
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deciphering chemical bonding in BnHn2− (n = 2–17): flexible multicenter bonding
Yan-Fang Shena,
Chang Xu*a and
Long-Jiu Cheng *ab
*ab
aDepartment of Chemistry, Anhui University, Hefei, Anhui 230601, P. R. China. E-mail: clj@ustc.edu; xuchang_1986@hotmail.com; Fax: +86-0551-63861435; Tel: +86-0551-63861435
bAnhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Hefei, Anhui 230601, P. R. China
First published on 25th July 2017
Abstract
It is well known that closo-borane dianions BnHn2− are stable aromatic cages and possess n + 1 valence electron pairs, in accordance with Wade's rules. However, the electronic structures of closo-BnHn2− cannot be explained by a Lewis structure because of their electron-deficient character, while recent theoretical and experimental studies reveal that multicenter bonding is a key part in their electronic structures. In this work, flexible multicenter bonding of BnHn2− (n = 2–17) is studied using the adaptive natural density partitioning (AdNDP) method in order to get insight into their stability and aromaticity. The large HOMO–LUMO gaps and negative NICS values indicate their close-shell electronic structure. Further chemical bonding analysis shows that all 2n + 2 delocalized valence electrons in closo-BnHn2− are involved in various delocalized σ and π multicenter bonding systems on their cage surface, well matched with different symmetric configurations. There are five types of multicenter bonds, including an open 3c–2e BBB bond, closed 3c–2e BBB bond, 4c–2e bond, 8c–2e bond and a fully delocalized bond, which are delocalized on a B3 broken-line, B3 triangle, B4 rhombus, B8 double-ring and the whole surface of the boron cage, respectively. Our work reveals the flexibility of multicenter bonding in diversified closo-BnHn2− clusters, giving new insights into the bonding nature of BnHn2−.
1. Introduction
The chemistry of boron is intriguing due to its electron-deficient character and delocalized bonding pattern. As a result, boron based clusters have rich forms of nanostructures, including quasi-planar, ring, two-dimensional sheet and cage.1–22 Over the past decade, small boron clusters have been systematically characterized both experimentally and theoretically to exhibit planar or quasi-planar structures in their ground states except for B14, but undergo the 2D-3D transition at B20 for neutral, at B27− for anionic and at B16+ for cationic clusters.5,23–29 The double-ring tubular B20 is confirmed to be stable and aromatic.30–32 The B40 cage is the first all-boron fullerene composed of hexagons and heptagons, according to the theoretical and experimental observation by photoelectron spectroscopy.33 In addition, there are also many fascinating sandwich-type complexes, metalloborospherenes, metal-centered boron molecular wheels and drums by doping metallic elements in boron clusters.25,34–49 Chemical bonding analyses reveal that there is numerous flexible multicenter bonding in the electronic structures of all these clusters.The polyhedral closo-borane BnHn2− clusters have attracted much interest from theoretical and experimental chemists since their synthesis in the early 20th century,50–58 especially for their bonding patterns. Lipscomb pointed out that the 3c–2e hydrogen bridge bond is the key part in the electronic structure of borane clusters, which was proved by experimental study. In 1971, Wade's rules gave the insight into topologic structures of deltahedral BnHn2− clusters. It revealed that there are (n + 1) electron pairs delocalized on their closo cage surface, where n is the number of vertexes.59 Mingos extended this principle to electron rich clusters to become popularly known as the Wade–Mingos rules.60,61 In 1977, King and co-workers investigated the three-dimensional aromaticity in BnHn2− using algebraic graph-theoretical methods, providing the theoretical basis for the stability of these clusters.62 Whereafter, O'Neill and Wade tried to apply the Kekulé-type structures which using chemical bonding instead of the delocalized structures to closo-borane dianions for suggesting the existence of 3c–2e bonds.63,64 In 2011, Lentz group obtained crystal structures of B10H102− and B12H122−, and revealed that both of them have 3c–2e bonds by electron density and atoms-in-molecules (AIM) analysis.65
Previous study reveals the structures and stability of closo-borane BnHn2− from their topologic structures according to Wade's rules, rather than in the view of chemical bonding. However, the chemical bonding pattern in the series of closo-borane BnHn2− has not been studied yet. As multicenter bonding is a popular language in the community of chemistry which is wildly used to interpret the electronic structure, stability and aromaticity of boron clusters, in this paper we try to reveal the nature of electronic structure in borane dianions BnHn2− (n = 2–17) series from this way.
2. Computational methods
In this article, the geometrical structures of borane dianions BnHn2− (n = 2–17) are optimized using TPSSh functional which has been proved to be reliable in the study of boron clusters.66–70 The optimization and the calculations of the nucleus-independent chemical shifts (NICS)71 are performed at the TPSSh/6-311+G* level, the HOMO–LUMO gaps are estimated using time-dependent density-functional theory (TD-DFT) method. NICS value is a popular measurement for aromaticity, where negative value indicates aromaticity, and positive value implies antiaromaticity. The vibrational frequencies are also checked to confirm the stability of structures at the same level of theory. All the DFT calculations are carried out in GAUSSIAN 09 package.72 Chemical bonding analysis are performed using the adaptive natural density partitioning (AdNDP) method.73 The AdNDP analysis is an effective tool developed by Boldyrev's group for deciphering the nature of the localized and delocalized bonds in boron clusters.14,74–76 Since the size of basis sets does not affect much of the AdNDP calculations, the 3-21G basis set is employed. Noncovalent interaction (NCI)77 are plotted using Multiwfn78 and VMD packages.79 The visualization of the AdNDP results are realized using the MOLEKEL 5.4.0.8 program.803. Results and discussion
3.1 Geometries and stability
The borane dianions BnHn2− (n = 2–17) series are optimized at TPSSh/6-311+G* level. The BnHn2− (n = 6–12) and (n = 4, 5, 13–17) have been observed experimentally and investigated theoretically, respectively.51–55,81,82 In order to grasp the structural features and assure the integrity in this family, BnHn2− with n = 2 and 3 are considered as well. The structures of borane dianions are taken from the previous studies of Lipscomb, Schleyer, Dreuw, and their co-workers.50,81,82 Fig. 1 plots the geometric structures of BnHn2− (n = 2–17). B2H22− and B3H32− are isovalent to C2H2 and C3H3+, as we expected, they possess alike linear and planar structures, respectively. B4H42− is neither planar nor tetrahedral, but somewhere in between, exhibiting a D2d-symmetric geometry. Borane dianions BnHn2− (n = 5–17) start to favor closo-structures with increasing cluster size, including n + 1 electron pairs, in accordance with Wade's rules. The smallest member of closo-borane family that has been synthesized is B6H62−, of which calculated bond length is 1.740 Å close to the experimental value (1.700 Å).83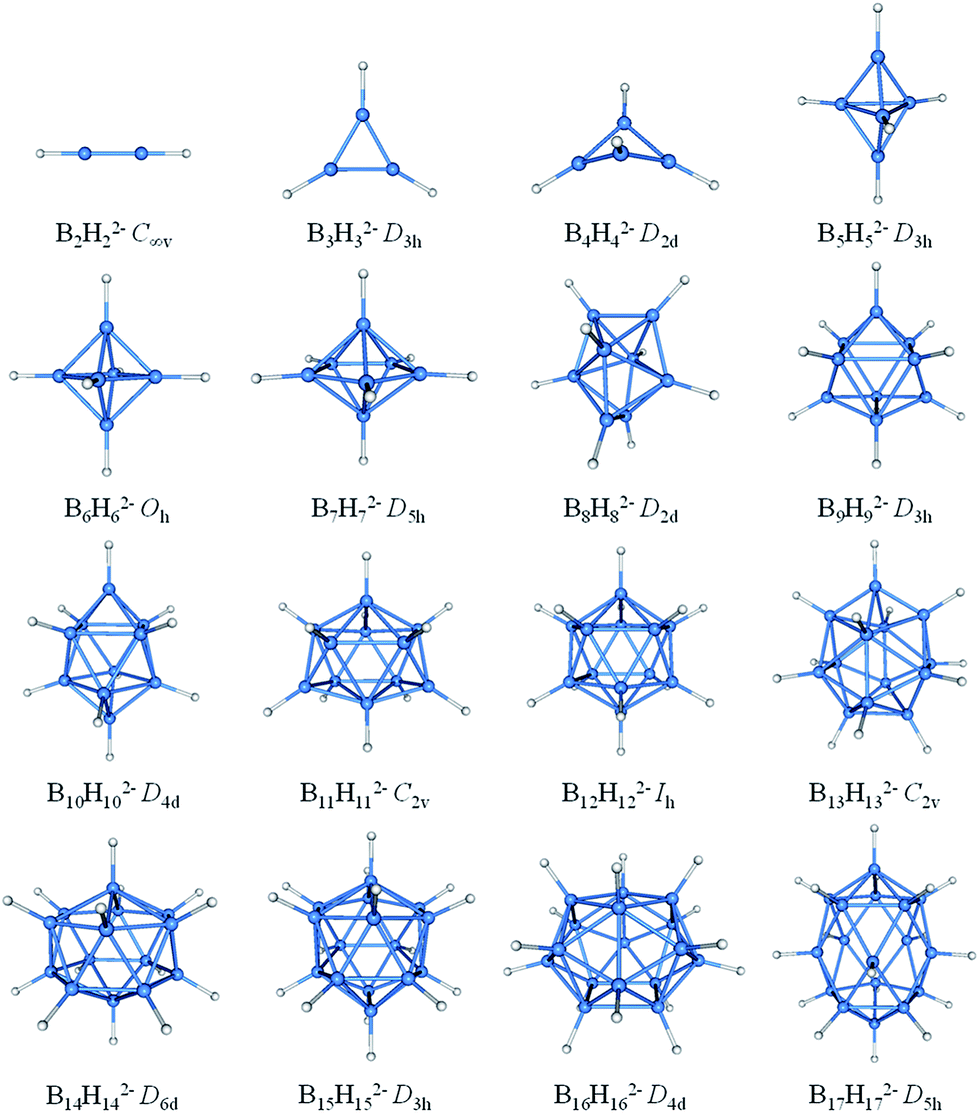 | ||
| Fig. 1 Optimized geometries for borane dianions BnHn2− (n = 2–17) at TPSSH/6-311+G* level of theory. B-blue; H-white. | ||
To investigate the stability of BnHn2− series, the HOMO–LUMO energy gaps (EHL) are computed using TPSSH/6-311+G* level. As shown in Fig. 2a, the HOMO–LUMO energy gaps at n ≤ 4 are lower than ones at n ≥ 5, the reason is supposed be that the structures are open-shaped at n ≤ 4, but getting closed at n ≥ 5. Moreover, for BnHn2− (n ≥ 5), there are four valleys at n = 8, 11, 13, 15, probably due to the lower symmetry of clusters comparing with other borane dianions. The relative stability of the system is further evaluated with the second-order difference in the total energies (Δ2E), which can be defined as: Δ2E(BnHn2−) = E(Bn−1Hn−12−) + E(Bn+1Hn+12−) − 2E(BnHn2−), where E(Bn−1Hn−12−), E(Bn+1Hn+12−), E(BnHn2−) are total energies of the clusters Bn−1Hn−12, Bn+1Hn+12−, BnHn2−. The second-order difference in energies is previously applied to study the relative stability of other boron clusters, and in its curves, positive peaks correspond to more relatively stable structures.84,85 The Δ2E plot in Fig. 2b shows that, four peaks present at B6H62−, B10H102−, B12H122−, B14H142−, suggesting their higher relative stability than their neighbours. In addition, it can be found that, each positive Δ2E value in Fig. 2b can correspond to a peak in Fig. 2a, the highest Δ2E and EHL value is found for B12H122−, indicating the highest one in the series of dianions considered.
