN and degassed using freeze–pump–thaw cycles more than three times before reaction at 60 °C for 4 h. The resulting solution was concentrated and poured into acetonitrile for reprecipitation; then it was dried in vacuum overnight. White powder was obtained (yield: 77%). 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, –CH3), 1.2–1.7 (br, side chain), 2.2 (br, main chain), 3.2 (br, –NHCH2–), and 3.6 (br, weak, CTA), and 6.5 ppm (br, NH).
2.5. Synthesis of pDDA-b-ptPA
pDDA macro-CTA (90.25 mg) and ptPA (83.95 mg) were dissolved in toluene (2 mL). To this solution was added 2 mL toluene solution of AIBN (0.243 mg mL−1). It was then degassed using freeze–pump–thaw cycles for three cycles before reaction at 60 °C for 20 h. After the reaction, the solution was cooled. Then 30 μL of tris(trimethylsilyl)silane and 4.51 mg of AIBN were added and degassed again using freeze–pump–thaw cycles for three cycles before reaction at 60 °C for 4 h. The resulting solution was concentrated and poured into acetonitrile for reprecipitation. It was then dried in vacuum overnight. White powder was obtained (yield: 61%). The polymer was reprecipitated two more times before monolayer studies. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz), 1.26 (br), 1.47 (br), 1.71 (s), 1.92 (s), 2.01 (s), 3.14 (br), and 6.30 ppm (br).
2.6. Monolayer formation and measurements
Amphiphilic polymer monolayer behaviour was examined using the Langmuir–Blodgett method.4,11,14–16 Surface pressure (π)–area (A) isotherm measurements were conducted using automatically controlled Langmuir troughs (HBM-AP; Kyowa Interface Science Co. Ltd. and FSD-21; USI Systems Inc.). A chloroform solution of the copolymer at a concentration of ca. 1 mM was spread on ultrapure water at 20 °C. After the solvent was evaporated, the monolayer was compressed at a rate of 15 cm2 min−1, with monitoring of surface pressure using a Wilhelmy plate.
3. Results and discussion
RAFT polymerizations were examined using two trithiocarbonate-based chain transfer agents (CTA-Me and CTA-Pr in Scheme 1), which have been used for controlled radical polymerizations of (meth)acrylamide monomers with short alkyl side chains.28–30 The conditions and results of polymerizations are presented in Table 1. Polymers were eluted with THF and were monitored using refractive index (RI) and UV-vis absorption detectors. The observation wavelength in the UV-vis detector was set at 300 nm, which is the absorption peak of CTA-Me (ε300 = 1.15 × 104 M−1 cm−1 in toluene) (Fig. 1(a)). Polymers with wide molecular weight distribution were obtained from reactions at the lowest CTA concentration (0.363 mM (Run 1)) and the highest AIBN concentration (1.70 mM (Run 2)). Fig. 1(b) shows SEC curves for Runs 2 and 6. In the case of Run 2, chromatograms obtained using UV and RI detectors show a different tendency: the chromatogram for UV has a minor shoulder in the low molecular weight region, suggesting that the reaction proceeds in multiple modes. DDA monomers undergo a non-controlled free radical polymerization reaction because the CTA concentration is too small compared with that of the propagating chain end. Reduction in the AIBN concentration (Runs 3 and 4) caused low conversion even with longer reaction time, suggesting that the side reaction including termination reaction is unavoidable in the polymerization. In contrast, polymers with a narrow molecular weight distribution were obtained from reactions with high CTA and AIBN concentrations (Runs 5–7 in Table 1). Especially in high CTA concentration cases (Runs 6 and 7), the molecular weight distribution reaches approximately 1.3. SEC chromatograms of RI and UV detectors have the same feature (Fig. 1(b)), indicating that the polymerization reaction proceeded in a single reaction mode, i.e., the polymerization reaction proceeds through the RAFT polymerization mechanism. At the optimum condition (Run 6), the [CTA]/[AIBN] ratio is calculated as 4.69, which is similar to that for RAFT polymerization of polar acrylamide monomers in highly polar solvents (ca. 5),28,33 but which is smaller than polymerizations of DMA and NIPAM in benzene (ca. 20).34 The result indicates that the higher concentration of active states is preferred to the active–dormant equilibrium of acrylamide polymers with longer alkyl side chains to compete with the termination reaction and non-controlled free radical polymerization.
