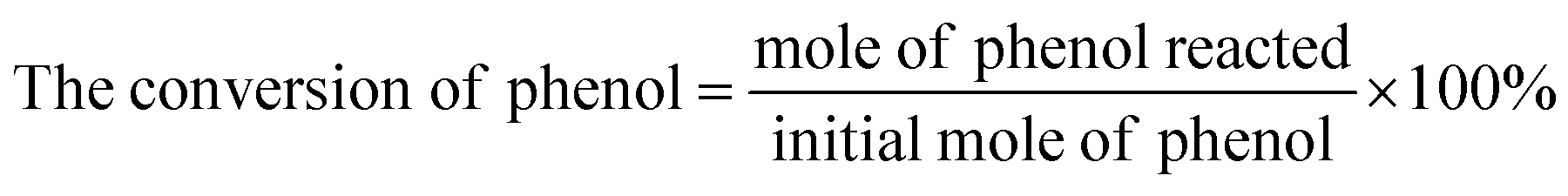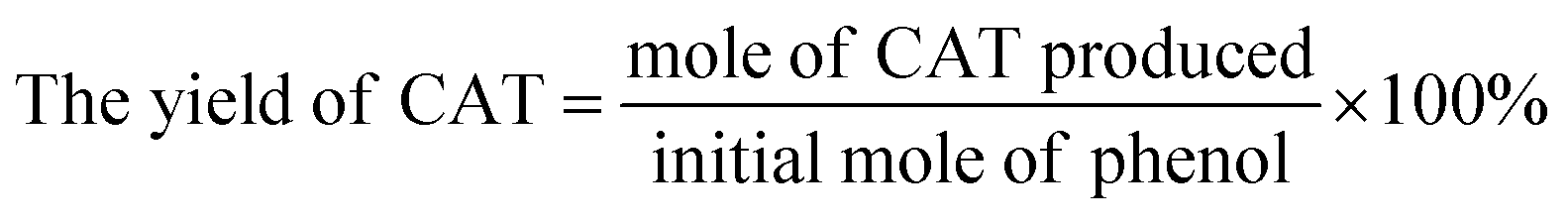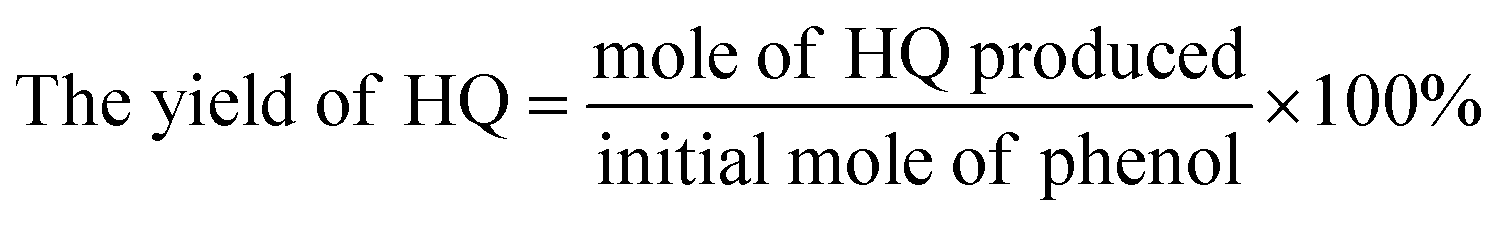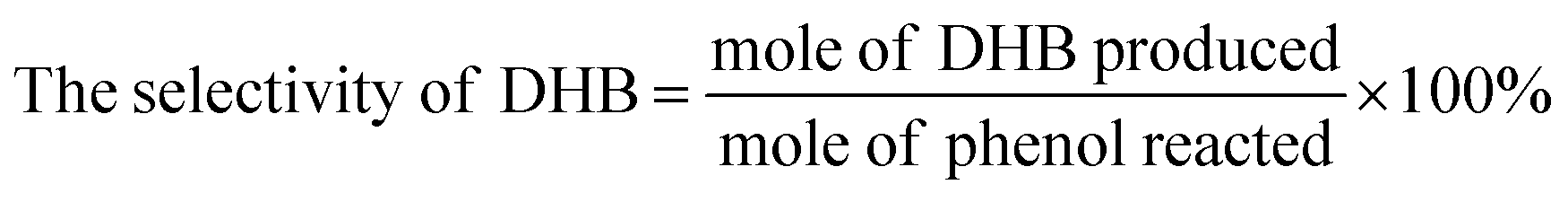DOI:
10.1039/C7RA06917K
(Paper)
RSC Adv., 2017,
7, 38691-38698
Ferrocene particles incorporated into Zr-based metal–organic frameworks for selective phenol hydroxylation to dihydroxybenzenes†
Received
21st June 2017
, Accepted 2nd August 2017
First published on 7th August 2017
Abstract
UiO-66 with high dispersibility and a cuboctahedron morphology was synthesized by an improved solvothermal method. The morphology of UiO-66 was adjusted using benzoic acid as a modulator. UiO-66 with a regular morphology was then used as the support to load ferrocene (Fc). A series of Fc@UiO-66 composites were prepared via a facile impregnation method. The composites were characterized by powder X-ray diffraction (XRD), high resolution transmission electron microscopy (HRTEM), scanning electron microscopy (SEM), SEM/EDX mapping, FT-IR, UV/vis, TG analysis, N2 adsorption–desorption, and X-ray photoelectron spectroscopy (XPS). The results showed that Fc was incorporated into UiO-66, thus preventing the agglomeration of Fc particles in water. The Fc@UiO-66 composites with a Fc loading of 5% (FU-5) exhibited the highest catalytic activity for hydroxylation of phenol with H2O2 at room temperature in water, which gave a phenol conversion of 38.5% and 87.4% selectivity for dihydroxybenzenes (DHB). UiO-66 played a crucial role in the enhancement of catalytic performance compared with conventional supports such as γ-Al2O3, etc. A hydroxyl radical mechanism was proposed for this catalytic hydroxylation process and the high selectivity was attributed to the interaction between Fc particles and UiO-66.
Introduction
Dihydroxybenzenes (DHB), including catechol (CAT) and hydroquinone (HQ) are important intermediates for preparing perfumes, dyes, medicines, pesticides, rubbers and other chemicals.1,2 The traditional method to synthesize DHB is the oxidation of aniline and diisopropylbenzene or the hydrolysis of o-chlorophenol and phenoldisulfonic acid.3 Although the selectivity of the target product in the traditional approaches was very high, DHB still faced the big challenge of being ideal choices in industry because of serious economic and environmental issues.4,5 The development of a new catalyst for more efficient phenol hydroxylation to obtain DHB has attracted more and more attention.6 The catalytic hydroxylation of phenol either driven by heat or light usually proceeded under rather mild conditions with H2O2 as a clean oxidant compared to the traditional routes.7,8 Among all of the transition metals, iron usually exhibits excellent catalytic activity for this transformation. Yang et al.9 have prepared Fe/wood-based activated carbon catalysts, which gave a phenol conversion of 51.1% and 80.6% selectivity for DHB. Adam et al.10 reported an iron modified zeolite nanocrystal (Fe/KL) catalyst with high selectivity to DHB (100%) and conversion of phenol (93.4%). Our group developed a Fe–Al-based photocatalyst, which give a phenol conversion of 64.9% and 95% of selectivity to DHB.11
Metal–organic frameworks (MOFs) have been widely explored in gas adsorption-storage, drug sustained release, catalysis, magnetic materials, etc., due to their large specific surface area and various choices of center metals and linker ligands.12–14 However, the instability to heat and acid of most MOFs always impede their application in many catalytic processes. UiO-66 is built up from hexanuclear zirconium clusters and the bridging ligand 1,4-benzenedicarboxylate (BDC), which was first reported by Lillerud's group.15 The strong Zr–O bonds make UiO-66 exhibit higher thermostability and chemical stability. Furthermore, UiO-66 also demonstrated the excellent stability of structure when soaked in water,16 which could still be retained after introduced with missing-linker defects and modified by active functional groups.17,18 Owing to the peculiarity of prominent structural stability, the development of UiO-66 based catalysts has attracted more and more research interests.19
The famous organometallic complex ferrocene (Fc) always exhibits good redox properties.20 However, it is hard to avoid the agglomeration when Fc is used in water due to its insolubility and hydrophobicity. Herein, we reported the preparation of a series of UiO-66 supported Fc composite via a simple impregnation method. The water dispersed Fc@UiO-66 was used as heterogeneous catalyst for the hydroxylation of phenol with H2O2 as oxidant, which showed high selectivity to DHB at room temperature in water.