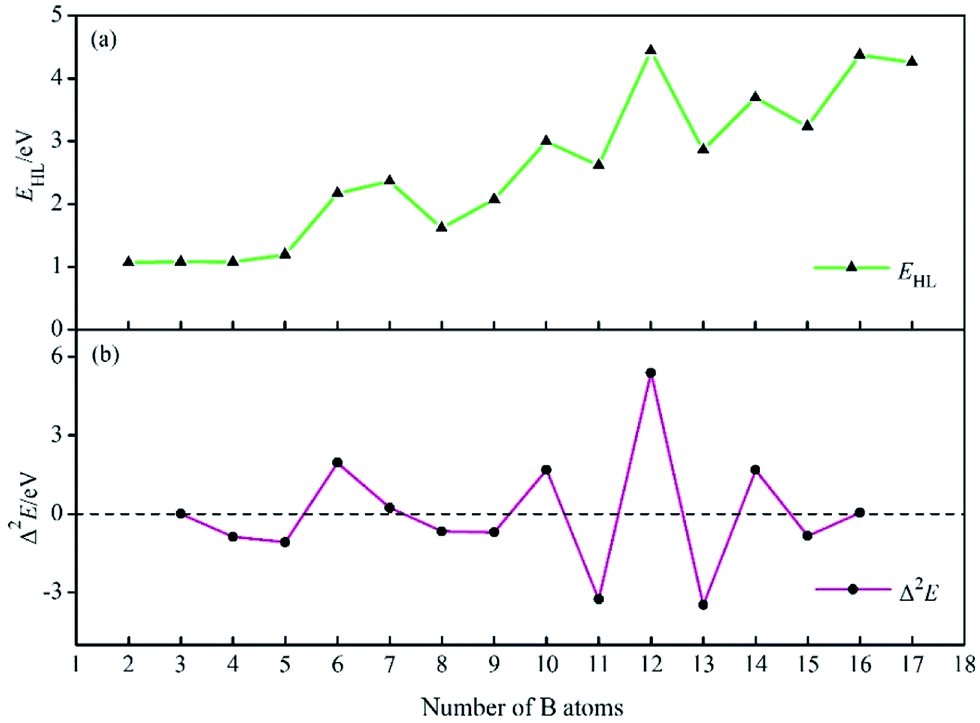 | ||
| Fig. 2 HOMO–LUMO energy gaps EHL (a) and the second-order differences in energy Δ2E (b) of BnHn2− (n = 2–17). | ||
3.2 Chemical bonding patterns
The theoretical study in 1963, Lipscomb's topological model involving styx numbers and rules inferred that closo-BnHn2− had (n − 2) 3c–2e BBB bonds and three 2c–2e BB bonds, which was somewhat limited in applicability.50,86 Until today, the bonding nature of closo-BnHn2− is still one of the many intrigued mysteries in chemistry because of their complicated structure and electron deficiency. Apart from theoretical calculations of geometries, energies and aromaticity for closo-borane, no attempt to characterize chemical bonding in this family by means of AdNDP method has been carried out. Herein, chemical bonding analysis by AdNDP gives a more straightforward evidence for the multicenter bonding in BnHn2− (n = 2–17). The AdNDP method is wildly applied for closed-shell species recovering both the classical Lewis bonding concepts (lone-pairs and 2c–2e bonds) and the delocalized nc–2e bonds.As shown in Fig. 3, B–B bond lengths in B2H22− is 1.507 Å somewhat longer than a typical B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) B bond, while the B–B distance in B3H32− is 1.634 Å between single B–B and double B
B bond, while the B–B distance in B3H32− is 1.634 Å between single B–B and double B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B bonds. In contrast with the previously reported C2H2 and C3H3+ both geometries and bonds are similar for B2H22− and B3H32−, respectively.87 Then, we succeed in finding all expected multicenter bonds in these two clusters. For B2H22−, there are two 2c–2e σ-BH bonds and three 2c–2e BB bonds (BB triple bonds, including one σ and two π bonds) with occupy number (ON) 1.99|e|, 2.00|e|, respectively. In B3H32−, there are three 2c–2e σ-BH bonds, three 2c–2e σ-BB bonds and one delocalized 3c–2e π-BBB bond (ON = 2.00|e|, 1.95|e| and 2.00|e|, respectively). Similarly, B4H42− has 10 valence electrons, featuring four 2c–2e σ-BH bonds, four 2c–2e σ-BB bonds and one 4c–2e delocalized over four boron atoms π-bond. Thus, bonding systems in boron framework of B3H32− and B4H42− are π-aromatic (2 electrons on one delocalized π-bond). B5H52− is the least stable member in closo-BnHn2− and has never been synthesized. It has a three-membered ring with two caps in D3h symmetry, the equatorial bonds are 1.827 Å and the axial bonds are 1.687 Å. Besides five 2c–2e σ-BH bonds, AdNDP analysis gives two bonding patters of the remained electrons of B5H52−. Pattern I includes three 3c–2e boron bridge σ bonds in the vertical direction and three delocalized 4c–2e σ bonds. Note that the open 3c–2e σ-BBB bond is formed by two apical B atoms and one of equatorial B atoms, while the apical B atom provides half of electron and the equatorial B atom provides one electron, respectively, such boron bridge bond was only observed previously in neutral borane (such as B10H14).50 Pattern II includes six 3c–2e σ-BBB bonds on six triangles with ON = 1.90|e| which is smaller than the open one. Thus, we think pattern I is more reasonable.
B bonds. In contrast with the previously reported C2H2 and C3H3+ both geometries and bonds are similar for B2H22− and B3H32−, respectively.87 Then, we succeed in finding all expected multicenter bonds in these two clusters. For B2H22−, there are two 2c–2e σ-BH bonds and three 2c–2e BB bonds (BB triple bonds, including one σ and two π bonds) with occupy number (ON) 1.99|e|, 2.00|e|, respectively. In B3H32−, there are three 2c–2e σ-BH bonds, three 2c–2e σ-BB bonds and one delocalized 3c–2e π-BBB bond (ON = 2.00|e|, 1.95|e| and 2.00|e|, respectively). Similarly, B4H42− has 10 valence electrons, featuring four 2c–2e σ-BH bonds, four 2c–2e σ-BB bonds and one 4c–2e delocalized over four boron atoms π-bond. Thus, bonding systems in boron framework of B3H32− and B4H42− are π-aromatic (2 electrons on one delocalized π-bond). B5H52− is the least stable member in closo-BnHn2− and has never been synthesized. It has a three-membered ring with two caps in D3h symmetry, the equatorial bonds are 1.827 Å and the axial bonds are 1.687 Å. Besides five 2c–2e σ-BH bonds, AdNDP analysis gives two bonding patters of the remained electrons of B5H52−. Pattern I includes three 3c–2e boron bridge σ bonds in the vertical direction and three delocalized 4c–2e σ bonds. Note that the open 3c–2e σ-BBB bond is formed by two apical B atoms and one of equatorial B atoms, while the apical B atom provides half of electron and the equatorial B atom provides one electron, respectively, such boron bridge bond was only observed previously in neutral borane (such as B10H14).50 Pattern II includes six 3c–2e σ-BBB bonds on six triangles with ON = 1.90|e| which is smaller than the open one. Thus, we think pattern I is more reasonable.