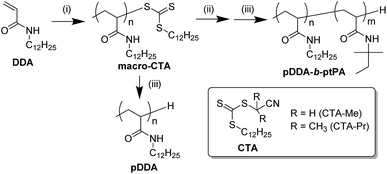 |
| | Scheme 1 Synthesis route of macro-CTA and block copolymers pDDA-b-ptPA: (i) AIBN, CTA, toluene or DMF, 60 °C, 24–48 h; (ii) tPA, AIBN, toluene, 60 °C, 20 h; (iii) tris(trimethylsilyl)silane, AIBN, toluene, 60 °C, 4 h. | |
Table 1 Results of polymerization of pDDA
| Run |
[DDA]/M |
[AIBN]/mM |
[CTA-Me]/mM |
Time/h |
Conv./% |
Mn/103 (Mw/Mn) |
| Runs F1 and F2 were executed without CTA. |
| 1 |
0.211 |
0.247 (0.12%) |
0.363 (0.17%) |
24 |
88 |
22.0 (2.01) |
| 2 |
0.216 |
1.70 (0.8%) |
2.62 (1.2%) |
24 |
>95 |
7.70 (1.77) |
| 3 |
0.172 |
0.267 (0.16%) |
2.09 (1.2%) |
48 |
88 |
10.1 (1.48) |
| 4 |
0.172 |
0.0267 (0.016%) |
2.09 (1.2%) |
48 |
55 |
7.90 (1.58) |
| 5 |
0.957 |
0.649 (0.07%) |
12.1 (1.3%) |
24 |
>95 |
9.44 (1.39) |
| 6 |
0.186 |
0.909 (0.49%) |
4.26 (2.3%) |
24 |
95 |
8.67 (1.34) |
| 7 |
0.186 |
0.909 (0.49%) |
9.86 (5.3%) |
24 |
94 |
4.17 (1.35) |
| F1a |
0.211 |
0.247 (0.12%) |
— |
24 |
79 |
23.7 (2.89) |
| F2a |
0.216 |
1.70 (0.8%) |
— |
24 |
>95 |
20.4 (3.28) |
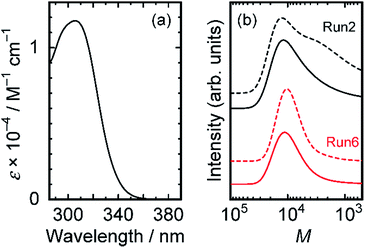 |
| | Fig. 1 (a) Molar extinction coefficient spectrum of CTA-Me in toluene. (b) SEC chromatogram monitored using RI (solid lines) and UV-vis absorption (at 300 nm) detectors (broken lines). Black and red lines respectively correspond to Runs 2 and 6. | |
We examined the polymerization reaction by varying the CTA agents as well as solvents at a concentration of 2 mol% CTA. Fig. 2 portrays a kinetic plot and molecular weight plots showing them as functions of the reaction conversion. In the case of the reaction in toluene with CTA-Me (black, Fig. 2(a)), a linear relation was observed, indicating that the polymerization reaction proceeds in pseudo-first-order kinetics. Therefore, the polymer radical concentration ([P˙]) remains constant throughout the reaction time. For that reason, the side reaction of the propagating ends is negligible during the reaction. The apparent pseudo-first-order propagation rate constant kapp (the product of the propagation rate constant kp and [P˙]) in toluene was determined as kapp = 5.7 × 10−5 s−1 (Table 2). The molecular weight, which increases linearly over the conversion range (black, Fig. 2(c)), shows good agreement with the calculated line, assuming 60% initiator efficiency (broken line in Fig. 2(c)). In the case of the polymerization with CTA-Me in DMF, the reaction also exhibits (1) pseudo-first-order reaction kinetics (red in Fig. 2(a)), (2) a linear relation between the molecular weight and the conversion (red in Fig. 2(c)), and (3) narrow molecular weight distribution as small as Mw/Mn = 1.23 (red in Fig. 2(b)), which show experimental evidence of RAFT polymerization. The kapp value was reduced to 1/3 of the kapp value in toluene: kapp = 1.7 × 10−5 s−1. The result implies that the trithiocarbonate radicals, which serve as dormant species, were deactivated because of the higher polarity of DMF. The effects of CTA agents were examined using CTA-Pr (blue in Fig. 2). Results showed that the kapp value