Experimental methods
Chemicals
Zirconium tetrachloride (ZrCl4), terephthalic acid, benzoic acid and ferrocene (Aladdin Industrial Corporation, China) were used for synthesizing Fc@UiO-66 composites. Phenol and 30 wt% H2O2 (Jiangtian Chemicals, China) were used for the test of catalytic activity. HQ and CAT (Guangfu Chemicals, China) were used for calculating the standard curve method. All the solvents and reagents were commercially available with analytical purity (purity >99%) (besides the purity of HQ and CAT was chromatographic purity).
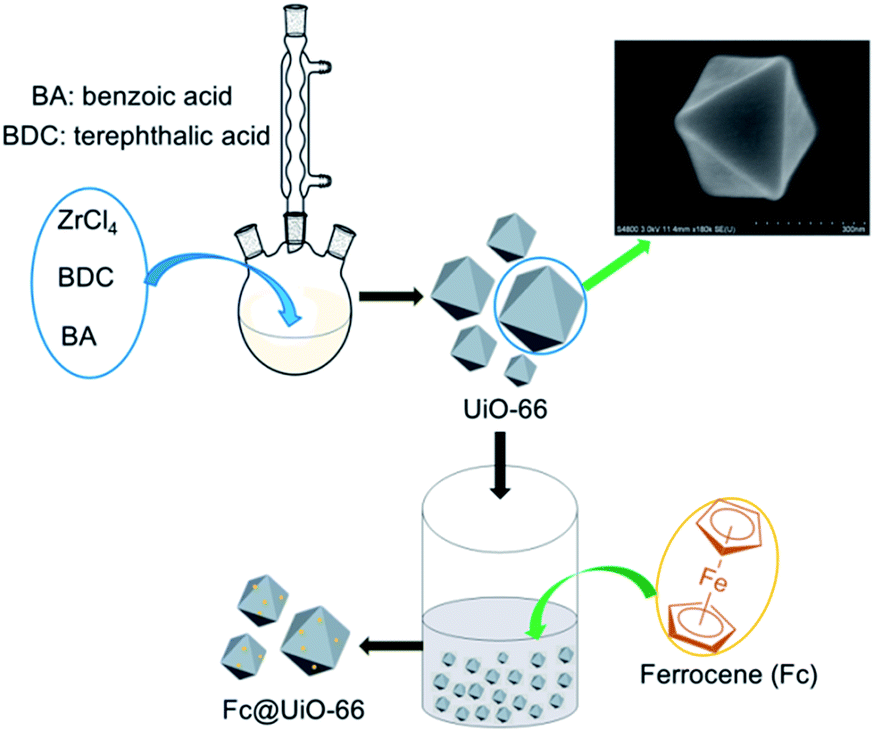
Preparation of UiO-66 support and Fc@UiO-66 composites
The UiO-66 support was synthesized by an improved solvothermal method, which was easier and safer compared with the traditional Teflon-lined autoclave. The yield of synthetic UiO-66 was as high as the traditional method, which could provide the source of assurance for the subsequent experiments. First, approximately 1.2 g ZrCl4 (5.1 mmol, 1 eq.) and 0.85 g terephthalic acid (5.1 mmol, 1 eq.) are dissolved in 240 mL N,N-dimethylformamide (DMF) in a 500 mL three-necked vial. Benzoic acid (0–50 eq.) was added to investigate the influence of modulator. The vial was heated to 120 °C in oil bath without stirring for 24 h. After cooling to the room temperature, the white precipitate (UiO-66) was collected by centrifugation and washed with 50 mL DMF and 50 mL ethanol, respectively. For further purification, UiO-66 was soaked in 50 mL ethanol overnight. After centrifugation, UiO-66 was placed in a vacuum oven at 100 °C for at least 8 h.
The Fc@UiO-66 composites were prepared by impregnation method. The preparation process of Fc@UiO-66 composite is shown in Scheme 1. Firstly, 1 g UiO-66 synthesized with 50 eq. benzoic acid was dispersed in 40 mL ethanol, then different dosage of Fc was dissolved in the suspension and the dosages of Fc were controlled by 10 mg, 50 mg, and 100 mg, respectively. Subsequently, the suspension was stirred for 2 h at room temperature. Finally, the suspension was heated to 100 °C until the ethanol was evaporated and the resultant samples were named as FU-1, FU-5, and FU-10.
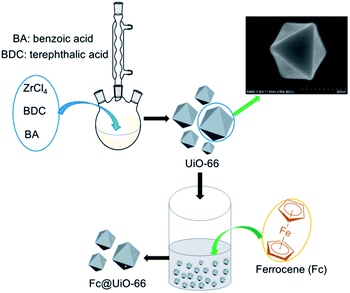 |
| | Scheme 1 The synthetic process of Fc@UiO-66 composites. | |
The preparation process of other supports composites catalyst was similar to FU-5.
Materials characterization
Powder X-ray diffraction (XRD) patterns were measured using a Rigaku D/max 2500V/PC diffractometer operated with Ge (111)-monochromatized Cu Kα radiation with a wavelength of λ = 0.154 nm in transmission, and the range of the diffraction angle was 5–50°; the scanning speed was 3° min−1. The reflections observed were indexed by using the Werner algorithm and were improved with least squares method. Scanning electron microscope (SEM) images were taken on a Hitachi S-4800 field emission SEM. The UV-vis absorption spectra were measured with the UV-vis diffuse reflectance spectra (UV-3600). Thermal gravimetric analyses (TGA) were obtained by a TA Q600 thermal analysis system under a continuous nitrogen flow with a heating rate of 10 °C min−1. High resolution transmission electron microscopy (HETEM) was conducted at 90 K using a JEOL-2011 equipped with a Bruker X-flash silicon drift detector (SDD) at 200 kV. FT-IR was measured on a Nicolet-380 spectrometer, and KBr was employed as the reference. N2 adsorption–desorption isotherms were performed on SSA4300 porosity and specific surface area analyzer with the samples degassed at 100 °C for 2 h before the test. XPS was measured on EscaLab 250Xi to analyze the surface composition.
Catalytic activity for phenol hydroxylation
The catalytic activities were evaluated by phenol hydroxylation. The experimental implementations were conducted in a 50 mL single-necked round bottom flask with a reflux condenser and a water bath kettle with magnetic stirrer. Typically, phenol (1 mmol) was dissolved in distilled water (20 mL), and the Fc@UiO-66 composites were added as catalyst, respectively. Then, the aqueous H2O2 (30 wt%, 4 mmol) was added all at once by pipettes. Water was used as the cheaper and more environment friendly solvent compared to the traditional organic solvents. When the reaction finished, the H2O2 in the mixture was detected with starch potassium iodide paper. The solutions after reaction were quantified by high performance liquid chromatography (HPLC, Agilent Zorbax SB-C18, and 4.6 mm × 250 mm, 5 μm) with a UV detector at 272 nm. The mobile phase was the mixed solvents of methanol and water (vMeOH/vH2O = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70). The flow rate of mobile phase was 1 mL min−1. The every injection volume was 10 μL.
:70). The flow rate of mobile phase was 1 mL min−1. The every injection volume was 10 μL.
The actual concentration of components was calculated by standard curve method. The conversion of phenol, the yield of CAT, HQ and the selectivity of the reaction were evaluated by the equations given below.