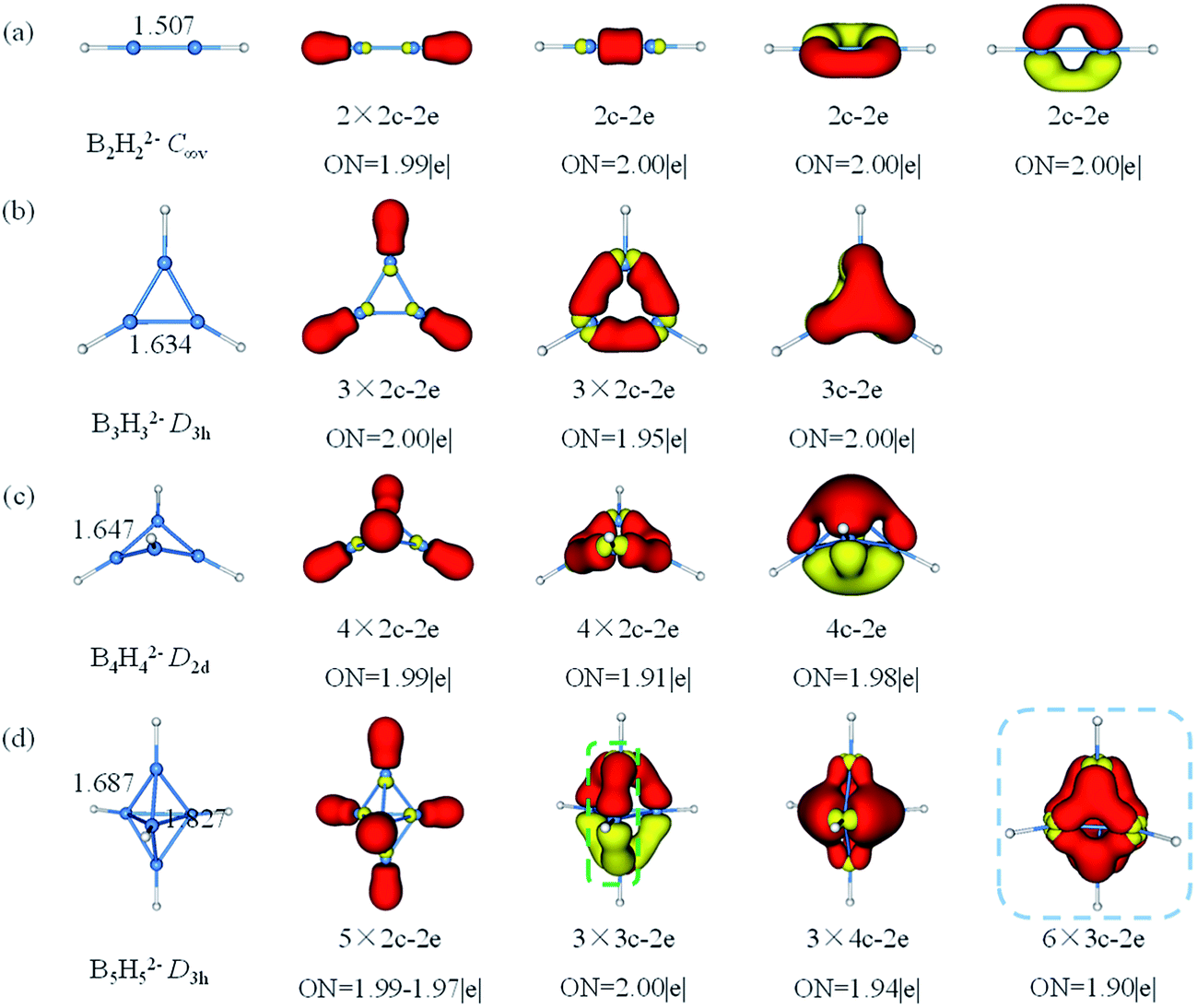 | ||
| Fig. 3 Structures and AdNDP localized natural bonding orbitals of borane dianions. (a) B2H22− (b) B3H32− (c) B4H42− (d) B5H52−. B-blue; H-white. | ||
The results of AdNDP analysis of BnHn2− (n = 6–8) are plotted in Fig. 4. B6H62− is octahedral resonance structure, two bonding patterns are given in consideration of resonance effect. Pattern I includes six 2c–2e σ-BH bonds, six 3c–2e σ-BBB bonds and one delocalized 6c–2e σ bond, which is selected one of three-fold axis as main axis on account of the resonance effect. From pattern II, these 6c–2e orbitals can be clearly viewed as super S, Px,y,z and Dxy,yz,xz orbitals. According to the spherical Jellium model,88 18-electron is the most stable magic number (S2P6D10). The five D orbitals split into two sets in the octahedral field, and energy level of the double-degenerate orbital (Dz2 and Dx2−y2) increase due to the repulsion of BH bonds. Thus, 14-electron becomes stable magic number. For D5h B7H72−, the equatorial bonds are 1.663 Å indeed smaller than the axial bonds (1.829 Å), which is prone to form 4c–2e bond. The σ-bonding framework includes seven 2c–2e σ-BH bonds and five delocalized 4c–2e σ bonds, the π-bonding framework consists of three delocalized 7c–2e π bonds. This type of π-bonding framework of B7H72− is analogous to that of benzene. The bonding in B8H82− can be explained by the formation of eight 2c–2e σ-BH bonds, four 3c–2e σ bonds, four 4c–2e σ bonds and one delocalized 8c–2e π bonds.
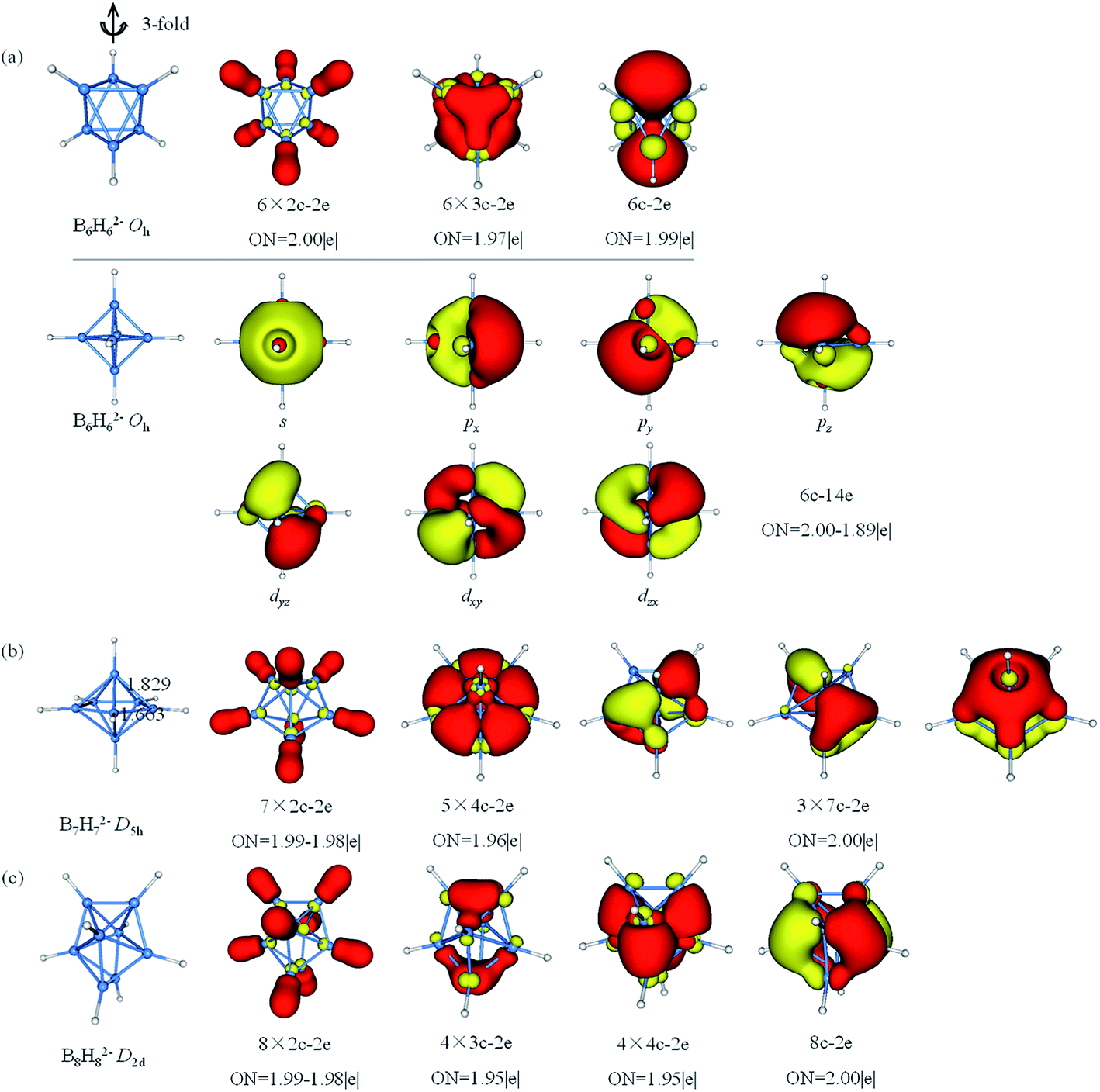 | ||
| Fig. 4 Structures and AdNDP localized natural bonding orbitals of borane dianions. (a) B6H62− (b) B7H72− (c) B8H82−. B-blue; H-white. | ||
The AdNDP analysis of BnHn2− (n = 9–11) are shown in Fig. 5. The B9H92− has a D3h symmetry which can be divided into three layers (each layer has three B atoms). The AdNDP results reveal that there are 2c–2e σ-BH bonds, six 3c–2e σ bonds and four delocalized 9c–2e bonds. The six 3c–2e σ-BBB bonds are formed by six triangular lattices of middle layer B atoms bonding with top and bottom B3 triangles. The four 9c–2e delocalized bonds can be classified into two sets including three 9c–2e delocalized π bonds and one 9c–2e delocalized σ bond, giving rise to double aromaticity (the delocalized π-system has 6 electrons following the (4n + 2) rule for π-aromaticity and similarly for the delocalized σ-system). B10H102− is square anti-prism geometry with two caps in D4d symmetry. Three different distances of two B atoms are given in Fig. 5b, the bond lengths of two equatorial B atoms in (1.837 Å) or between (1.820 Å) two four-membered rings are clearly longer than the B–B distance (1.708 Å) between apical and equatorial B atoms. Thus, we guess that there are multicenter bonds in B8 “double-ring”. The AdNDP analysis reveals ten 2c–2e σ-BH bonds, eight 3c–2e σ-BBB bonds and three delocalized 8c–2e σ bonds, leading to σ-aromaticity. There are 46 valence electrons altogether in B11H112− with C2v symmetry. The results of the AdNDP analysis include eleven 2c–2e σ-BH bonds and twelve 3c–2e σ-BBB bonds with occupation numbers ranging from 1.77–1.92|e|.