was reduced to 2/3 of that of the reaction with CTA-Me in DMF and that the reaction had an induction period ti = 4.3 h. This result might derive from the slow chain transfer reaction of more stable 3° cyanopropyl radicals generated from CTA-Pr in the active/dormant equilibrium.35,36 Nevertheless, this reaction affords controlled radical polymerization with narrow molecular weight distribution. In the case of polymerization of acrylamide monomers with short-branched side chain (ptPA), the kapp value was higher than that for pDDA (kapp = 6.4 × 10−5 s−1 in toluene, Fig. S1†). The result indicates that steric hindrance of long alkyl chains of DDA monomers is an origin of slow polymerization reaction. It also indicates that pDDA polymerization with cyanododecyl trithiocarbonates proceeds in the controlled RAFT mechanism to afford pDDA macro-CTAs by competing with the termination reaction and the free radical polymerization reaction. Therefore, we conclude that this polymerization reaction proceeds by the RAFT mechanism, which is rarely reported for the polymerization of acrylamide monomers with long alkyl chains.
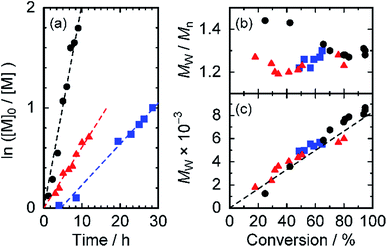 |
| | Fig. 2 (a) First-order kinetic plot of ln([M]0/[M]) versus time, (b) evolution of polydispersity index, and (c) number-average molecular weight for the RAFT polymerization of DDA. The broken line in (c) represents the theoretical prediction. Reaction conditions: (black circles) [DDA]/[CTA-Me]/[AIBN] = 204/4.7/1 in toluene, (red triangles) [DDA]/[CTA-Me]/[AIBN] = 210/4.8/1 in DMF, and (blue squares) [DDA]/[CTA-Pr]/[AIBN] = 210/4.8/1 in DMF. | |
Table 2 Results of RAFT polymerization of pDDA and ptPA homopolymers
| Monomer |
CTA |
Solvent |
kapp/s−1 |
Final conversion/% |
Final Mn/103 (Mw/Mn) |
| DDA |
Me |
Toluene |
5.7 × 10−5 |
95 |
8.67 (1.28) |
| DDA |
Me |
DMF |
1.7 × 10−5 |
72 |
5.98 (1.23) |
| DDA |
Pr |
DMF |
0.92 × 10−5 |
65 |
5.68 (1.27) |
| tPA |
Me |
Toluene |
6.4 × 10−5 |
82 |
5.33 (1.20) |
The end group in the macro-CTAs was transformed by several chemical modifications. Some examples are end group removal by radical-induced reduction, block copolymer polymerization, and modification by hetero Diels–Alder reaction.37 For functional nanoscale self-assemblies, end group removal is necessary to apply RAFT polymers, thereby enhancing their thermal and chemical stability. The trithiocarbonate CTA groups in the chain ends were removed via radical-induced reduction using tris(trimethylsilyl)silane.38 After the reaction, polymers were purified by reprecipitation and were analysed using NMR and SEC measurements. As a result, the NMR signal of the CTA groups and UV absorption at 300 nm disappeared, indicating that the CTA groups were removed from polymers. The SEC chromatograms before and after the reaction show a negligible change, indicating that the polymer backbone underwent no cleavage through the terminal treatment. Therefore, we conclude that CTA group removal by tris(trimethylsilyl)silane proceeds with no polymer chain damage.