Results and discussion
Characterization of the catalyst
The SEM images of the as-synthesized UiO-66 crystalsare showed in Fig. 1, which were prepared in the presence of different amounts of modulator. The crystallinities and compositions of the as-synthesized UiO-66 were characterized by XRD and all the diffractions were high in accordance with the simulated XRD (ESI, Fig. S1†). Without BA modulator, the image of the UiO-66 crystals (Fig. 2a) exhibited disordered and agglomerative. With increasing amount of benzoic modulator, the UiO-66 crystals became more and more regular and dispersive. Finally, the UiO-66 crystals synthesized with 50 eq. benzoic acid formed the morphology of a regular cuboctahedron. This crystal growth tendency corresponded with the coordination modulation mechanism, in which the benzoic acid modulator influenced the growth of UiO-66 crystals by coordination with secondary building units during the formation of the framework. This may be due to the formation of intermediate complexes between benzoic ions and Zr clusters. When the amount of benzoic acid was increased, the competition between benzoic ions and linker molecules for the coordination of the Zr clusters became distinctly, resulting in slowing down the growth of UiO-66 crystals. Otherwise, some benzoic ions were selectively adsorbed on the (111) facets, which could reduce the surface energy of the (111) facets, resulting in the exposure of (111) facets. Therefore, UiO-66 crystals with the morphology of a cuboctahedron were obtained.21
 |
| | Fig. 1 SEM images of as-synthesized UiO-66. (a), (b), (c), (d): with 0 eq., 12.5 eq., 25 eq. and 50 eq. of modulator (benzoic acid), respectively. | |
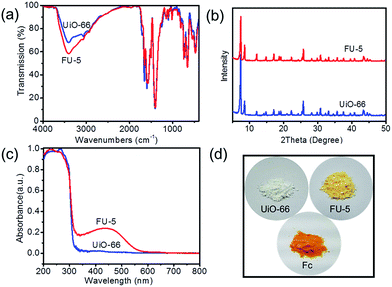 |
| | Fig. 2 (a) FT-IR spectra, (b) XRD patterns and (c) UV/vis spectra of as-synthesized UiO-66 and FU-5; (d) photograph of UiO-66, FU-5 and Fc. | |
FT-IR spectra of UiO-66 and FU-5 were given in Fig. 2a. There was no significant change when the Fc was loaded on the UiO-66 due to the similarity of molecular structure. The characteristic peaks of infrared spectrum of Fc were overlaid by UiO-66 such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C band, Fe–Cp band, C–H band, etc. XRD patterns (Fig. 2b) indicate the diffractions of FU-5 are the same as UiO-66 attributed to the fact that Fc is in amorphous phase without obvious diffractions. It is noted that FU-5 has an obvious optical band compared to UiO-66 peaking at 440 nm, characteristic absorption of Fc, other than the similar 200 nm–300 nm band (Fig. 2c). The color of FU-5 changed obviously compared with UiO-66 and Fc, as shown in Fig. 2d.
C band, Fe–Cp band, C–H band, etc. XRD patterns (Fig. 2b) indicate the diffractions of FU-5 are the same as UiO-66 attributed to the fact that Fc is in amorphous phase without obvious diffractions. It is noted that FU-5 has an obvious optical band compared to UiO-66 peaking at 440 nm, characteristic absorption of Fc, other than the similar 200 nm–300 nm band (Fig. 2c). The color of FU-5 changed obviously compared with UiO-66 and Fc, as shown in Fig. 2d.
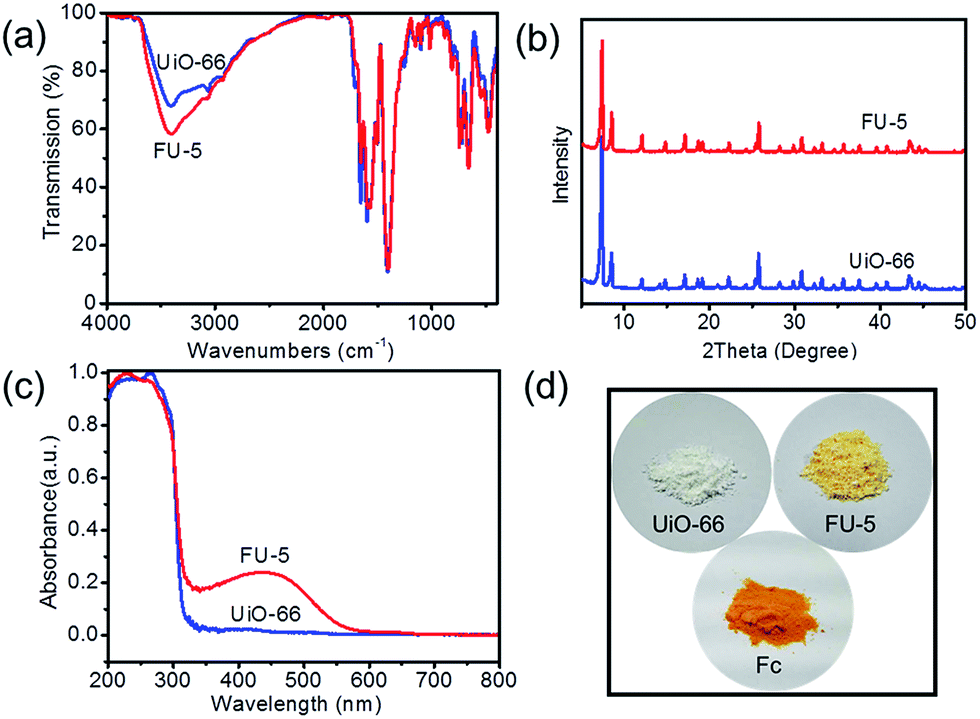
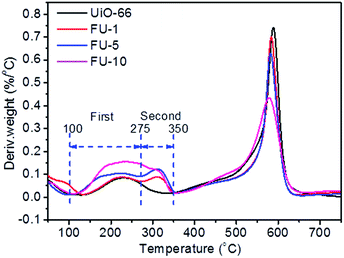
To investigate the existent forms of Fc, the as-synthesized samples were analyzed by TG (ESI, Fig. S2–S5†). TG curves changed less compared with DTG curves (Fig. 3). Seen from Fig. 3, there were distinct differences between the temperatures from 100 °C to 350 °C. Two processes in the DTG curve were observed. The first process was the temperature from 100 °C to 275 °C, which showed the decomposition and sublimation of Fc particles on the surface of UiO-66 because the Fc particles were easily sublimate when the temperature was over 100 °C; the second process was from 275 °C to 350 °C, which showed the decomposition of Fc particles in the micropores of UiO-66. With the protection of the frameworks, the decomposition of Fc was postponed. The DTG curve of UiO-66 was very similar to FU-1. It could be credited that the very low loading Fc was largely constrained in the micropores of UiO-66. The DTG curves of FU-5 and FU-10 had two more similar processes between the temperatures from 100 °C to 350 °C compared with FU-1. It may be due to that the increasing loading led to the Fc particles adsorbed not only in the micropores, but also on the surface. Furthermore, FU-10 had more amounts of Fc particles which loaded on the surface of UiO-66 than FU-5.