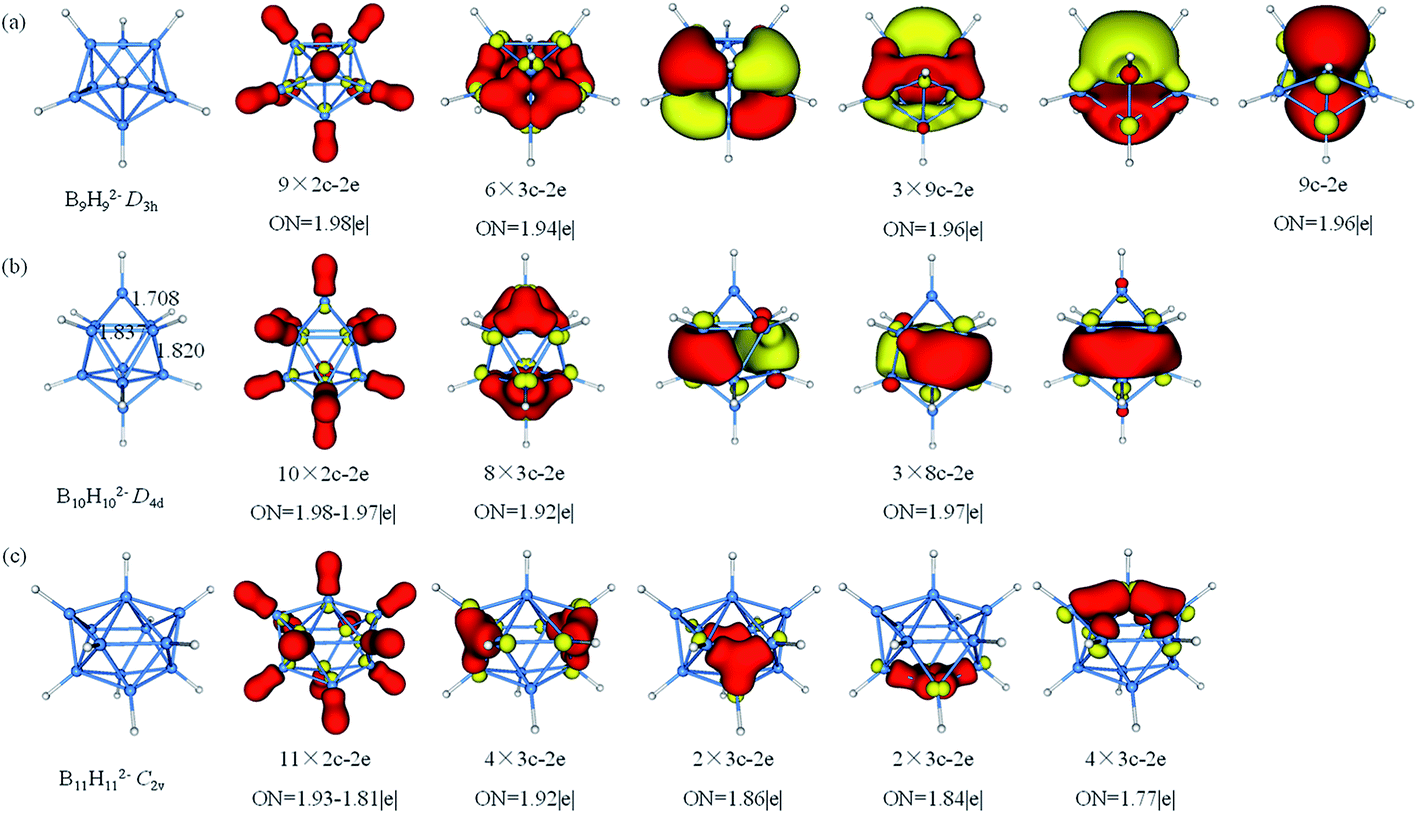 | ||
| Fig. 5 Structures and AdNDP localized natural bonding orbitals of borane dianions. (a) B9H92− (b) B10H102− (c) B11H112−. B-blue; H-white. | ||
The AdNDP analysis of BnHn2− (n = 12–14) are displayed in Fig. 6. The icosahedral B12H122− cluster is the most stable molecule among polyhedral boranes observed experimentally up to now.52,89,90 All of B atoms in Ih B12H122− are equivalent, therefore, it can be treated for AdNDP analysis in similar way as B6H62−. Pattern I includes twelve 2c–2e σ-BH bonds, six 3c–2e σ-BBB bonds, six 4c–2e σ bonds and one delocalized 12c–2e bond. From pattern II, these orbitals are fulfilled with 26 valence electrons (S2P6D10F8), the B12H122− possesses thirteen 12c–2e bonds and forms the closed-shell spherical structure. Similarly, seven F orbitals split into two sets in an icosahedral field, and only the lower quadruple-degenerate orbitals are filled. Both B13H132− and B11H112− are in lowest symmetry and have a relatively simple bonding pattern in this family. The B13H132− dianion has 54 valence electrons that form thirteen 2c–2e σ-BH bonds and fourteen delocalized 3c–2e σ-BBB bonds distributing on the left and right, top and bottom, front and back, in an overall symmetry of C2v. B14H142− is composed of two six-membered rings and two caps with D6d symmetry. In contrast to the bond lengths in B10H102−, B14H142− tends to bond between two six-membered rings. B14H142− possesses fourteen 2c–2e σ-BH bonds, twelve 3c–2e σ-BBB bonds on twelve B3 triangles forming a “double-ring”, and three delocalized 14c–2e delocalized σ bonds, making the contribution to the σ-aromaticity of B14H142−.