Next, we synthesized diblock copolymer using pDDA macro CTA. Block copolymer with the second block composed of tPA units (pDDA-b-ptPA) was synthesized, following the procedure of the pDDA first block polymerization described above. The polymerization reaction was conducted at 60 °C for 20 h, followed by chain end removal using tris(trimethylsilyl)silane. pDDA-b-ptPA was soluble in chloroform and THF, but insoluble in acetonitrile and water. Fig. 3 shows SEC chromatograms of pDDA-macro CTA and pDDA-b-ptPA. The monomer content in the diblock copolymer pDDA-b-ptPA was estimated using NMR spectra: DDA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :tPA = 76:24 (Fig. S2†). The SEC curves show a single peak even after the second block polymerization and the end-group removal (Table 3). Consequently, RAFT polymerizations with high [CTA]/[AIBN] ratios enable us to demonstrate controlled radical polymerization of narrow molecular weight distribution amphiphilic acrylamide-based block copolymers with long alkyl side chains.
:tPA = 76:24 (Fig. S2†). The SEC curves show a single peak even after the second block polymerization and the end-group removal (Table 3). Consequently, RAFT polymerizations with high [CTA]/[AIBN] ratios enable us to demonstrate controlled radical polymerization of narrow molecular weight distribution amphiphilic acrylamide-based block copolymers with long alkyl side chains.
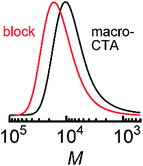 |
| | Fig. 3 SEC chromatograms of (black) pDDA-macroCTA and (red) pDDA-b-ptPA measured using an RI detector. | |
Table 3 Results of block copolymerization of pDDA-b-ptPA
| |
Mn/103 (Mw/Mn) |
Conversion/% |
| pDDA macro-CTA |
8.75 (1.37) |
95 |
| pDDA-b-ptPA |
13.0 (1.43) |
93 |
Finally, Langmuir monolayer properties of the amphiphilic diblock copolymer pDDA-b-ptPA were examined. Fig. 4(a) shows π–A isotherms for several combinations of homopolymers pDDA and ptPA, and diblock copolymer pDDA-b-ptPA. The π–A isotherms of pDDA (black in Fig. 4(a)) and ptPA (grey in Fig. 4(a)) exhibit a monotonic rise in surface pressure. The collapse pressure (πc) was determined respectively as 32.9 mN m−1 and 51.9 mN m−1 for ptPA and pDDA homopolymers. However, pDDA-b-ptPA and pDDA:ptPA blend monolayers have plateau regions in their π–A isotherms, indicating that the monolayers have phase-separated structures. In fact, the AFM image of pDDA-b-ptPA LB films proved the phase-separated structures on a ten-nanometre scale (Fig. S3†). The pDDA:ptPA blend monolayer (green in Fig. 4(a)) shows a short plateau region at around π = 35 mN m−1, which is attributed to the collapse of the ptPA domain. The surface pressure increases immediately after reaching the plateau region, suggesting that the collapse of the ptPA domains does not influence pDDA monolayer formation. The monolayer collapses at 51.8 mN m−1, which is identical to pDDA homopolymer. The π–A isotherm of pDDA-b-ptPA (red in Fig. 4(a)) shows a more expanded plateau region at 33.1 mN m−1 and a lower collapse pressure at 41.4 mN m−1, implying that the collapse of the ptPA domains makes the pDDA-b-ptPA monolayer less stable. In terms of the monolayer stability, the pDDA-b-ptPA monolayer is stable over 1000 min at surface pressure of 30 mN m−1, suggesting a steady monolayer structure (Fig. S4†). However, the stability worsened above 33.1 mN m−1: the surface area decreases drastically at surface pressure of 40 mN m−1 as time proceeds, indicating the collapse of the pDDA-b-ptPA monolayer at higher pressures.