 |
| | Fig. 3 DTG of as-synthesized samples. | |
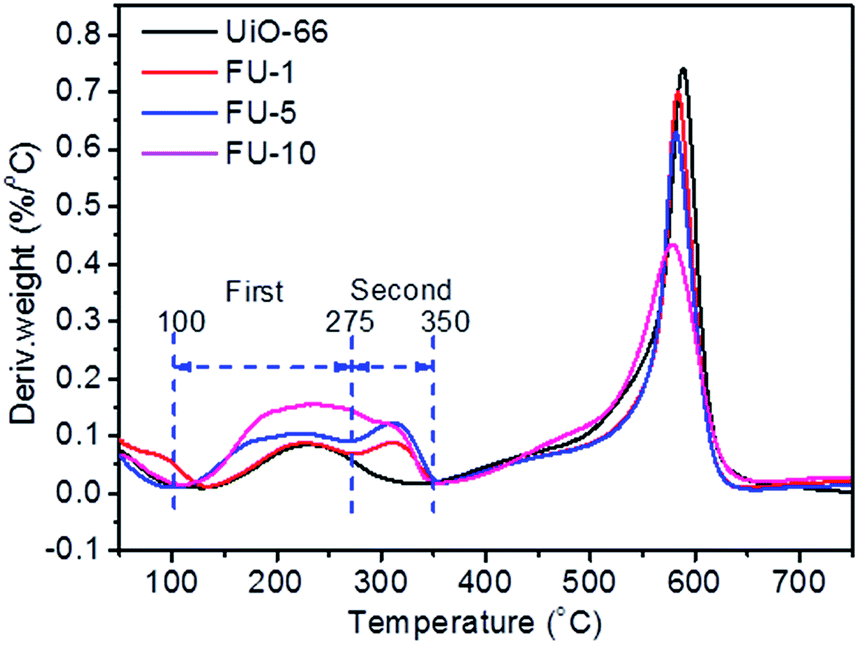
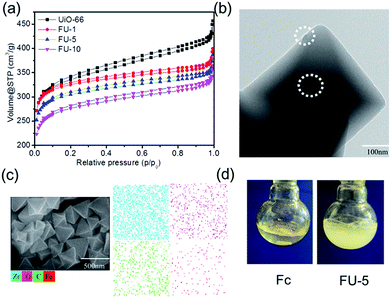
The N2 adsorption–desorption isotherms of as-prepared samples are given in Fig. 4a. All samples exhibited type I characteristics which indicated that the resultant samples were abundant of micropore structures.22 The pore diameter distribution of samples was largely under 2 nm (ESI, Fig. S6†). Type I characteristics was indicative of adsorption in micropores due to strong adsorbent–adsorbate interactions.23 Table 1 lists the surface area and pore volume of as-prepared samples. The BET specific surface area and the pore volume of UiO-66 were 1287.6 m2 g−1 and 0.706 cm3 g−1 (Table 1, entry 1), respectively, larger than those reported before, 1080 m2 g−1 and 0.52 cm3 g−1, respectively.24 This may be ascribed to the influence of modulator benzoic acid. The BET specific surface area of other samples (FU-1, FU-5, FU-10) were 1287.4 m2 g−1, 1180.3 m2 g−1, 1060.5 m2 g−1, respectively. The pore volume of other samples (FU-1, FU-5, FU-10) were 0.618 cm3 g−1, 0.594 cm3 g−1, 0.564 cm3 g−1, respectively. The BET surface area and the pore volume of Fc@UiO-66 composites (FU-1, FU-5, and FU-10) decreased with the increasing dosage of Fc. Two possible reasons for that, one was that the molecule of Fc particles diffused into the micropore of UiO-66 and occupied its space; the other one was that the aggregated Fc particles loaded the surface of UiO-66 and blocked the micropore (Fig. 4b). With the increasing dosage of Fc, the surface area and pore volume of UiO-66 crystal's micropore structure was more occupied and blocked, bringing down the BET surface area and the pore volume of Fc@UiO-66 composites (FU-1, FU-5 and FU-10). Furthermore, we sought to visualize positions of Fc on the UiO-66. Fig. 4c, SEM/EDX mapping of FU-5 showed the loading Fc particles which aggregated on the surface of UiO-66 were well distributed. Fig. 4d showed the photographs of Fc and FU-5 in the reaction solution, indicating the good dispersibility of FU-5 in phenol solution. It may be ascribed to the feature of UiO-66, which has the large surface area and good dispersibility in the water.
 |
| | Fig. 4 (a) N2 adsorption–desorption isotherms and (b) TEM image of FU-5, (c) SEM/EDX mapping of FU-5, (d) photographs of Fc and FU-5 in the reaction solution. | |
Table 1 Surface area and pore volume of as-prepared samples
| Entry |
Materials |
Surface area (m2 g−1) |
Pore volume (cm3 g−1) |
| 1 |
UiO-66 |
1287.6 |
0.706 |
| 2 |
FU-1 |
1287.4 |
0.618 |
| 3 |
FU-5 |
1180.3 |
0.598 |
| 4 |
FU-10 |
1060.5 |
0.564 |
Hydroxylation of phenol
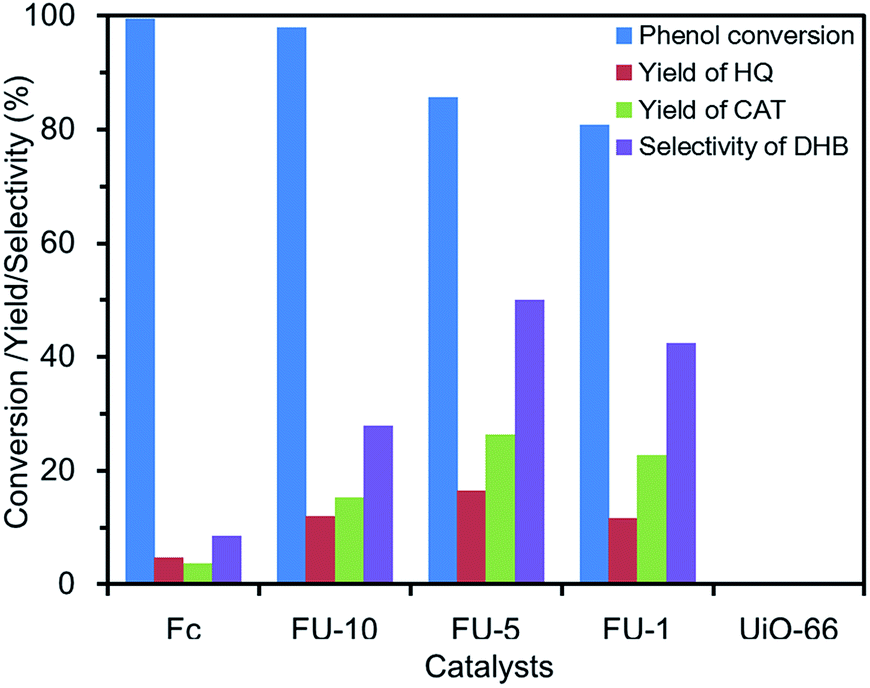
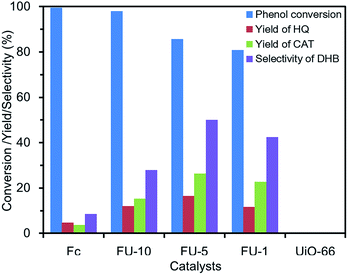
Catalytic phenol hydroxylation by Fc@UiO-66. Before the phenol hydroxylation, a blank reaction had been implemented, in which there were phenol and H2O2 (phenol/H2O2 = 1:4) but no catalyst. DHB were not detected even when the reaction time was prolonged for 24 h.With UiO-66 as the catalyst, HQ and CAT were not detected in the solution after reaction (Fig. 5). It indicated that UiO-66 had no catalytic activity for phenol hydroxylation. Although pure Fc had high phenol conversion in the reaction, the yield and selectivity to products were too low. Therefore, considering UiO-66 had the feature of large surface area and easy dispersibility in the water and the Fc had the feature of high catalytic activity of conversion and easy aggregation, the Fc@UiO-66 composites were designed to integrate the advantages of both. FU-1, FU-5 and FU-10 composites catalyst had different loading amounts of Fc. With the increasing loading amount of Fc, the phenol conversion increased from 80.8% to 98%. The Fc@UiO-66 composites catalyst with higher loading amount of Fc had more catalytic active sites, which would facilitate phenol conversion in the reaction. However, the degree of catalytic oxidation would be difficult to control with too many catalytic active sites. Hence, FU-10 had a lower yield and selectivity than FU-1 and FU-5. FU-5 had a high phenol conversion and the highest yield and selectivity. FU-5 was then chosen as the catalyst to investigate the influences of various reaction conditions such as catalyst amount, the molar ratio of phenol/H2O2, and reaction time.