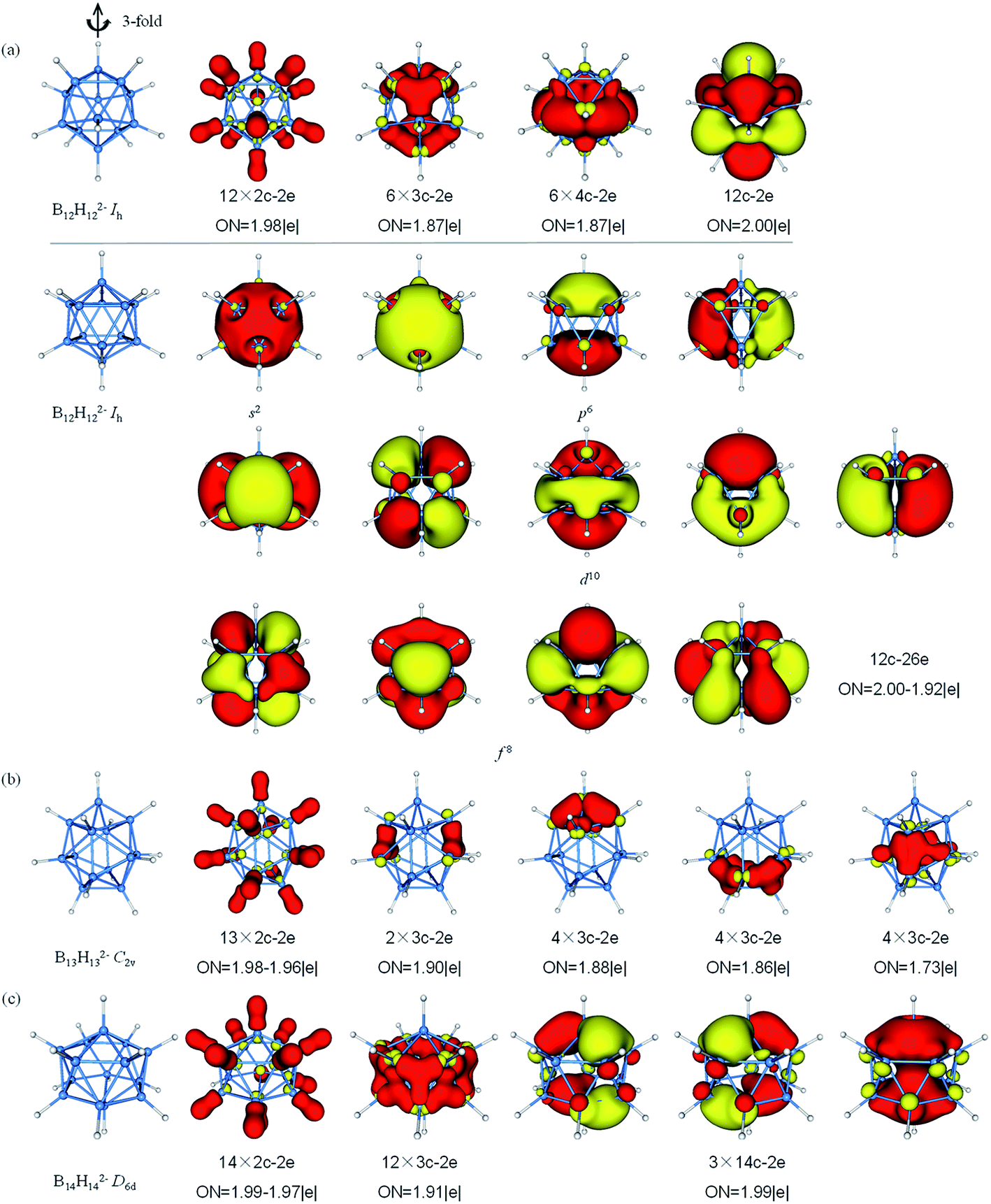 | ||
| Fig. 6 Structures and AdNDP localized natural bonding orbitals of borane dianions. (a) B12H122− (b) B13H132− (c) B14H142−. B-blue; H-white. | ||
The fifteen B atoms of D5h B15H152− can be classified into five three-membered rings of which can be available for bonding. There are 62 valence electrons in B15H152−, AdNDP analysis (Fig. 7) yields fifteen 2c–2e σ-BH bonds involving all atoms, six 3c–2e σ-BBB bonds which are shared by six B3 triangles, three delocalized 4c–2e σ bonds around the middle of cage, six delocalized 4c–2e σ bonds covering on surface and one delocalized 15c–2e bond. The D4d B16H162− features a similar bonding pattern of B12H122−, with sixteen 2c–2e σ-BH bonds, eight 3c–2e σ-BBB bonds, eight delocalized 4c–2e σ bonds (are in a cycle just like a lantern) and one delocalized 16c–2e bond. B17H172− is regard as “a hot candidate” for synthesis due to its high stability, aromaticity and symmetry through the theoretical study.81 The 70 valence electrons in B17H172− can be divided into three sets. The first set consists of localized bonding elements, seventeen 2c–2e σ-BH bonds, while the other two sets are composed of delocalized bonding elements, fifteen 4c–2e σ bonds (on the top, around the waist and at the bottom) and three 17c–2e delocalized bonds. The delocalized bonding can be viewed as three concentric σ-systems and one π-system, satisfying the (4n + 2) rule for aromaticity, the similar delocalized bonding has been investigated in planar B13+ which has two concentric σ-systems and one π-system.91
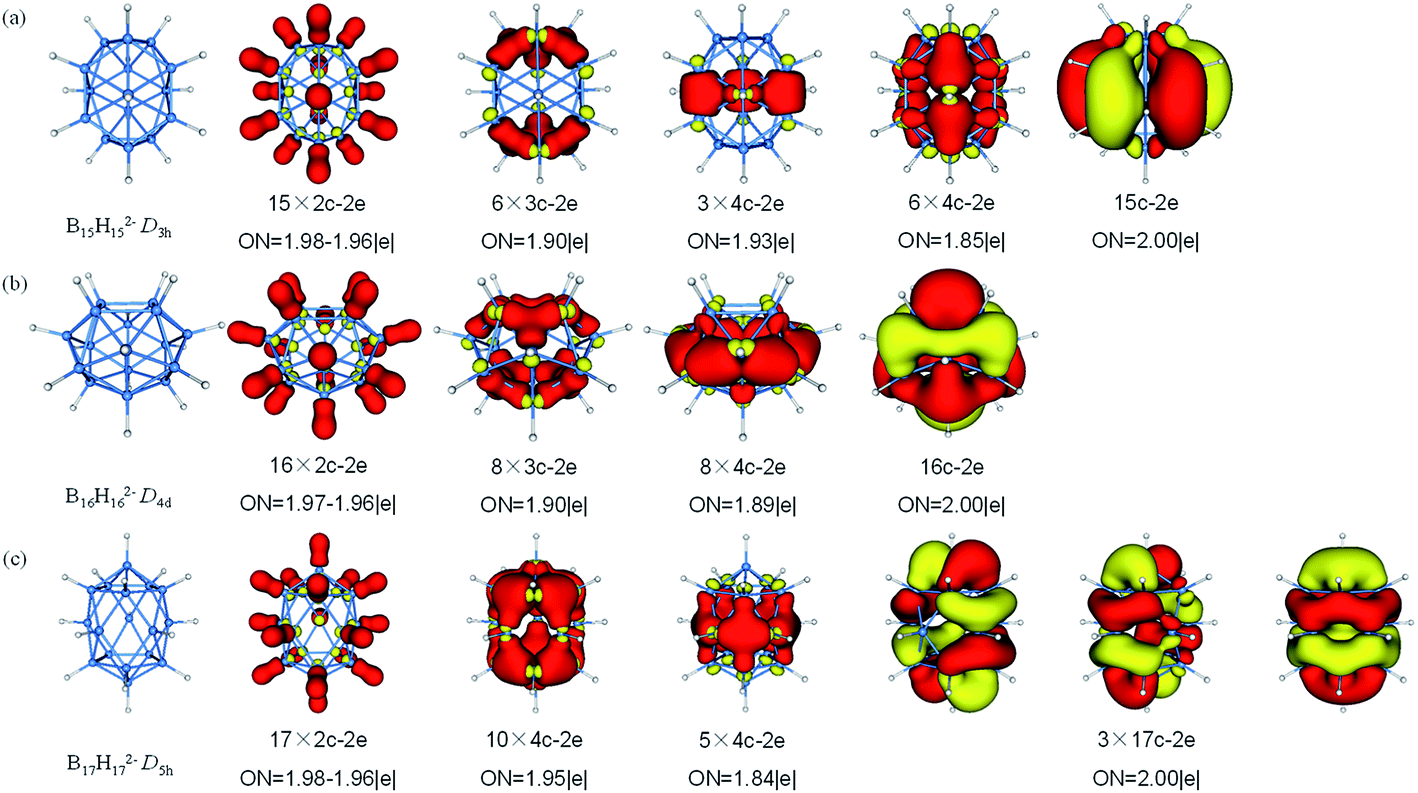 | ||
| Fig. 7 Structures and AdNDP localized natural bonding orbitals of borane dianions. (a) B15H152− (b) B16H162− (c) B17H172−. B-blue; H-white. | ||
3.3 Aromaticity
From all above, it is believed that borane dianions with cage structure are highly stable from their relative energies. The stability and aromaticity can also be analyzed with the aid of a noncovalent interaction (NCI) method. The NCI index is based on the electron density (ρ) and the reduced density gradient (RDG, s = 1/[2(3π2)1/3]|∇ρ|/ρ4/3), which can indicate the presence of noncovalent interactions with low-s low-ρ and covalent bonds with low-s high-ρ. The second ingredient of the NCI index adopts the second density Hessian eigenvalue (λ2) to distinguish between bonding interactions (λ2 < 0) and non-bonding interactions (λ2 > 0). We believe that the NCI characteristic peak with disparate values of borane cage depends on their bonding patterns and all delocalized valence electrons distribute on the cage surface.Fig. 8 plots the reduced density gradient (s) versus electron density (ρ) multiplied by the sign of λ2 and the low-gradient (s = 0.10) NCI isosurfaces of the cage structure B12H122− and B10H102−. As is illustrated in Fig. 8a, the high density, low-gradient spikes lie at about sign (λ2) ρ = −0.160 and −0.126 a.u. for B12H122−, which represent strong covalent attractions. The more negative spike is clearly associated to the B–H covalent bonds which can be seen from NCI isosurfaces in the right part of Fig. 8, whereas the other strong attractive spike is the strong covalent interaction between boron atoms, indicating high stability and σ delocalization of B12H122− cage. Similarly, there are one attractive spike (−0.160 a.u.) and the other two attractive spike (−0.136 and −0.108 a.u.) associated to the B–H covalent bonds and strong covalent interaction between boron atoms for B10H102−, which correspond to the blue regions of isosurfaces at the top (or bottom) and in the middle, respectively. Moreover, for B12H122− and B10H102−, there are positive spikes at sign (λ2) ρ = 0.119 and 0.103 a.u., respectively, which represent steric interactions among the boron atoms with the red regions of the isosurfaces. The results in Fig. 8 reveal that the spikes of B–H covalent interactions lie at about sign (λ2) ρ = −0.160, and the rest of negative spikes with more intensive dots are because all the skeletal bonding electrons (in Wade's rules) of B10H102− and B12H122− are delocalized on the cage surface causing high stability. In addition, the number of NCI characteristic peak with negative values of B10H102− is one more than that of B12H122−, which is due to the two different kinds of boron atoms in the former one, whereas the boron atoms of B12H122− are equivalent.