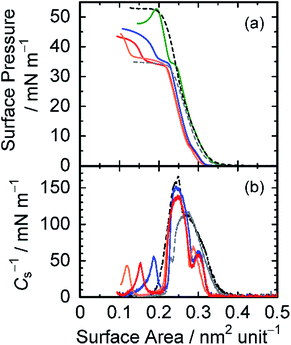 |
| | Fig. 4 (a) π–A isotherms and (b) Cs−1–A isotherms of pDDA-b-ptPA (red), pDDA (black), ptPA (grey), and blend of pDDA and ptPA (pDDA:ptPA = 76:24 (mol)) (green), pDDA and pDDA-b-ptPA (1:1 mol) (blue), and ptPA and pDDA-b-ptPA (1:1 mol) (orange) measured at 20 °C. | |
From these curves, the compressibility modulus Cs−1 was calculated using the equation Cs−1 = −A(dπ/dA).39 In the Cs−1–A isotherms (Fig. 4(b)), each Langmuir film has multiple peaks and bottoms. It is noteworthy that pDDA-b-ptPA and pDDA:ptPA blend monolayers have a shoulder peak at a larger surface area, indicating the effects of two-component mixing. Table 4 presents mechanical parameters of monolayers including π*, which is defined as the highest surface pressure at the corresponding peak position. The Cs−1 values of the pDDA-b-ptPA monolayer at each π* are determined as 58.8 mN m−1 (0.30 nm−2), 138 mN m−1 (0.25 nm−2), and 46.8 mN m−1 (0.15 nm−2) from the larger surface area. The value at 0.25 nm−2 is larger than the corresponding value for the ptPA homopolymer (118 mN m−1 at 0.27 nm−2), but smaller than that for pDDA (165 mN m−1 at 0.25 nm−2), suggesting disordered packing of the pDDA domain in the pDDA-b-ptPA monolayer. Compared to the pDDA:ptPA blend monolayer, all these values are smaller than the relevant values (101 mN m−1, 154 mN m−1, and 155 mN m−1). Especially for the smallest area (0.15 nm−2 for pDDA-b-ptPA), the value decreased to 1/3, suggesting instability of the pDDA-b-ptPA monolayer at higher pressures. It is particularly interesting that the compressibility moduli of pDDA-b-ptPA monolayers are influenced by the mixing component. The Cs−1 value at around 0.25 nm−2 increases selectively from 138 mN m−1 to 152 mN m−1 by blending with pDDA (blue in Fig. 4(b)), although the value is not changed by blending with ptPA (orange in Fig. 4(b)). The plateau region is also influenced by blending: expanding (blending with ptPA) and contracting (blending with pDDA). Results suggest that each domain in the pDDA-b-ptPA monolayer is enhanced by mixing with the corresponding component, and that the Cs−1 value is dependent on the introduced counterpart. Introduction of the homopolymer pDDA, which has high Cs−1, enhances the Cs−1 value of the blend monolayer up to a similar level to the pDDA:ptPA blend monolayer. This result indicates that the pDDA block in pDDA-b-ptPA takes a disordered structure because of the covalent junction between two blocks. As a consequence, several DDA units from the junction might be involved in the disordered ptPA domains. Therefore, the monolayer stability was reduced at higher pressures than 33.1 mN m−1. Compared with the pDDA:ptPA blend monolayer, the plateau region expands such that the surface-limiting area of pDDA domains becomes smaller after the ptPA domains collapse.
Table 4 Cs−1 peak values, corresponding surface pressure and surface area
| Polymer |
S*/nm−2 |
π*/mN m−1 |
Cs−1/mN m−1 |
| pDDA-b-ptPA |
0.30 |
3.03 |
58.8 |
| 0.25 |
20.3 |
138 |
| 0.15 |
37.9 |
46.8 |
| pDDA:ptPA = 76:24 |
0.30 |
7.84 |
101 |
| 0.27 |
26.5 |
154 |
| 0.22 |
39.5 |
155 |
| pDDA:pDDA-b-ptPA = 1:1 |
0.30 |
3.52 |
63.7 |
| 0.24 |
23.4 |
152 |
| 0.19 |
38.6 |
56.2 |
| ptPA:pDDA-b-ptPA = 1:1 |
0.29 |
4.64 |
70.0 |
| 0.25 |
19.1 |
140 |
| 0.12 |
40.0 |
43.5 |
| pDDA |
0.25 |
30.3 |
165 |
| ptPA |
0.27 |
17.5 |
118 |
4. Conclusions
We demonstrated controlled polymerization of acrylamide monomers with long alkyl chains and block copolymer synthesis with different amphiphilic acrylamide comonomers. RAFT polymerization of DDA monomer was conducted by varying the type of CTA, and the concentration ratio of CTA agent to AIBN initiator. The reaction kinetics were tracked using NMR and SEC measurements, proving that the reaction proceeded in the RAFT mechanism only in the low [CTA]/[AIBN] ratios, thereby suggesting competition with the termination reaction and the importance of suppressing the non-controlled free radical polymerization path. The apparent pseudo-first-order propagation rate constant of pDDA in toluene was found to be kapp = 5.7 × 10−5 s−1, which was slightly smaller than that of ptPA in toluene with a short alkyl chain (kapp = 6.4 × 10−5 s−1). One plausible explanation for the difference is the steric hindrance of long dodecyl chains. The trithiocarbonate groups in the obtained pDDA macro-CTAs were removed easily by treatment with tris(trimethylsilyl)silane, resulting in no damage of the polymer chains. Amphiphilic block copolymer, pDDA-b-ptPA was obtained in high conversion by polymerizing tPA as the second block of pDDA macro-CTA. The condensation and collapse processes of the block copolymer monolayer were investigated using an LB trough. The obtained block copolymer, pDDA-b-ptPA, formed a stable monolayer at the air–water interface with collapse pressure as high as πc = 33.1 mN m−1. The pDDA domain in the block copolymer monolayer takes a disordered structure compared with that in the pDDA:ptPA blend monolayer. Results suggest that amphiphilic acrylamide block copolymers are effective materials for phase-separated structure formations. This study presents the possibility of pDDA-based block copolymers in terms of self-assembled structures on a nanometre scale. The monolayer behaviour depends on the molecular weight of amphiphilic polymer.40 Additional work on block copolymer synthesis and film formation as well as amphiphilic acrylamide block copolymer nano-assemblies is expected to provide important insights into nanoscale bottom-up technologies.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partly supported by Grants-in-Aid for Young Scientists ((B) 26810111) from JSPS and for Scientific Research on Innovative Areas (New Polymeric Materials Based on Element-Blocks, 15H00719) from MEXT and the Murata Science Foundation.
References
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39, 1877–2336 RSC.
- X. Zhao, F. Pan, H. Xu, M. Yaseen, H. Shan, C. A. E. Hauser, S. Zhang and J. R. Lu, Chem. Soc. Rev., 2010, 39, 3480–3498 RSC.
- M. Guardingo, E. Bellido, R. Miralles-Llumà, J. Faraudo, J. Sedó, S. Tatay, A. Verdaguer, F. Busqué and D. Ruiz-Molina, Small, 2014, 10, 1594–1602 CrossRef CAS PubMed.
- S. Yamamoto, S. Uchiyama, T. Miyashita and M. Mitsuishi, Nanoscale, 2016, 8, 5912–5919 RSC.
- R. Lund, J. Ang, J. Y. Shu and T. Xu, Biomacromolecules, 2016, 17, 3964–3972 CrossRef CAS PubMed.
- R. A. Segalman, B. McCulloch, S. Kirmayer and J. J. Urban, Macromolecules, 2009, 42, 9205–9216 CrossRef CAS.
- H. G. Yoo, M. Byun, C. K. Jeong and K. J. Lee, Adv. Mater., 2015, 27, 3982–3998 CrossRef CAS PubMed.
- S. Yamamoto, H. Yasuda, H. Ohkita, H. Benten, S. Ito, S. Miyanishi, K. Tajima and K. Hashimoto, J. Phys. Chem. C, 2014, 118, 10584–10589 CAS.
- Y. He, T. Tu, M. Su, C. Yang, K. V. Kong and Y. Chan, J. Am. Chem. Soc., 2017, 139, 4218–4224 CrossRef CAS PubMed.