 |
| | Fig. 5 Phenol hydroxylation with different catalysts. (20 mg of catalyst, 20 mL of distilled water, 100 mg of phenol (1 mmol), 0.46 mL of 30 wt% H2O2 (4 mmol), 30 °C, 3 h). | |
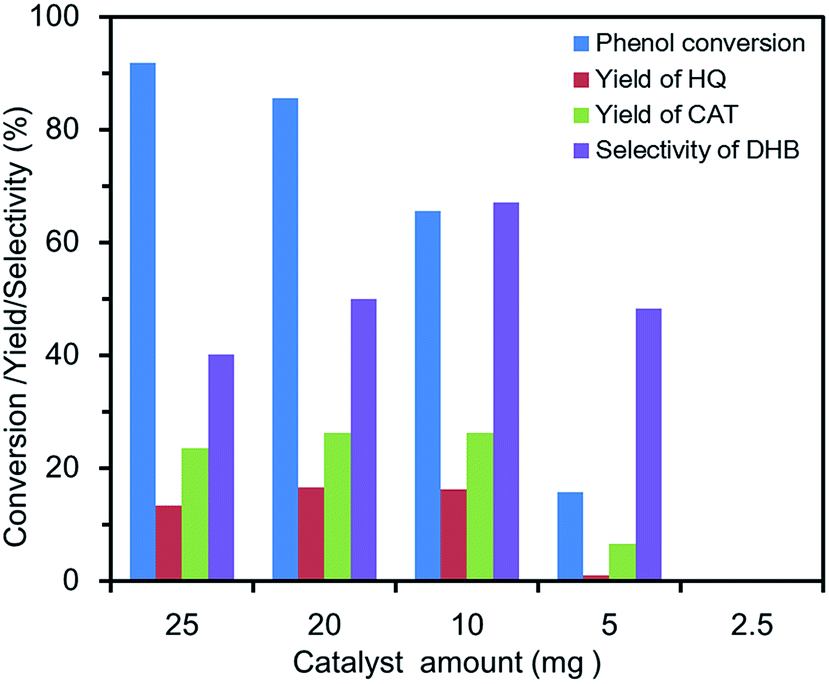
Effect of catalyst amount. The influence of the catalyst amount on the conversion, yield and selectivity of phenol are displayed in Fig. 6. No significant DHB was detected with 2.5 mg FU-5 due to too few catalytic active sites. When the amount of catalyst increased from 5 mg to 25 mg, the phenol conversion increased from 15.9% to 92%, but the yield and selectivity fell off when the amount of catalyst was 20 mg. Generally, the more amount of catalyst would provide the more active sites, which accelerated not only the hydroxylation of phenol, but also the decomposition of H2O2. Thus, the decreasing tendency about catalytic properties might be due to the reduction of active component for hydroxylation, in which the excess catalysts accelerated the decomposition of H2O2 to oxygen and water. Therefore, the optimum catalytic amount for phenol hydroxylation was 10 mg.
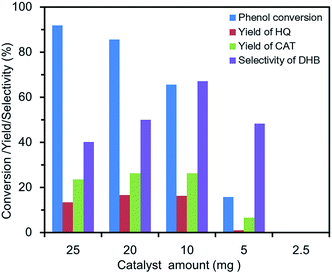 |
| | Fig. 6 Effect of catalyst amount on the phenol hydroxylation. (20 mL of distilled water, 100 mg of phenol (1 mmol), 0.46 mL of 30 wt% H2O2 (4 mmol), 30 °C, 3 h). | |
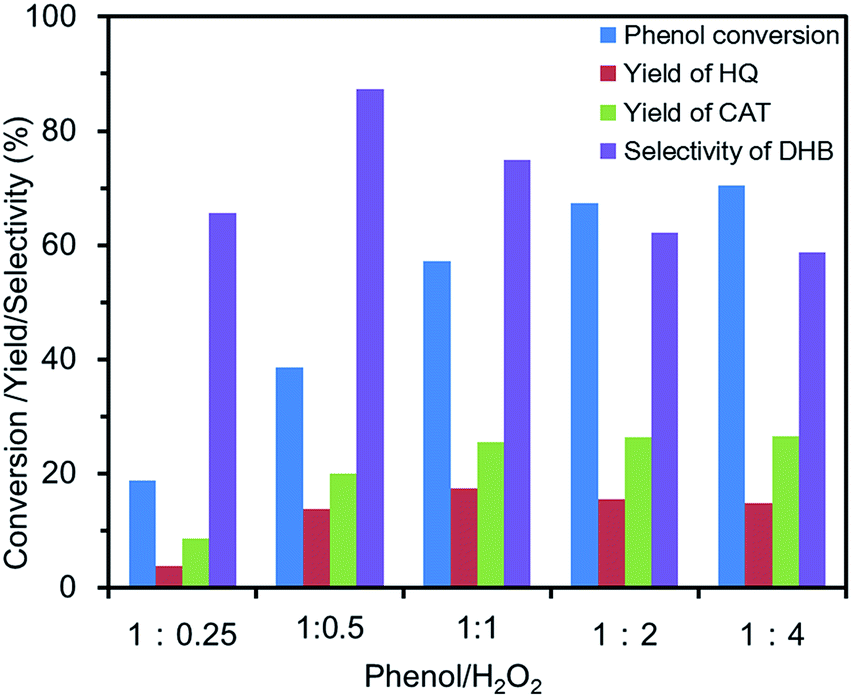
Effect of phenol/H2O2. The effects of phenol/H2O2 in the phenol hydroxylation on the conversion, yield and selectivity are illustrated in Fig. 7. The phenol conversion increased from 18.7% to 70.4% when the phenol/H2O2 ratio changed from 1:0.25 to 1:4. As we all know that the low phenol conversion was beneficial for improvement of DHB selectivity.25 When the phenol conversion was low, most of phenol would get to DHB. With increasement of H2O2, the excess H2O2 would accelerate oxidization of DHB to produce benzoquinones. DHB have one more hydroxyl group and the higher electronic cloud density on the phenyl ring than phenol, which make themselves easier to be oxidized. Benzoquinones would continue being oxidized to become the tar or cis-butenedioic acid and trans-butenedioic acid, etc. In order to obtain a high yield and selectivity, the optimum ratio of the phenol/H2O2 was at 1:0.5.
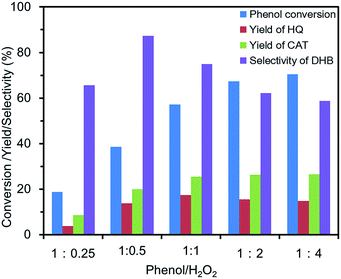 |
| | Fig. 7 Effect of the phenol/H2O2 on the phenol hydroxylation. (10 mg of FU-5, 20 mL of distilled water, 100 mg of phenol (1 mmol), 30 °C, 3 h). | |
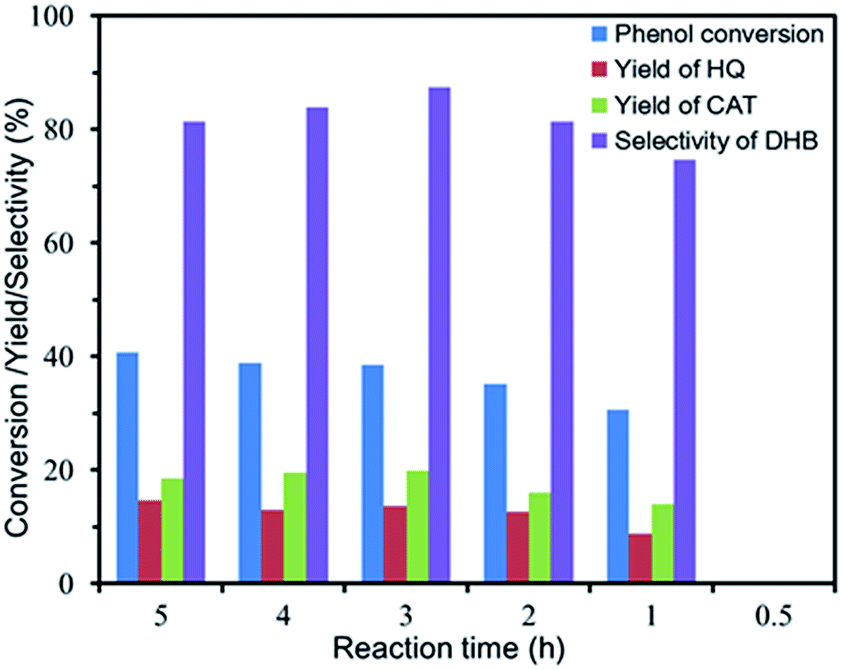
Effect of reaction time. Fig. 8 illustrated the influence of the reaction time on the conversion, yield and selectivity. DHB were not detected when the reaction time was 0.5 h. The phenomenon indicated that the start of hydroxylation reaction needed a certain time, which was attributed to the contact between the catalyst and H2O2. It was noted that the conversion of phenol ascended rapidly at the beginning, but the conversion increased slowly when the reaction time was prolonged from 1 h to 5 h. Furthermore, the selectivity fell down slightly at the highest point (87.4%), which could be attributed to the further oxidation of phenol and products (DHB) to the byproducts.
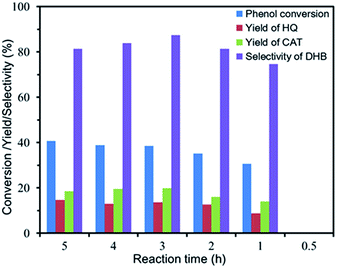 |
| | Fig. 8 Effect of reaction time on the phenol hydroxylation. (10 mg of FU-5, 20 mL of distilled water, 100 mg of phenol (1 mmol), 0.06 mL of 30 wt% H2O2 (0.5 mmol), 30 °C). | |
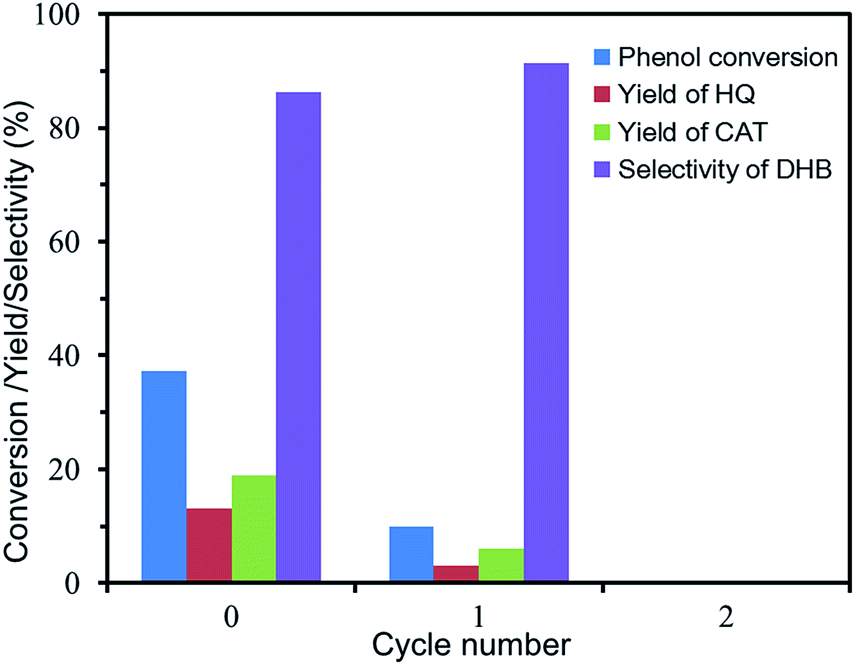
Reusability of Fc@UiO-66 composites in phenol hydroxylation. The reusability of Fc@UiO-66 composite in phenol hydroxylation is shown in Fig. 9. Although the catalytic activity of FU-5 declined distinctly when reused, the selectivity to DHB increased a little bit to ca. 90%. It is probably because the generated by-products like tar in hydroxylation absorb on the surface and cover up some of the active site and some of the catalytic active sites in the surface drop off a little bit easily. However, the Fc particles kept in micropores of the frameworks still show excellent selectively catalytic activity, but some loss of phenol conversion and DHB yield.
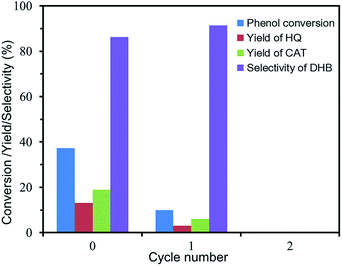 |
| | Fig. 9 Reusability of FU-5 in the phenol hydroxylation. (Reaction 0: 10 mg of FU-5, 20 mL of distilled water, 100 mg of phenol (1 mmol), 0.06 mL of 30 wt% H2O2 (0.5 mmol), 30 °C). (Reaction 1: the reaction conditions were the same as reaction0 except using the FU-5 after reaction0 as catalyst). (Reaction 2: the reaction conditions were the same as reaction0 except using the FU-5 after reaction1 as catalyst). | |
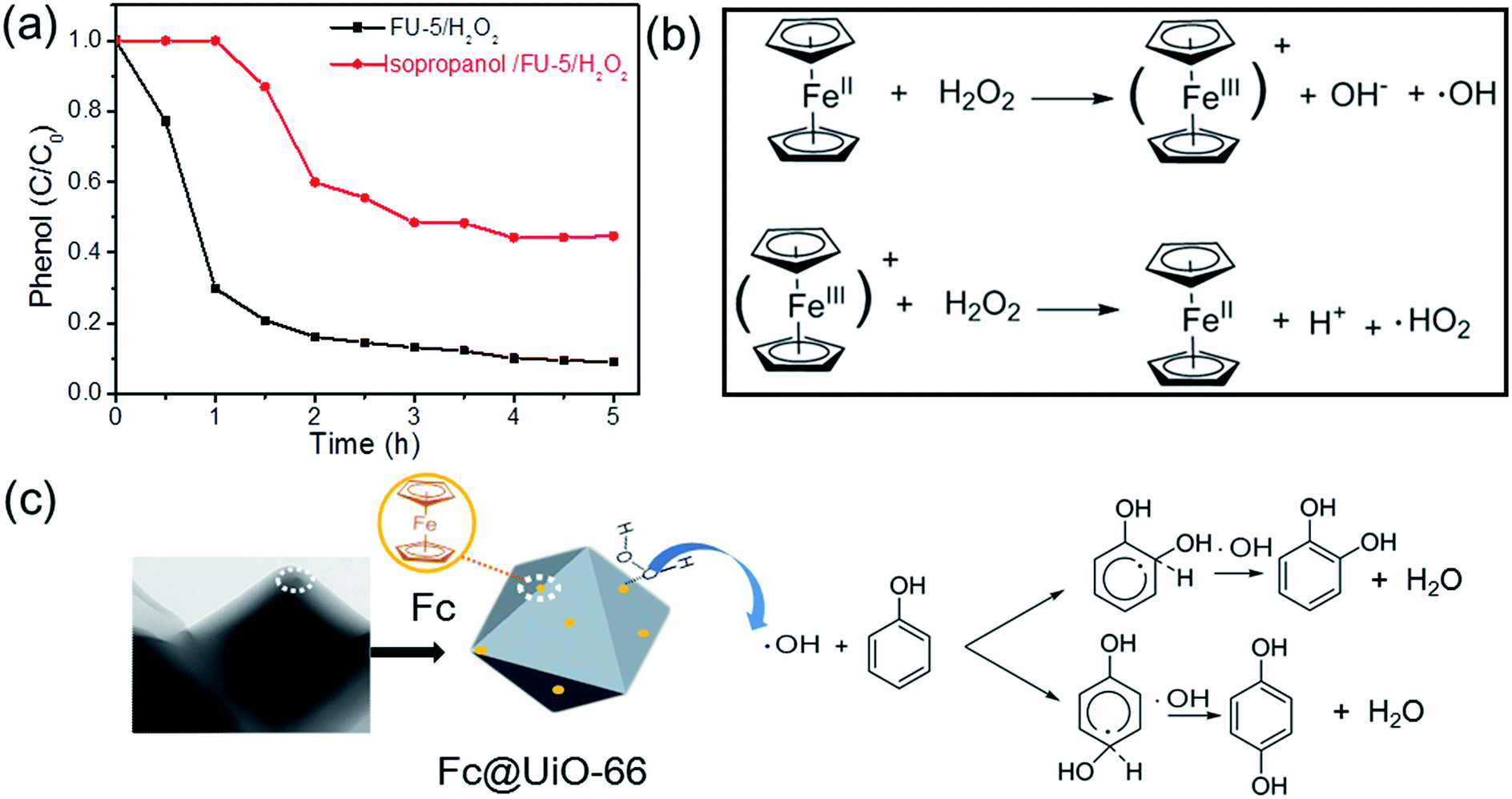
Reaction mechanism consideration. In order to explore the reaction mechanism for phenol hydroxylation with Fc@UiO-66 composites, a radical trapping experiment was carried to investigate the reactive radical species involved in the phenol hydroxylation. It is well known that the hydroxyl radical (˙OH) could be produced in the Fenton reaction, which has an important role in the Fenton reaction.26 Thus, isopropanol was applied as the scavenger of ˙OH for investigating the effect of ˙OH in the hydroxylation of phenol. The phenol conversions in the normal reaction condition and in the presence of isopropanol are shown in Fig. 10a. It demonstrated that the phenol conversion changed dramatically in the presence of isopropanol, where the phenol conversion reduced signally and the start time was prolonged. It could be credited to that the amount of ˙OH decreased obviously in the presence of isopropanol so that the reaction was restricted. The influences of different oxidants (O2, mCPBA and TBHP) on the phenol hydroxylation with FU-5 as catalyst were also investigated (Table 2). As we know, mCPBA and TBHP were widely used in various organic synthesis, they easily form the catalyst-oxidant intermediates as oxygen transfer reagent rather than ˙OH.27,28 However, no DHB was detected except H2O2 in the same reaction condition. This indicates that a hydroxyl radical mechanism probably was involved here.
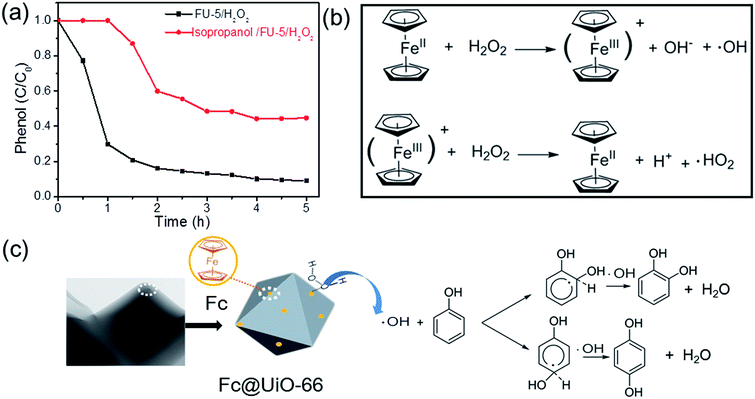 |
| | Fig. 10 (a) Changes of phenol conversion in the normal reaction condition* and in the presence of isopropanol**; (b) the process of the formation of the hydroxyl radical (˙OH); (c) proposed reaction mechanism for the phenol hydroxylation with Fc@UiO-66 composites as catalyst. (*: 20 mg of FU-5, 20 mL of distilled water, 100 mg of phenol (1 mmol), 0.46 mL of 30 wt% H2O2 (4 mmol), 30 °C). (**: the same as the reaction condition* unless the addition of 0.6 mL isopropanol). | |
Table 2 Different oxidants used in phenol hydroxylation with FU-5 as catalysta
| Entry |
Oxidant |
Conversion (%) |
Yield (%) |
Selectivity (%) |
| HQ |
CAT |
| N.D.: not detect; 20 mg of FU-5, 20 mL of distilled water, 100 mg of phenol (1 mmol), phenol/oxidant = 1:1, 30 °C. |
| 1 |
H2O2 |
57.2 |
17.3 |
25.5 |
74.9 |
| 2 |
O2 |
— |
N.D. |
N.D. |
— |
| 3 |
m-Chloroperbenzoic acid (mCPBA) |
— |
N.D. |
N.D. |
— |
| 4 |
tert-Butyl hydroperoxide (TBHP) |
— |
N.D. |
N.D. |
— |
Taking into account the above results and referred literatures,29–31 a tentative Fenton-like mechanism is suggested and illustrated in Fig. 10b and c. The Fc@UiO-66 composite catalysts can react with H2O2 to form ˙OH and ferrocenium ion. Then ferrocenium ion react with another H2O2 to form the Fc and ˙HO2. The process (Fig. 10b) could produce ˙OH like Fenton reaction quickly. Then, the ˙OH added onto the carbon at ortho and para positions of hydroxyl group on the phenol led to the aromatic ring to form a delocalized electron and subsequently came into being an adduct intermediate. Furthermore, another ˙OH added onto the certain carton atom, which had the hydrogen abstraction (H-abst) at the same position from the previous addition. Finally, it generated a water molecule and DHB (Fig. 10c).
Comparison between different supports
The comparisons between different support composite catalysts for the phenol hydroxylation are illustrated in Table 3. When the supports of composites catalyst were γ-Al2O3 and talcum powder, the DHB products were not detected in the reaction solution at 3 h and 24 h reaction time. This may be credited to that these supports could not promote the touch between Fc particles and H2O2, causing not enough ˙OH to react with phenol. The start time of the reaction was prolonged when the supports were diatomite, atlapulgite and active carbon. These phenol conversions and the selectivity were lower than the reaction with FU-5 as a catalyst. When UiO-66 was used as the support of Fc particles, the start time of the phenol hydroxylation reaction was shortened. In conclusion, UiO-66 played a vital role in the hydroxylation with Fc@UiO-66 as catalyst. On the one hand, the large surface area of UiO-66 contributed to the good dispersion of Fc particles on the UiO-66; on the other hand a possible interaction between Fc particles and UiO-66 can improve the selectivity.
Table 3 Comparisons between different support composite catalysts in the phenol hydroxylationa
| Entry |
Support |
Reaction time (h) |
Phenol conversion (%) |
Yield (%) |
Selectivity (%) |
| HQ |
CAT |
| (10 mg of catalyst, 20 mL of distilled water, 100 mg of phenol (1 mmol), 0.06 mL of 30 wt% H2O2 (0.5 mmol), 30 °C). |
| 1 |
γ-Al2O3 |
3 |
— |
N.D. |
N.D. |
— |
| 2 |
Talcum powder |
3 |
— |
N.D. |
N.D. |
— |
| 3 |
Diatomite |
3 |
— |
N.D. |
N.D. |
— |
| 4 |
Atlapulgite |
3 |
— |
N.D. |
N.D. |
— |
| 5 |
Active carbon |
3 |
— |
N.D. |
N.D. |
— |
| 6 |
UiO-66 (FU-5) |
3 |
38.5 |
13.7 |
19.9 |
87.4 |
| 7 |
γ-Al2O3 |
24 |
— |
N.D. |
N.D. |
— |
| 8 |
Talcum powder |
24 |
— |
N.D. |
N.D. |
— |
| 9 |
Diatomite |
24 |
32 |
8.3 |
12.1 |
62.8 |
| 10 |
Atlapulgite |
24 |
15 |
2.1 |
4.4 |
43 |
| 11 |
Active carbon |
24 |
8 |
1.1 |
0.6 |
21.2 |
X-ray photoelectron spectroscopy (XPS) analysis
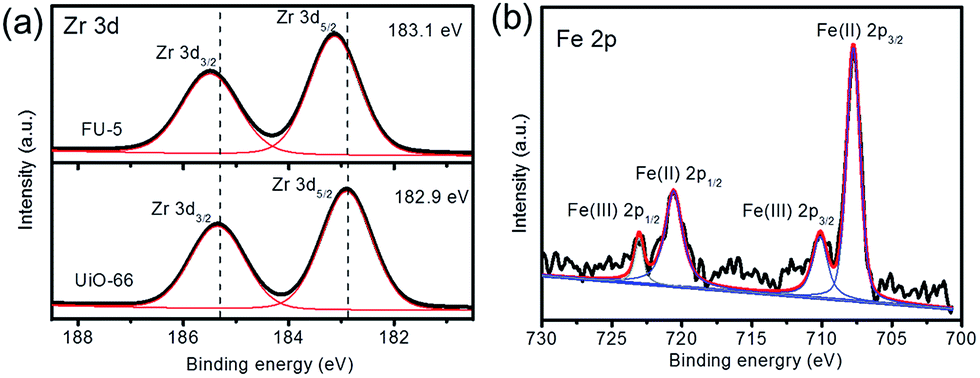
To investigate the high selectivity of Fc@UiO-66 catalyst, XPS analysis (Fig. 11) was used to probe the electronic properties of the composite materials. However, the depth of Fc in the micropores of UiO-66 was over the measurement depth of the X-rays used in XPS.32 We then investigated the Fc on the surface of UiO-66 and applied it as substitute for the Fc in the micropores because it is reasonable that they had the same electronic properties and interaction. Fig. 10a shows Zr 3d spectra of FU-5 and UiO-66, the two main peaks corresponded to the spin–orbit of Zr 3d3/2 and Zr 3d5/2. Besides, the peak of Zr 3d5/2 increased from 182.9 eV to 183.1 eV when the Fc loaded on UiO-66, illustrating that the Fc had the interaction with UiO-66.33 Fig. 11b shows the Fe 2p spectra of FU-5, the main peaks were at 707.81 eV, 710.13 eV, 720.59 eV and 723.6 eV, respectively. The peaks at 707.81 eV and 720.59 eV belonged to FeIII 2p3/2 and FeIII 2p1/2 and 710.13 eV and 723.6 eV belonged to FeII 2p3/2 and FeII 2p1/2.34 However, the Fc was mainly formed by FeII, it may be credited to that some Fc was oxidized to FeIII.
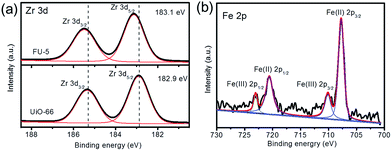 |
| | Fig. 11 (a) XPS Zr 3d spectra of FU-5 and UiO-66 (b) XPS Fe 2p spectra of FU-5. | |
Conclusions
In summary, UiO-66 with cuboctahedron morphologies was synthesized in large scale in the ambient atmosphere. Fc particles were incorporated into the Zr-based MOFs, which facilitated the dispersion of Fc@UiO-66 composite in water. The phenol hydroxylation catalyzed by FU-5 proceeded efficiently at room temperature in water, which yield the highest DBH selectivity of 87.4% while the phenol conversion was 38.5%. UiO-66 played a unique role in the composite as compared to the usual catalyst supports. The good catalytic activity of Fc@UiO-66 in hydroxylation was attributed to the interaction between Fc particles and UiO-66. A hydroxyl radical mechanism was demonstrated in this Fc@UiO-66 catalyzed hydroxylation with H2O2.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21276187) and the Tianjin Municipal Natural Science Foundation (16JCYBJC20800).
Notes and references
- S. Ikurumi, S. Okada, K. Nakatsuka, T. Kamegawa, K. Mori and H. Yamashita, J. Phys. Chem. C, 2015, 118, 575–581 Search PubMed.
- G. Q. Wu, J. H. Xiao, L. Zhang, W. J. Wang, Y. Q. Hong, H. J. Huang, Y. Jiang, L. Lia and C. R. Wang, RSC Adv., 2016, 6, 101071–101078 RSC.
- Z. He, J. Wu, B. Y. Gao and H. Y. He, ACS Appl. Mater. Interfaces, 2015, 7, 2424–2432 CAS.
- A. Hoffmann, C. Citek, S. Binder, A. Goos, M. Rbhausen, O. Troeppner, I. Ivanović-Burmazović, E. C. Wasinger and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2013, 52, 5398–5401 CrossRef CAS PubMed.
- J. Tekla, K. A. Tarach, Z. Olejniczak, V. Girman and K. Góra-Marek, Microporous Mesoporous Mater., 2016, 233, 16–25 CrossRef CAS.
- E. A. Karakhanov, A. L. Maximov, Y. S. Kardasheva, V. A. Skorkin, S. V. Kardashev and V. V. Predeina, Appl. Catal., A, 2010, 385, 62–72 CrossRef CAS.
- O. Osegueda, A. Dafinov, J. Llorca, F. Medina and J. Sueiras, Chem. Eng. J., 2015, 262, 344–355 CrossRef CAS.
- Y. L. Dong, X. Y. Niu, W. N. Song, D. Wang, L. Q. Chen, F. L. Yuan and Y. J. Zhu, Catalysts, 2016, 6, 74–90 CrossRef.
- R. G. Yang, G. Y. Li and C. W. Hu, Catal. Sci. Technol., 2015, 5, 2486–2495 CAS.
- F. Adam, J. T. Wong and E. P. Ng, Chem. Eng. J., 2013, 214, 63–67 CrossRef CAS.
- H. X. Shi, T. Y. Zhang, B. Li, X. Wang, M. He and M. Y. Qiu, Catal. Commun., 2011, 12, 1022–1026 CrossRef CAS.
- B. An, J. Z. Zhang, K. Cheng, P. F. Ji, C. Wang and W. B. Lin, J. Am. Chem. Soc., 2017, 139, 3834–3840 CrossRef CAS PubMed.
- S. A. Burgess, A. Kassie, S. A. Baranowski, K. J. Fritzsching, K. Schmidt-Rohr, C. M. Brown and C. R. Wade, J. Am. Chem. Soc., 2016, 138, 1780–1783 CrossRef CAS PubMed.
- Q. Xu, Z. G. Guo, M. Zhang, Z. G. Hu, Y. H. Qian and D. Zhao, CrystEngComm, 2016, 18, 4046–4052 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- B. Wang, Q. Yang, C. Guo, Y. X. Sun, L. H. Xie and J. R. Li, ACS Appl. Mater. Interfaces, 2017, 9, 10286–10295 CAS.
- Z. G. Hu and D. Zhao, CrystEngComm, 2017, 19, 4066–4081 RSC.
- Y. Bai, Y. B. Dou, L. H. Xie, W. Rutledge, J. R. Li and H. C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2014, 43, 5750–5765 RSC.
- F. Xu, H. Li, Y. L. Luo and W. Tang, ACS Appl. Mater. Interfaces, 2017, 9, 5181–5192 CAS.
- Z. Y. Li, A. W. Peters, J. Liu, X. Zhang, N. M. Schweitzer, J. T. Hupp and O. K. Farha, Inorg. Chem. Front., 2017, 4, 820–824 RSC.
- Y. M. Sun, X. L. Hu, W. Luo and Y. H. Huang, J. Phys. Chem. C, 2012, 116, 20794–20799 CAS.
- Y. L. Min, X. F. Qi, Q. J. Xu and Y. C. Chen, CrystEngComm, 2014, 16, 1287–1295 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- W. Qian, H. Q. Wang, J. Chen and Y. Kong, Materials, 2015, 8, 1752–1765 CrossRef.
- K. T. Lee, Y. J. Lu, F. L. Mi, T. Burnouf, Y. T. Wei, S. C. Chiu, E. Y. Chuang and S. Y. Lu, ACS Appl. Mater. Interfaces, 2017, 9, 1273–1279 CAS.
- Y. H. Wang, T. Y. Zhang, B. Li, S. Jiang and L. Sheng, RSC Adv., 2015, 5, 29022–29031 RSC.
- P. Cancino, V. Paredes-garcía, C. Aliaga, P. Aguirre, D. Aravenabd and E. Spodine, Catal. Sci. Technol., 2017, 7, 231–242 CAS.
- W. Chen, X. L. Yang, J. F. Huang, Y. H. Zhu, Y. Zhou, Y. F. Yao and C. Z. Li, Electrochim. Acta, 2016, 200, 75–83 CrossRef CAS.
- X. Wang, T. Y. Zhang, Q. S. Yang, S. Jiang and B. Li, Eur. J. Inorg. Chem., 2015, 5, 817–825 CrossRef.
- C. Xu and L. M. Wang, J. Phys. Chem. A, 2013, 117, 2358–2364 CrossRef CAS PubMed.
- J. Yang, Y. Dai, X. Y. Zhu, Z. Wang, Y. S. Li, Q. X. Zhuang, J. L. Shi and J. L. Gu, J. Mater. Chem. A, 2015, 3, 7445–7452 CAS.
- B. Rungtaweevoranit, J. Baek, J. R. Araujo, B. S. Archanjo, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2016, 16, 7645–7649 CrossRef CAS PubMed.
- G. Pilatos, A. V. Perdikaki, A. Sapalidis, G. S. Pappas, T. Giannakopoulou, D. Tsoutsou, E. Xenogiannopoulou, N. Boukos, A. Dimoulas and C. Trapalis, J. Appl. Phys., 2016, 119, 3284–3288 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06917k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ac,
Wubin Wua,
Tianyong Zhang*abc,
Shuang Jiang*a,
Xingwei Chena,
Guanghui Zhanga and
Xia Zhanga
ac,
Wubin Wua,
Tianyong Zhang*abc,
Shuang Jiang*a,
Xingwei Chena,
Guanghui Zhanga and
Xia Zhanga