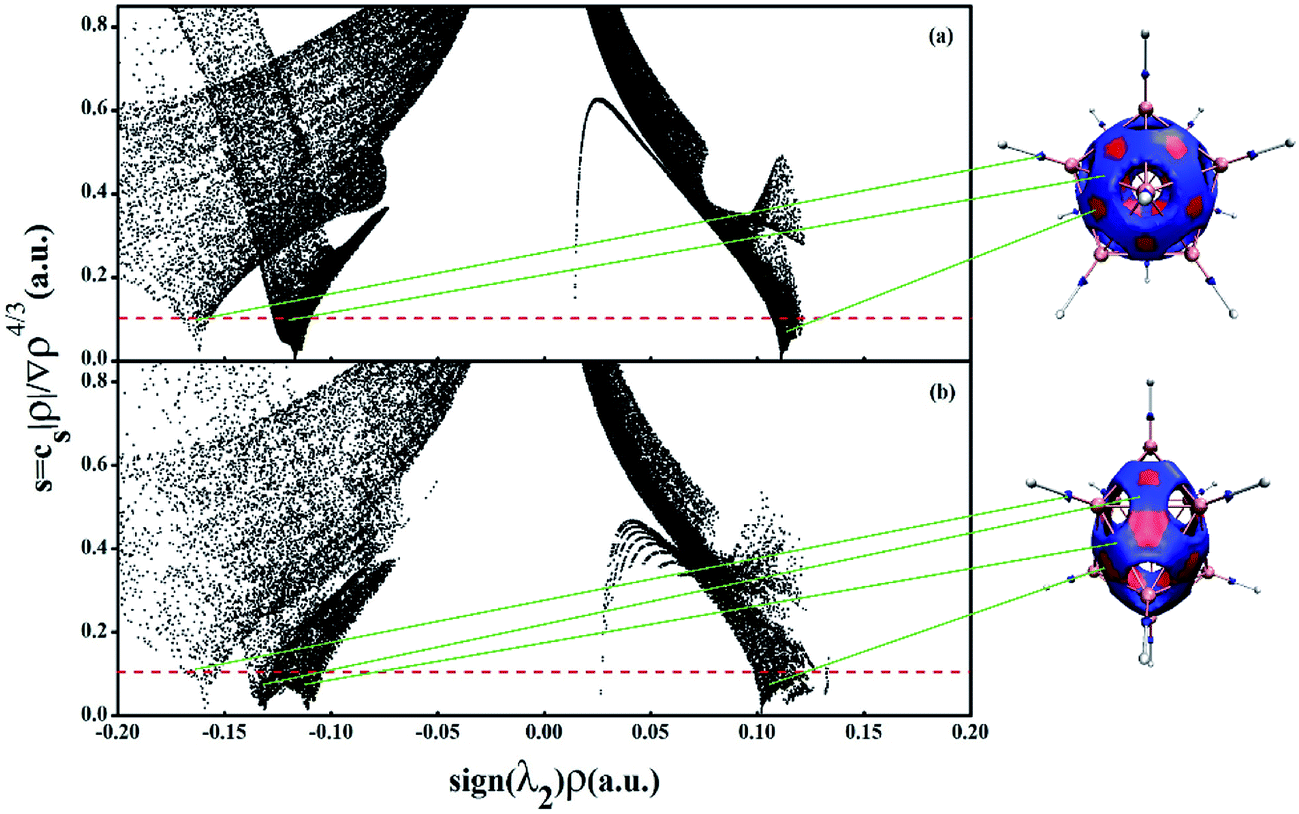 | ||
| Fig. 8 Plots of the reduced density gradient versus the electron density multiplied by the sign of the second Hessian eigenvalue sign (λ2) (left) and NCI isosurfaces using a blue-green-red color scale at s = 0.10 for B12H122− (a) and B10H102− (b), respectively. | ||
In 2016, Poater and coworkers applied Hückel (4n + 2) rule and Wade's rules in closo-BnHn2− to establish a connection between three-dimensional aromatic closo boron hydride clusters and planar aromatic annulenes.92 Herein, NCI is a direct evidence to identify the aromaticity of clusters, and the aromatic characteristic of BnHn2− can obtain from their NICS value and electronic structures. We calculate the NICS values of above dianions except B2H22− out of consideration for its line structure. As shown in Fig. 9, the negative NICS values suggest that all structures are aromatic. It should be noted that the tendency is accordant with EHL gaps and NICS values (one wanes and the other one waxes), which indicates their close-shell electronic structures. For B3H32− and B4H42−, both of them have two π electrons obeying (4n + 2) Hückel rule with π-aromaticity. BnHn2− with n = 5–17 possess large negative NICS values from −17.91 to −29.13 ppm as a comparison with that of benzene of −11.92 ppm. B5H52− is a watershed of structure and property in this system, which includes two categories of six delocalized σ electrons causing double σ-aromaticity in vertical and vertical direction, respectively. The Oh B6H62− and Ih B12H122− have two delocalized σ electrons along their five and ten 3-fold axis of symmetry as a consequence of resonance, respectively, showing large negative NICS values and EHL gaps, which display high stability and strong aromaticity. The D5h B7H72− has ten σ and six π delocalized electrons leading to (σ + π) aromaticity. B8H82− is less stable with lower EHL gap and smaller negative NICS value in the lower D2d symmetry, the reason is probably that, there are two π and sixteen σ delocalized electrons, rendering π-aromaticity and σ-antiaromatic in B8H82−, according to the (4n + 2) rule for aromaticity and 4n rule for antiaromaticity, respectively, but finally the former is dominated and counteract the latter. Likewise, B9H92− and B10H102− are (σ + π) aromaticity and σ-aromaticity with 6πdelo–2σdelo (six π delocalized and two σ delocalized) and 2σdelo electrons, respectively. Both B11H112− and B13H132− have lower EHL gaps and smaller negative NICS values than their neighbors, which perhaps on account of their lower symmetry, but are aromatic with close-shell electronic structures. The aromaticity of B14H142− derives from its six delocalized σ electrons. The delocalized electrons of B15H152− can be segmented into six parts, which include 5 × 6σdelo electrons from top to bottom and 2σdelo electrons around whole structure, according to Hückel rule. The result of B16H162− is similar to that of B8H82−, but allowing higher aromaticity with higher symmetry. B17H172− also has high σ-aromaticity with four types of delocalized electrons (3 × 10σdelo and 6σdelo).
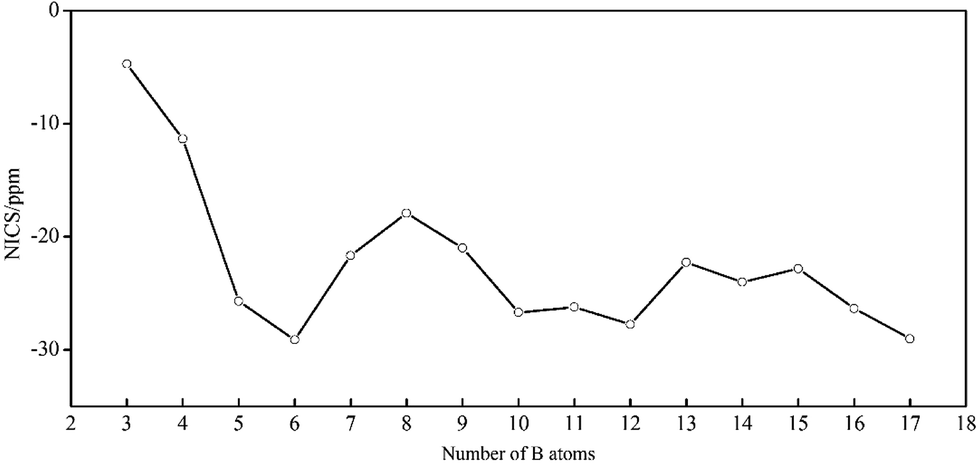 | ||
| Fig. 9 Plot of Nucleus-Independent Chemical Shifts (NICS) for borane dianions BnHn2− (n = 3–17) (in ppm). | ||
3.4 Flexibility of multicenter bonding
According to the bonding patterns of closo-BnHn2−, their available topologic structures are exhibited in different lines and colors. As depicted in Fig. 10, there are different types of delocalized multicenter bonds, including open 3c–2e bond, close 3c–2e bond, 4c–2e bond, 8c–2e bond and fully delocalized bond, which corresponds to B3 broken-line, B3 triangle, B4 rhombus, B8 double-ring and the whole surface of boron cage in closo-BnHn2−, respectively. The location of multicenter bond is related to the relative sizes of multi-membered rings and caps of clusters. In B10H102−, the 3c–2e bonds distribute on B3 triangles of two caps at the top and bottom but of two multi-membered rings for B14H142−. Moreover, the bonding pattern of closo-BnHn2− are diversified both with their symmetry and with different B–B bond lengths. In B8H82−, B12H122− and B16H162−, there are four, six and eight 3c–2e bonds on B3 triangles, respectively, lying on the top and bottom, while the 4c–2e bonds distribute on B4 rhombuses with longer B–B bond length. Thus, the flexibility of multicenter bonding can adapt well to all borane cages with different configurations. However, it is difficult to grasp clear generalizations of closo-borane clusters which is part of the attraction for study and distinct from carbon clusters.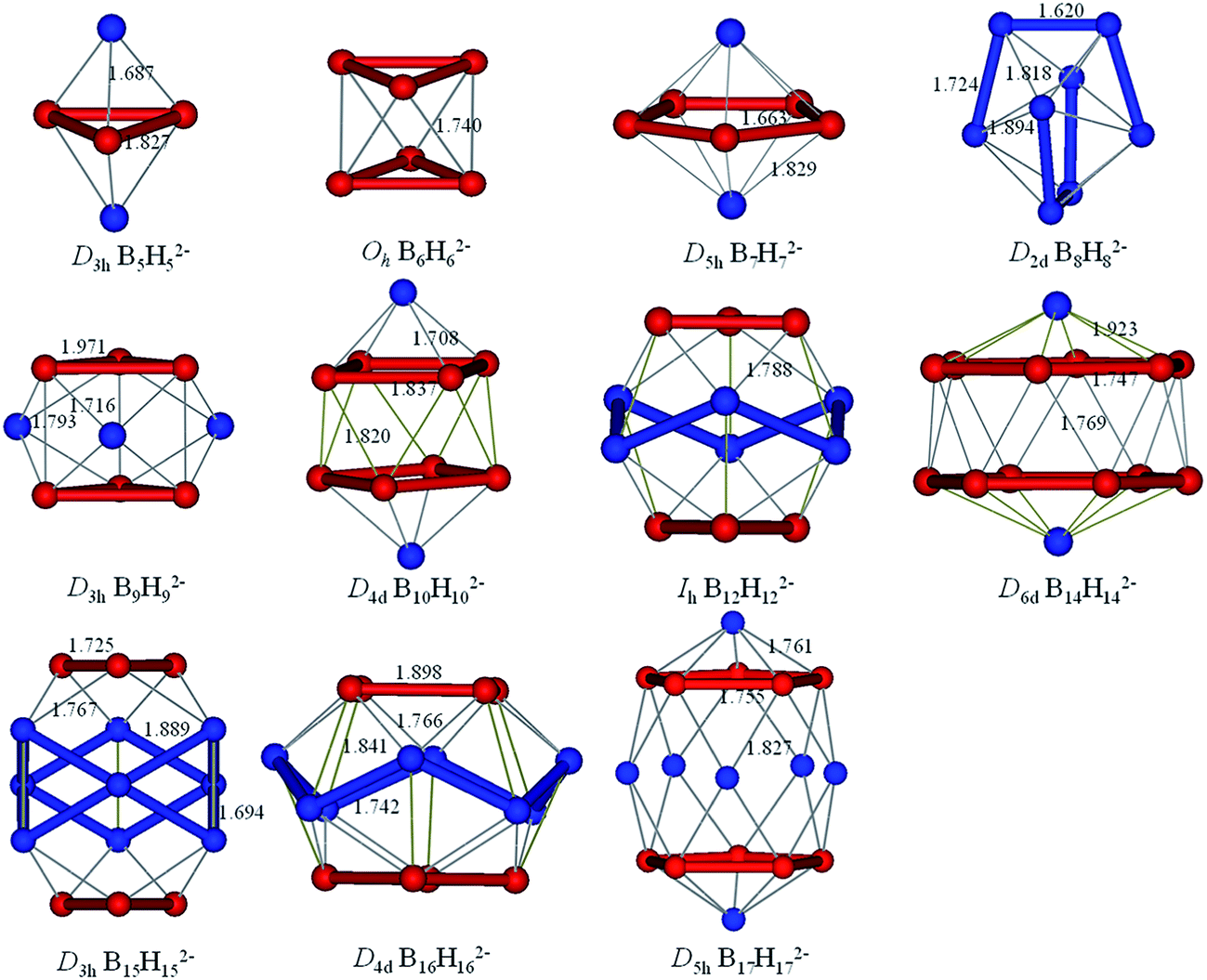 | ||
| Fig. 10 Topology structures for closo-borane dianions, BnHn2− (n = 5–10, 12, 14–17). | ||
4. Conclusion