- H. Abdalla, S. Fabiano and M. Kemerink, Phys. Rev. B, 2017, 95, 85301 CrossRef.
- M. Mitsuishi, J. Matsui and T. Miyashita, Polym. J., 2006, 38, 877–896 CrossRef CAS.
- J. Y. Park and R. C. Advincula, Soft Matter, 2011, 7, 9829–9843 RSC.
- K. Ariga, Y. Yamauchi, T. Mori and J. P. Hill, Adv. Mater., 2013, 25, 6477–6512 CrossRef CAS PubMed.
- T. Miyashita, Y. Mizuta and M. Matsuda, Br. Polym. J., 1990, 22, 327–331 CAS.
- T. Miyashita, Prog. Polym. Sci., 1993, 18, 263–294 CrossRef CAS.
- T. Taniguchi, Y. Yokoyama and T. Miyashita, Macromolecules, 1997, 30, 3646–3649 CrossRef CAS.
- J. Kumaki and T. Hashimoto, J. Am. Chem. Soc., 1998, 120, 423–424 CrossRef CAS.
- H. Aoki, Y. Kunai, S. Ito, H. Yamada and K. Matsushige, Appl. Surf. Sci., 2002, 188, 534–538 CrossRef CAS.
- J. Y. Park, N. Koenen, M. Forster, R. Ponnapati, U. Scherf and R. Advincula, Macromolecules, 2008, 41, 6169–6175 CrossRef CAS.
- I. I. Perepichka, Q. Lu, A. Badia and C. G. Bazuin, Langmuir, 2013, 29, 4502–4519 CrossRef CAS PubMed.
- P. C. dos S. Claro, M. E. Coustet, C. Diaz, E. Maza, M. S. Cortizo, F. G. Requejo, L. I. Pietrasanta, M. Ceolín and O. Azzaroni, Soft Matter, 2013, 9, 10899–10912 RSC.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087–1097 RSC.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1996, 28, 1721–1723 CrossRef.
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. A. N. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed.
- D. B. Thomas, A. J. Convertine, L. J. Myrick, C. W. Scales, A. E. Smith, A. B. Lowe, Y. A. Vasilieva, N. Ayres and C. L. McCormick, Macromolecules, 2004, 37, 8941–8950 CrossRef CAS.
- Y. Cao, X. X. Zhu, J. Luo and H. Liu, Macromolecules, 2007, 40, 6481–6488 CrossRef CAS.
- M. T. Savoji, S. Strandman and X. X. Zhu, Macromolecules, 2012, 45, 2001–2006 CrossRef CAS.
- W. M. Wan, P. D. Pickett, D. A. Savin and C. L. McCormick, Polym. Chem., 2014, 5, 819–827 RSC.
- P. Raffa, D. A. Z. Wever, F. Picchioni and A. A. Broekhuis, Chem. Rev., 2015, 115, 8504–8563 CrossRef CAS PubMed.
- A. Graillot, S. Monge, C. Faur, D. Bouyer and J.-J. Robin, Polym. Chem., 2013, 4, 795–803 RSC.
- D. Roy, A. Ullah and B. S. Sumerlin, Macromolecules, 2009, 42, 7701–7708 CrossRef CAS.
- M. Drache, G. Schmidt-Naake, M. Buback and P. Vana, Polymer, 2005, 46, 8483–8493 CrossRef CAS.
- V. J. Cunningham, Y. Ning, S. P. Armes and O. M. Musa, Polymer, 2016, 106, 189–199 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polym. Int., 2011, 60, 9–25 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- B. Li, Y. Wu, M. Liu and A. R. Esker, Langmuir, 2006, 22, 4902–4905 CrossRef CAS PubMed.
- F. Q. Fan and T. Miyashita, Chem. Lett., 1999, 28, 669–670 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Kinetic plot showing ptPA synthesis and monolayer stability at a constant pressure. See DOI: 10.1039/c7ra06788g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Tokuji Miyashita and
Masaya Mitsuishi
*,
Tokuji Miyashita and
Masaya Mitsuishi