The closo-borane dianion BnHn2− (n = 6–12) are proven to be stable and aromatic through the joint experimental and theoretical study. However, the bonding nature of closo-BnHn2− is still one of the mysteries in borane chemistry. In present work, we investigate the evolution rules of structures in BnHn2− (n = 2–17) and decipher their stability and aromaticity in the view of chemical bonding. BnHn2− with n = 2 and 3 are liner and planar structures, respectively, but undergo a transition between 2D and 3D structures at B4H42−. BnHn2− (n = 5–17) are inclined cage structures which have been explained by Wade's rules. There are (4n + 2) valence electrons in BnHn2−, the bonding in B2H22− and B3H32− are very similar to that in C2H2 and C3H3+, respectively. Moreover, in closo-BnHn2−, 2n valence electrons are involved in localized σ-BH bonds, the remaining (2n + 2) valence electrons are evenly delocalized in either multicenter σ/π bonds, forming an effective σ and π delocalized system causing high stability, and σ/π aromaticity. The bonding in B5H52− exits the 3c–2e open boron bridge bonds which was only observed in neutral borane preciously. For the high symmetry B6H62− and B12H122−, one type of bonding patterns is given in a particular way in which any 3-fold axis has two delocalized valence electrons due to their resonance, while the other type of bonding patterns are listed in terms of superatom, both leading to high stability and aromaticity. In addition, we extend the concentric aromaticity of planar B13+ to 3D B15H152− and B17H172− to explain their aromaticity. Moreover, it is found that the tendency of EHL gaps in closo-BnHn2− are in accordance with their NICS values which indicates their close-shell electronic structures. In short, the highlight of our work is deciphering chemical bonding in closo-BnHn2−, and the flexibility of multicenter bonding can adapt well to all borane cages with different configurations, making their stable and aromatic.Acknowledgements
This work is financed by the National Natural Science Foundation of China (21573001), and by the Foundation of Distinguished Young Scientists of Anhui Province. The calculations were carried out at the High-Performance Computing Center of Anhui University.References
- R. Kawai and J. H. Weare, J. Chem. Phys., 1991, 95, 1151–1159 CrossRef CAS.
- I. Boustani, Chem. Phys. Lett., 1995, 240, 135–140 CrossRef CAS.
- A. Ricca and C. W. Bauschlicher Jr, Chem. Phys., 1996, 208, 233–242 CrossRef CAS.
- I. Boustani, Phys. Rev. B, 1997, 55, 426–438 CrossRef.
- E. Oger, N. R. Crawford, R. Kelting, P. Weis, M. M. Kappes and R. Ahlrichs, Angew. Chem., Int. Ed., 2007, 46, 8503–8506 CrossRef CAS PubMed.
- J. J. Zhao, L. Wang, F. Y. Li and Z. F. Chen, J. Phys. Chem. A, 2010, 114, 9969–9972 CrossRef CAS PubMed.
- R. X. He and X. C. Zeng, Chem. Commun., 2015, 51, 3185–3188 RSC.
- T. B. Tai and M. T. Nguyen, Phys. Chem. Chem. Phys., 2015, 17, 13672–13679 RSC.
- T. B. Tai, L. V. Duong, H. T. Pham, D. T. Mai and M. T. Nguyen, Chem. Commun., 2014, 50, 1558–1560 RSC.
- C. B. Kah, M. Yu, P. Tandy, C. S. Jayanthi and S. Y. Wu, Nanotechnology, 2015, 26, 405701 CrossRef CAS PubMed.
- X. Y. Zhao, Q. Chen, H. R. Li, Y. W. Mu, H. G. Lu and S. D. Li, Phys. Chem. Chem. Phys., 2017, 19, 10998–11003 RSC.
- Q. Chen, W. L. Li, Y. F. Zhao, S. Y. Zhang, H. S. Hu, H. Bai, H. R. Li, W. J. Tian, H. G. Lu, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, ACS Nano, 2015, 9, 754–760 CrossRef CAS PubMed.
- Q. Chen, S. Y. Zhang, H. Bai, W. J. Tian, T. Gao, H. R. Li, C. Q. Miao, Y. W. Mu, H. G. Lu, H. J. Zhai and S. D. Li, Angew. Chem., 2015, 127, 8278–8282 CrossRef.
- Y. J. Wang, Y. F. Zhao, W. L. Li, T. Jian, Q. Chen, X. R. You, T. Ou, X. Y. Zhao, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, J. Chem. Phys., 2016, 144, 064307 CrossRef PubMed.
- T. B. Tai and M. T. Nguyen, Nanoscale, 2015, 7, 3316–3317 RSC.
- Z. A. Piazza, H. S. Hu, W. L. Li, Y. F. Zhao, J. Li and L. S. Wang, Nat. Commun., 2014, 5, 3113 Search PubMed.
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Nat. Chem., 2016, 8, 563 CrossRef CAS PubMed.
- W. L. Li, Y. F. Zhao, H. S. Hu, J. Li and L. S. Wang, Angew. Chem., Int. Ed., 2014, 53, 5540–5545 CrossRef CAS PubMed.
- W. L. Li, Q. Chen, W. J. Tian, H. Bai, Y. F. Zhao, H. S. Hu, J. Li, H. J. Zhai, S. D. Li and L. S. Wang, J. Am. Chem. Soc., 2014, 136, 12257–12260 CrossRef CAS PubMed.
- A. P. Sergeeva, Z. A. Piazza, C. Romanescu, W. L. Li, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2012, 134, 18065–18073 CrossRef CAS PubMed.
- A. P. Sergeeva, D. Y. Zubarev, H. J. Zhai, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2008, 130, 7244–7246 CrossRef CAS PubMed.
- W. Huang, A. P. Sergeeva, H. J. Zhai, B. B. Averkiev, L. S. Wang and A. I. Boldyrev, Nat. Chem., 2010, 2, 202–206 CrossRef CAS PubMed.
- A. N. Alexandrova, A. I. Boldyrev, H. J. Zhai and L. S. Wang, Coord. Chem. Rev., 2006, 250, 2811–2866 CrossRef CAS.
- L. J. Cheng, J. Chem. Phys., 2012, 136, 104301 CrossRef PubMed.
- I. A. Popov, T. Jian, G. V. Lopez, A. I. Boldyrev and L. S. Wang, Nat. Commun., 2015, 6, 8654 CrossRef CAS PubMed.
- D. Z. Li, H. Bai, T. Ou, Q. Chen, H. J. Zhai and S. D. Li, J. Chem. Phys., 2015, 142, 014302 CrossRef PubMed.
- A. P. Sergeeva, I. A. Popov, Z. A. Piazza, W. L. Li, C. Romanescu, L. S. Wang and A. I. Boldyrev, Acc. Chem. Res., 2014, 47, 1349–1358 CrossRef CAS PubMed.
- B. Kiran, G. G. Kumar, M. T. Nguyen, A. K. Kandalam and P. Jena, Inorg. Chem., 2009, 48, 9965–9967 CrossRef CAS PubMed.
- T. B. Tai, D. J. Grant, M. T. Nguyen and D. A. Dixon, J. Phys. Chem. A, 2010, 114, 994–1007 CrossRef CAS PubMed.
- B. Kiran, S. Bulusu, H. J. Zhai, S. Yoo, X. C. Zeng and L. S. Wang, PNAS, 2005, 102, 961–964 CrossRef CAS PubMed.
- T. B. Tai, A. Ceulemans and M. T. Nguyen, Chem.–Eur. J., 2012, 18, 4510–4512 CrossRef CAS PubMed.
- M. P. Johansson, J. Phys. Chem. C, 2009, 113, 524–530 CAS.
- H. J. Zhai, Y. F. Zhao, W. L. Li, Q. Chen, H. Bai, H. S. Hu, Z. A. Piazza, W. J. Tian, H. G. Lu, Y. B. Wu, Y. W. Mu, G. F. Wei, Z. P. Liu, J. Li, S. D. Li and L. S. Wang, Nat. Chem., 2014, 6, 727–731 CAS.
- S. D. Li, J. C. Guo, C. Q. Miao and G. M. Ren, Angew. Chem., Int. Ed., 2005, 44, 2158–2161 CrossRef CAS PubMed.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2011, 50, 9334–9337 CrossRef CAS PubMed.
- B. P. Fokwa and M. Hermus, Angew. Chem., Int. Ed., 2012, 51, 1702–1705 CrossRef CAS PubMed.
- Y. Yuan and L. J. Cheng, J. Chem. Phys., 2013, 138, 024301 CrossRef PubMed.
- L. F. Li, C. Xu, B. K. Jin and L. J. Cheng, Dalton Trans., 2014, 43, 11739–11744 RSC.
- M. Mbarki, R. St Touzani and B. P. Fokwa, Angew. Chem., Int. Ed., 2014, 53, 13174–13177 CrossRef CAS PubMed.
- B. Mondal, B. Mondal, K. Pal, B. Varghese and S. Ghosh, Chem. Commun., 2015, 51, 3828–3831 RSC.
- H. J. Zhai, A. N. Alexandrova, K. A. Birch, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2003, 42, 6004–6008 CrossRef CAS PubMed.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, J. Chem. Phys., 2013, 138, 134315 CrossRef PubMed.
- S. Li, Z. Zhang, Z. Long and S. Qin, Sci. Rep., 2017, 7, 40081 CrossRef CAS PubMed.
- Q. Chen, H. R. Li, C. Q. Miao, Y. J. Wang, H. G. Lu, Y. W. Mu, G. M. Ren, H. J. Zhai and S. D. Li, Phys. Chem. Chem. Phys., 2016, 18, 11610–11615 RSC.
- H. Bai, Q. Chen, H. J. Zhai and S. D. Li, Angew. Chem., Int. Ed., 2015, 54, 941–945 CrossRef CAS PubMed.
- Q. Chen, T. Gao, W. J. Tian, H. Bai, S. Y. Zhang, H. R. Li, C. Q. Miao, Y. W. Mu, H. G. Lu, H. J. Zhai and S. D. Li, Phys. Chem. Chem. Phys., 2015, 17, 19690–19694 RSC.
- W. L. Li, T. Jian, X. Chen, H. R. Li, T. T. Chen, X. M. Luo, S. D. Li, J. Li and L. S. Wang, Chem. Commun., 2017, 53, 1587–1590 CAS.
- W. L. Li, C. Romanescu, T. R. Galeev, Z. A. Piazza, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2012, 134, 165–168 CrossRef CAS PubMed.
- W. J. Tian, Q. Chen, X. X. Tian, Y. W. Mu, H. G. Lu and S. D. Li, Sci. Rep., 2016, 6, 8 CrossRef PubMed.
- W. N. Lipscomb, Boron Hydrides, ed. W. A. Benjamin, New York, 1963 Search PubMed.
- M. F. Hawthorne and A. R. Pitochelli, J. Am. Chem. Soc., 1959, 81, 5519 CrossRef CAS.
- A. R. Pitochelli and F. M. Hawthorne, J. Am. Chem. Soc., 1960, 82, 3228–3229 CrossRef CAS.
- J. L. Boone, J. Am. Chem. Soc., 1964, 86, 5036 CrossRef CAS.
- F. Klanberg and E. Muetterties, Inorg. Chem., 1966, 5, 1955–1960 CrossRef CAS.
- F. Klanberg, D. R. Eaton, L. J. Guggenberger and E. L. Muetterties, Inorg. Chem., 1967, 6, 1271–1281 CrossRef CAS.
- F. Schluter and E. Bernhardt, Inorg. Chem., 2011, 50, 2580–2589 CrossRef PubMed.
- N. Zint, A. Dreuw and L. S. Cederbaum, J. Am. Chem. Soc., 2002, 124, 4910–4917 CrossRef CAS PubMed.
- L. D. Brown and W. N. Lipscomb, Inorg. Chem., 1977, 16, 2989–2996 CrossRef CAS.
- K. Wade, J. Chem. Soc. D, 1971, 549, 792–793 RSC.
- D. M. P. Mingos, Nature (London), Phys. Sci., 1972, 236, 99–102 CrossRef CAS.
- D. M. P. Mingos, Acc. Chem. Res., 1984, 17, 311–319 CrossRef CAS.
- R. B. King and D. H. Rouvray, J. Am. Chem. Soc., 1977, 9, 7834–7840 CrossRef.
- M. E. O'Neill and K. Wade, THEOCHEM, 1983, 103, 259–268 CrossRef.
- M. E. O'Neill and K. Wade, Polyhedron, 1984, 3, 199–212 CrossRef.
- S. Mebs, R. Kalinowski, S. Grabowsky, D. Forster, R. Kickbusch, E. Justus, W. Morgenroth, C. Paulmann, P. Luger, D. Gabel and D. Lentz, Inorg. Chem., 2011, 50, 90–103 CrossRef CAS PubMed.
- Y. Yuan and L. J. Cheng, J. Chem. Phys., 2012, 137, 044308 CrossRef PubMed.
- L. F. Li, C. Xu and L. J. Cheng, Comput. Theor. Chem., 2013, 1021, 144–148 CrossRef CAS.
- L. F. Li, C. Xu, B. K. Jin and L. J. Cheng, J. Chem. Phys., 2013, 139, 174310 CrossRef PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- C. Xu, L. J. Cheng and J. L. Yang, J. Chem. Phys., 2014, 141, 124301 CrossRef PubMed.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- M. Frisch, G. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian 09, Revision B. 01, Gaussian Inc., Wallingford, CT, 2010 Search PubMed.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- W. L. Li, T. Jian, X. Chen, T. T. Chen, G. V. Lopez, J. Li and L. S. Wang, Angew. Chem., 2016, 128, 7484–7489 CrossRef.
- E. Osorio, J. K. Olson, W. Tiznado and A. I. Boldyrev, Chem.–Eur. J., 2012, 18, 9677–9681 CrossRef CAS PubMed.
- A. P. Sergeeva, B. B. Averkiev, H. J. Zhai, A. I. Boldyrev and L. S. Wang, J. Chem. Phys., 2011, 134, 224304 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- U. Varetto, Molekel 5.4.0.8, Swiss National Supercomputing Centre, Manno, Switzerland Search PubMed.
- P. v. R. Schleyer, K. Najafian and A. M. Mebel, Inorg. Chem., 1998, 37, 6765–6772 CrossRef CAS PubMed.
- A. Dreuw, N. Zint and L. S. Cederbaum, J. Am. Chem. Soc., 2002, 124, 10903–10910 CrossRef CAS PubMed.
- R. Schaeffer, Q. Johnson and G. S. Smith, Inorg. Chem., 1965, 4, 917–918 CrossRef CAS.
- J. F. Jia, L. J. Ma, J. F. Wang and H. S. Wu, J. Mol. Model., 2013, 19, 3255–3261 CrossRef CAS PubMed.
- D. T. Mai, L. V. Duong, T. B. Tai and M. T. Nguyen, J. Phys. Chem. A, 2016, 120, 3623–3633 CrossRef CAS PubMed.
- A. J. Welch, Chem. Commun., 2013, 49, 3615–3616 RSC.
- D. Y. Zubarev and A. I. Boldyrev, J. Org. Chem., 2008, 73, 9251–9258 CrossRef CAS PubMed.
- B. K. Teo, J. Cluster Sci., 2014, 25, 883–891 CrossRef CAS.
- W. S. Hopkins, P. J. Carr, D. Huang, K. P. Bishop, M. Burt, T. B. McMahon, V. Steinmetz and E. Fillion, J. Phys. Chem. A, 2015, 119, 8469–8475 CrossRef CAS PubMed.
- G. S. Nevill and C. J. Tymczak, Nanoscale Res. Lett., 2012, 7, 236 CrossRef PubMed.
- W. Huang, A. P. Sergeeva, H. J. Zhai, B. B. Averkiev, L. S. Wang and A. I. Boldyrev, Nat. Chem., 2010, 2, 202–206 CrossRef CAS PubMed.
- J. Poater, M. Sola, C. Vinas and F. Teixidor, Chem.–Eur. J., 2016, 22, 7437–7443 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |