
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cimifrigines A–G, cytotoxic triterpenes with an oxime group from the flowers of Cimicifuga frigida†
Yin Nian,
Hui Yan,
Xiao-Nian Li,
Lin Zhou and
Ming-Hua Qiu*
State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, People's Republic of China. E-mail: mhchiu@mail.kib.ac.cn
First published on 7th August 2017
Abstract
Seven new dahurinol-type triterpene derivatives, including three aglycones, cimifrigines A–C (1–3), and four glycosides, cimifrigines D–G (4–7), were purified from the flowers of Cimicifuga frigida. These triterpenoids are characterized by an oxime group at C-15. Spectroscopic analyses and X-ray crystallography were used to determine the new structures. In the in vitro cytotoxicity screening, glycosides (4–7) exhibited more noticeable activities than the aglycones (1–3) against human HL-60, SMMC-7721, A549, MCF-7, and SW-480 cell lines. Interestingly, compounds 5 and 7, bearing a 2′-O-acetyl moiety on the sugar unit, showed comparable cytotoxicities to the positive control, cisplatin (IC50: 0.5 to 5.4 μM). Whereas, analogues 4 and 6, without the 2′-O-acetyl group, indicated weaker activities with IC50 values ranging from 8.9 to 14.3 μM.
Introduction
Plants of the Cimicifuga genus (Ranunculaceae) are time-honored herbal medicines worldwide.1–3 C. racemosa, namely black cohosh, is a popular food supplement in Europe and the United States for relief of menopausal disorders.4,5 In East Asia, several Cimicifuga spp., for instance, C. foetida, C. dahurica, C. heracleifolia, C. simplex, and C. japonica, have been used to alleviate fever, pain, and inflammation since ancient times.6,7Extensive efforts have been attracted to investigate chemical constituents of this genus. 9,19-Cycloartane triterpenoids (CTs) is the major chemical component of Cimicifuga spp. and more than 300 CTs2,3,7–27 have been reported (among them, our research group reported over 100 ones2,3,8–24). Pharmaceutical studies revealed these CTs possessed various bioactivities, for instance, antiosteoporotic,28 cytotoxicity,2,14 anti-AIDS,29 anti-Alzheimer,30 and immunosuppression.31 Nevertheless, aforementioned CTs were mainly from the rhizomes or roots of C. racemosa, C. foetida, C. dahurica, C. heracleifolia, and C. simplex.7–27 Therefore, we carried out successive investigations on the roots, aerial parts, and fruits of C. yunnanensis, an indigenous species in southwest China.2,3,15 Consequently, a number of bioactive CTs were discovered. Among them, cimyunnin A, with an unprecedented carbon skeleton from the fruits of this plant, was considered as an anti-angiogenic leading structure. It showed comparable in vitro and ex vivo activities to those of a first-line clinical medicine, sunitinib.3 Therefore, aforementioned studies exemplified that expansion of research objects in genus Cimicifuga may led to discovery of novel active CTs.
The taxonomic treatment of the Sino-Himalayan species C. frigida has a very long and controversial history.32,33 However, recent cytological evidence showed that this plant is the only tetraploid (2n = 32) currently known in the genus, which together with morphological features confirmed its independent species status.34 Thus, it is of interest to explore whether there are novel bioactive compounds in this plant. In the present study, we initially carried out a study on the flowers of C. frigida from Litang County, Sichuan Province. Consequently, seven new 9,19-cycloartane triterpene derivatives (Fig. 1), cimifrigines A–G (1–7), were isolated. Their structures were elucidated by the help of MS, NMR, and single-crystal X-ray diffraction techniques. The unique characteristic of these analogues is that containing an oxime group at C-15 based on the dahurinol–skeleton. In addition, the isolated compounds showed potent to week cytotoxicities against human HL-60, SMMC-7721, A549, MCF-7, and SW-480 cell lines.
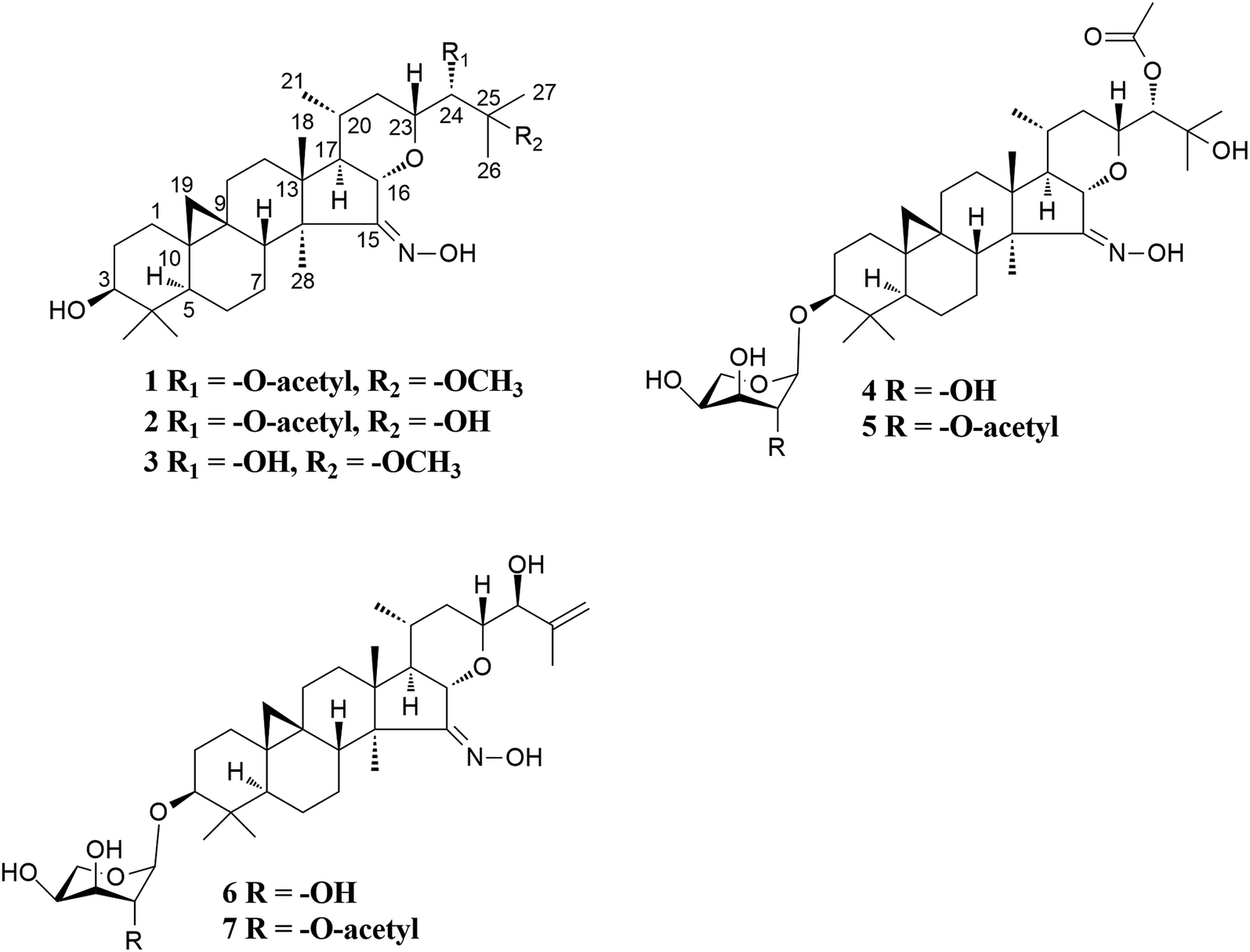 | ||
| Fig. 1 Structures of compounds 1–7. | ||
Results and discussion
Cimifrigine A (1), purified as colorless crystals. The HREIMS ion peak at m/z 559.3864 [M]+ (calcd 559.3873) determined its molecular formula as C33H53NO6, requiring 8 double-bond equivalents. The 1H NMR spectrum (Table 1) showed characteristic cyclopropane methylene signals at δH 0.29 and 0.52 (each 1H, brs), a secondary methyl signal at δH 0.88 (d, J = 5.9 Hz), five tertiary methyl groups at δH 1.04–1.59, and an active hydrogen signal at δH 12.32, respectively. In the 13C NMR spectrum (Table 2), an olefinic (or a carbonyl) carbon signal at δC 164.34 (C-15), and five oxygenated carbons at δC 78.01 (C-3, d), 80.55 (C-16, d), 78.76 (C-23, d), 77.13 (C-24, d), and 77.53 (C-25, s) were also observed. Aforementioned data suggested that 1 was a highly oxygen-bearing 9,19-cycloartane triterpenoid with a six-ring carbon skeleton.| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| a Signals overlapped. | |||||||
| 1 | 1.52a | 1.57 m | 1.55 m | 1.54 m | 1.52 m | 1.58 m | 1.51 m |
| 1.12a | 1.21a | 1.18a | 1.18 m | 1.15a | 1.23 m | 1.16 m | |
| 2 | 1.96 m | 2.02 m | 1.97 m | 2.36 m | 2.29 m | 2.42 m | 2.27 m |
| 1.82 m | 1.91 m | 1.89 m | 1.94 m | 1.87 m | 1.99 m | 1.87 m | |
| 3 | 3.51 m | 3.57 dd (11.2, 3.4) | 3.54 m | 3.49 dd (11.6, 4.2) | 3.39 dd (11.6, 4.4) | 3.54 dd (11.6, 4.3) | 3.37 dd (11.2, 3.8) |
| 4 | |||||||
| 5 | 1.27 m | 1.31 dd (12.4, 4.1) | 1.32 dd (12.5, 4.3) | 1.30 dd (12.6, 4.3) | 1.30 dd (12.4, 4.1) | 1.37 dd (12.5, 4.2) | 1.31 m |
| 6 | 1.52a | 1.56 m | 1.59a | 1.50 m | 1.51 m | 1.59 m | 1.55 m |
| 0.71 m | 0.73 q (12.7) | 0.76 m | 0.65 q (12.4) | 0.68 q (12.7) | 0.75 q (12.5) | 0.73 m | |
| 7 | 2.54 m | 2.57 m | 2.60 m | 2.53 m | 2.55 m | 2.60 m | 2.59 m |
| 1.01a | 1.07a | 1.08a | 1.10a | 1.02 m | 1.13 m | 1.08a | |
| 8 | 1.82 m | 1.84 m | 1.86 m | 1.80 m | 1.80 m | 1.87 dd (12.6, 3.5) | 1.84 m |
| 9 | |||||||
| 10 | |||||||
| 11 | 1.99 m | 2.04 m | 2.03 m | 1.98 m | 1.97 m | 2.05a | 2.00a |
| 1.03a | 1.07a | 1.06a | 1.10a | 0.99 m | 1.05a | 1.00 m | |
| 12 | 1.52a (2H) | 1.58 m (2H) | 1.55 m (2H) | 1.55 m (2H) | 1.54 m (2H) | 1.62 m (2H) | 1.58 m (2H) |
| 13 | |||||||
| 14 | |||||||
| 15 | |||||||
| 16 | 4.24 d (10.1) | 4.41 brd (10.3) | 4.37 d (10.2) | 4.23 brd (10.4) | 4.27 d (10.4) | 4.40 brd (10.1) | 4.38 d (9.9) |
| 17 | 1.60a | 1.66 brd (11.0) | 1.58a | 1.62 m | 1.63 m | 1.65 m | 1.62 m |
| 18 | 1.07 s | 1.14 s | 1.09 s | 1.07 s | 1.09 s | 1.09 s | 1.08 s |
| 19 | 0.52 brs | 0.56 d (3.3) | 0.55 d (3.7) | 0.48 d (3.3) | 0.45 d (3.5) | 0.53 d (3.4) | 0.47 d (3.3) |
| 0.29 brs | 0.34 d (3.8) | 0.32 d (4.0) | 0.26 d (3.9) | 0.23 d (3.9) | 0.30 d (3.9) | 0.24 d (3.8) | |
| 20 | 1.77 m | 1.84 m | 1.80 m | 1.80 m | 1.80 m | 1.76 m | 1.75 m |
| 21 | 0.88 d (5.9) | 0.93 d (6.3) | 0.91 d (6.1) | 0.89 d (6.3) | 0.90 d (6.3) | 0.93 d (6.3) | 0.92 d (6.3) |
| 22 | 1.65 m | 1.66 brd (11.0) | 1.84 m | 1.65 m | 1.68 m | 1.68 m | 1.65 m |
| 1.42 m | 1.50a | 1.62a | 1.41 m | 1.44 m | 1.32a | 1.28a | |
| 23 | 4.11 d (11.1) | 4.39 brd (10.1) | 4.25 brd (11.3) | 4.23 brd (10.4) | 4.26 m | 4.02 m | 3.99 m |
| 24 | 5.34 brs | 5.26 brs | 3.64 s | 5.55 brs | 5.59 s | 4.37 d (6.3) | 4.34 d (6.3) |
| 25 | |||||||
| 26 | 1.59 s | 1.68 s | 1.57 s | 2.02 s | 2.05 s | 5.30 s | 5.27 s |
| 5.04 s | 5.01 s | ||||||
| 27 | 1.56 s | 1.50 s | 1.61 s | 2.09 s | 2.13 s | 2.05 s | 2.02 s |
| 28 | 1.14 s | 1.19 s | 1.16 s | 1.14 s | 1.15 s | 1.29 s | 1.27 s |
| 29 | 1.04 s | 1.09 s | 1.08 s | 1.00 s | 0.96 s | 1.05 s | 0.96 s |
| 30 | 1.16 s | 1.21 s | 1.20 s | 1.27 s | 1.09 s | 1.32 s | 1.08 s |
| 3-Ara | |||||||
| 1′ | 4.79 d (7.1) | 4.75 d (7.7) | 4.83 d (7.1) | 4.74 d (7.5) | |||
| 2′ | 4.45 t (7.5) | 5.95 dd (9.5, 7.8) | 4.50 t (7.9) | 5.92 t (8.1) | |||
| 3′ | 4.16 dd (8.8, 3.2) | 4.21 m | 4.20 dd (8.8, 3.0) | 4.19 dd (9.6, 3.0) | |||
| 4′ | 4.32 brs | 4.30 brs | 4.35 brs | 4.29 brs | |||
| 5′ | 4.29 m | 4.29 brd (11.4) | 4.34 m | 4.28 m | |||
| 3.78 d (11.4) | 3.77 d (11.3) | 3.83 d (10.9) | 3.77 d (11.3) | ||||
24-OCOC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3 3 |
2.16 s | 2.23 s | 2.22 s | 2.13 s | |||
| 25-OC3 |
3.24 s | 3.29 s | |||||
| 2′-OCOC3 |
2.24 s | 2.11 s | |||||
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–O N–O |
12.32 s | 12.73 s | 12.31 s | 12.43 s | 12.43 s | 12.56 s | |
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 1 | 33.96 CH2 | 33.29 CH2 | 32.92 CH2 | 32.70 CH2 | 32.57 CH2 | 32.64 CH2 | 32.57 CH2 |
| 2 | 31.53 CH2 | 31.98 CH2 | 31.47 CH2 | 30.09 CH2 | 30.44 CH2 | 30.14 CH2 | 29.98 CH2 |
| 3 | 78.01 CH | 78.37 CH | 78.00 CH | 88.59 CH | 89.07 CH | 88.51 CH | 88.69 CH |
| 4 | 41.16 C | 41.58 C | 41.14 C | 41.35 C | 41.47 C | 41.34 C | 41.06 C |
| 5 | 47.69 CH | 47.99 CH | 47.67 CH | 47.78 CH | 48.02 CH | 47.71 CH | 47.67 CH |
| 6 | 21.70 CH2 | 21.99 CH2 | 21.42 CH2 | 21.38 CH2 | 21.76 CH2 | 21.37 CH2 | 21.42 CH2 |
| 7 | 26.98 CH2 | 27.36 CH2 | 227.08 CH2 | 26.84 CH2 | 26.12 CH2 | 26.68 CH2 | 26.90 CH2 |
| 8 | 46.43 CH | 46.74 CH | 46.34 CH | 46.33 CH | 46.70 CH | 46.21 CH | 46.26 CH |
| 9 | 20.06 C | 20.38 C | 20.02 C | 20.03 C | 20.46 C | 19.98 C | 20.13 C |
| 10 | 27.16 C | 27.59 C | 26.99 C | 26.84 C | 27.25 C | 26.85 C | 26.70 C |
| 11 | 25.83 CH2 | 26.30 CH2 | 25.75 CH2 | 25.66 CH2 | 26.02 CH2 | 25.55 CH2 | 25.62 CH2 |
| 12 | 30.99 CH2 | 31.26 CH2 | 30.94 CH2 | 30.89 CH2 | 31.21 CH2 | 30.83 CH2 | 30.93 CH2 |
| 13 | 42.50 C | 43.01 C | 42.44 C | 42.48 C | 42.85 C | 42.61 C | 42.52 C |
| 14 | 51.67 C | 51.93 C | 51.65 C | 51.62 C | 51.98 C | 51.62 C | 51.71 C |
| 15 | 164.34 C | 164.63 C | 164.64 C | 164.10 C | 164.53 C | 164.37 C | 164.50 C |
| 16 | 80.55 CH | 81.17 CH | 80.09 CH | 80.42 CH | 80.83 CH | 80.25 CH | 80.55 CH |
| 17 | 54.60 CH | 54.61 CH | 54.66 CH | 54.36 CH | 54.70 CH | 54.88 CH | 54.99 CH |
| 18 | 19.86 CH3 | 20.36 CH3 | 19.83 CH3 | 19.77 CH3 | 20.27 CH3 | 19.80 CH3 | 19.86 CH3 |
| 19 | 31.44 CH2 | 31.85 CH2 | 31.42 CH2 | 31.36 CH2 | 31.62 CH2 | 31.20 CH2 | 31.19 CH2 |
| 20 | 33.28 CH | 33.48 CH | 33.23 CH | 33.18 CH | 33.57 CH | 32.85 CH | 32.93 CH |
| 21 | 20.09 CH3 | 20.38 CH3 | 20.17 CH3 | 19.93 CH3 | 20.40 CH3 | 20.08 CH3 | 20.13 CH3 |
| 22 | 39.18 CH2 | 38.74 CH2 | 39.94 CH2 | 38.83 CH2 | 39.14 CH2 | 38.16 CH2 | 38.26 CH2 |
| 23 | 78.76 CH | 80.00 CH | 78.59 CH | 78.77 CH | 79.17 CH | 81.89 CH | 82.01 CH |
| 24 | 77.13 CH | 79.07 CH | 78.34 CH | 80.25 CH | 80.65 CH | 78.74 CH | 78.82 CH |
| 25 | 77.53 C | 73.11 C | 78.18 C | 74.15 C | 74.79 C | 146.53 C | 146.57 C |
| 26 | 23.21 CH3 | 28.86 CH3 | 23.08 CH3 | 29.57 CH3 | 30.08 CH3 | 113.13 CH2 | 113.18 CH2 |
| 27 | 21.70 CH3 | 27.73 CH3 | 20.42 CH3 | 29.57 CH3 | 29.97 CH3 | 18.55 CH3 | 18.59 CH3 |
| 28 | 19.56 CH3 | 20.59 CH3 | 19.39 CH3 | 19.55 CH3 | 20.07 CH3 | 19.29 CH3 | 19.34 CH3 |
| 29 | 14.89 CH3 | 15.38 CH3 | 15.25 CH3 | 15.39 CH3 | 15.69 CH3 | 15.36 CH3 | 15.25 CH3 |
| 30 | 26.19 CH3 | 26.62 CH3 | 25.46 CH3 | 25.75 CH3 | 25.89 CH3 | 25.67 CH3 | 25.46 CH3 |
| 3-Ara | |||||||
| 1′ | 107.26 CH | 105.01 CH | 107.59 CH | 104.50 CH | |||
| 2′ | 72.92 CH | 74.83 CH | 72.93 CH | 74.37 CH | |||
| 3′ | 74.59 CH | 72.89 CH | 74.68 CH | 72.52 CH | |||
| 4′ | 69.39 CH | 70.28 CH | 69.61 CH | 69.79 CH | |||
| 5′ | 66.56 CH2 | 67.76 CH2 | 66.91 CH2 | 67.18 CH2 | |||
24-O![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) OCH3 OCH3 |
170.83 C | 171.74 C | 170.62 C | 170.52 C | |||
| 24-OCOH3 |
21.05 CH3 | 21.99 CH3 | 20.75 CH3 | 21.78 CH3 | |||
| 25-OH3 |
49.27 CH3 | 49.23 CH3 | |||||
| 2′-OOCH3 |
171.14 C | 170.17 C | |||||
| 2′-OCOH3 |
21.28 CH3 | 21.35 CH3 |
Study of the 1H–1H COSY (Fig. 2) spectrum of 1 revealed the existence of partial structures C1/C-2/C3 (–CH2–CH2–CH–), C-5/C-6/C-7/C8 (–CHCH2CH2CH–), C-11/C-12 (–CH2CH2–), and C-16/C-17/C-20(C-21)/C-22/C-23 (–CHCHCH–(CH3)CH2CH–), which consistent with typical rings A–E of a dahurinol-type triterpene.11,13,35,36 Based on HMBC correlation from H-16 at δH 4.24 to the oxygenated methine at δC 78.76 (C-23), and the existence of the spin system C-16/C-17/C-20(C-21)/C-22/C-23, the ring E was established as shown. 1H–1H COSY association between H-23 (δH 4.11) and H-24 (δH 5.34) indicated the linkage of C-23 and C-24. In addition, the connection of C-24 and C-25 was elucidated from the HMBC correlation of H-24 (δH 5.34) to quaternary carbon resonance at δC 77.53 (C-25). Similarly, an acetoxy group was attached to C-24, and CH3-26 (δH 1.59) and CH3-27 (δH 1.56), and the methoxy group at δH 3.24 were linked to C-25 based on the HMBC correlations (Fig. 2). Thus, the side chain of 1 was constructed. Further analyses of HMBC cross-peaks from H-16 (δH 4.24) to C-14 (δC 51.67), and the olefinic (or carbonyl) carbon signal (C-15) at δC 164.34; H-17 (δH 1.60) to C-13 (δC 42.50) and C-14 (δC 51.67); CH3-18 (δH 1.07) to C-13 (δC 42.50), C-14 (δC 51.67) and C-17 (δC 54.60); CH3-28 (δH 1.14) to C-14 (δC 51.67) and C-15 (δC 164.34), suggested the presence of a five-membered ring D with an oxime unit at C-15 (Fig. 2). In the HMBC spectrum, the active hydrogen (δH 12.32) coupled with C-15 (δC 164.34) further supported this elucidation. Thus, the planar structure of 1 was established as shown (Fig. 2).
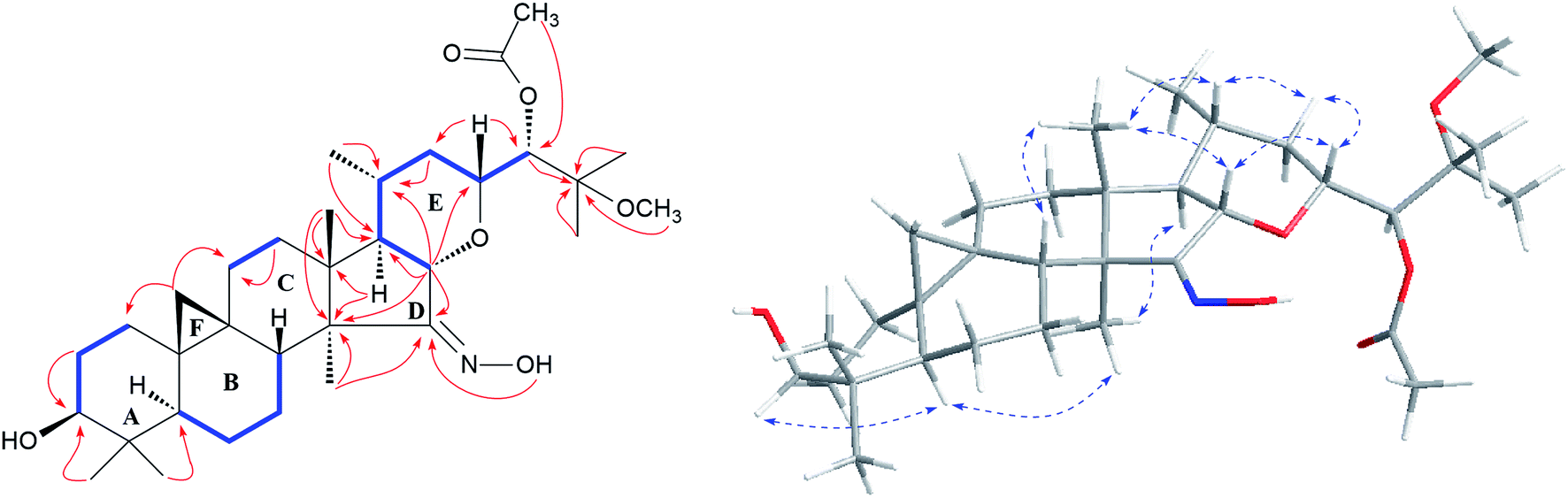 | ||
Fig. 2 Major HMBC ( ), 1H–1H COSY ( ), 1H–1H COSY ( ), and ROESY ( ), and ROESY ( ) correlations of compound 1. ) correlations of compound 1. | ||
The orientations of core structure of 1 was established by the ROESY correlations (Fig. 2) between H-5 (biogenetically α-oriented) and H-3, Me-28 (biogenetically α-oriented) and H-17, Me-18 (biogenetically β-oriented) and H-16, Me-18 and H-20, and H-23 and H-16. In addition, due to its similar coupling constant of H-24 (brs) as that of isodahurinyl-type molecules (<2 Hz), the configuration of C-24 of 1 was deduced as S (the coupling constants of H-24 of dahurinyl-type compounds is around 6–9 Hz).11,13,35,36 Finally, X-ray diffraction analysis (Fig. 3) allowed to confirm the oxime group at C-15, the relative configurations, and the stereochemistry of S at C-24 of 1 due to the Hooft parameter 0.13(8) for 1536 Bijvoet pairs.37 Therefore, the structure of 1 was constructed as shown.
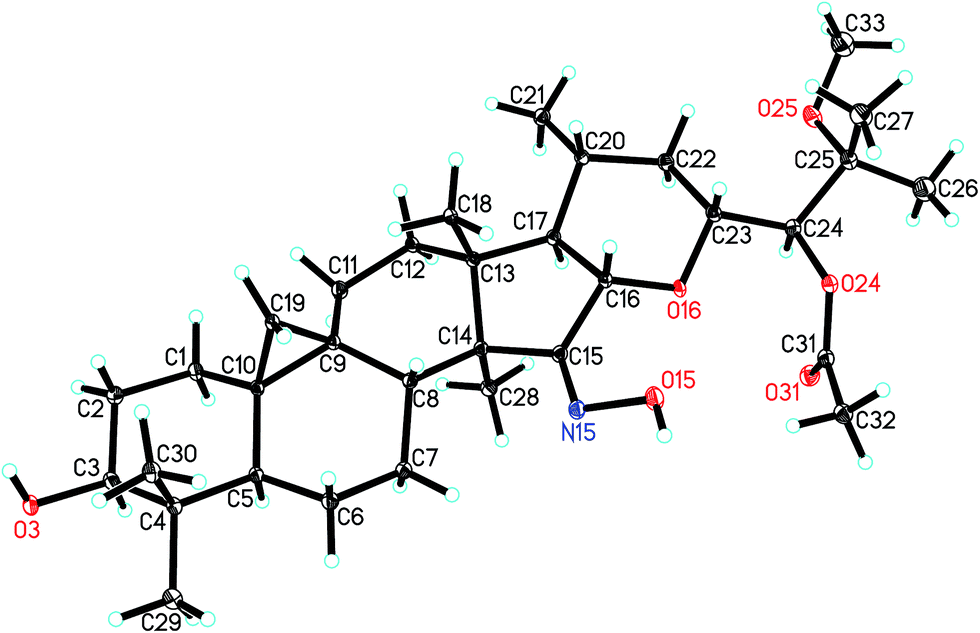 | ||
| Fig. 3 X-ray crystal structure of 1. | ||
The molecular formulas of cimifrigines B (2) and C (3) were determined as C33H53NO6 and C31H51NO5 by the HREIMS ([M]+ m/z 545.3723, calcd 545.3716, and [M]+ m/z 517.3776, calcd 517.3767, respectively). The NMR spectra (Tables 1 and 2) of 2 and 3 resembled to those of 1. The main differences were the substituent groups at C-25 and C-24, respectively. For compound 2, the methoxy group was replaced by a hydroxyl group at C-25. This elucidation was supported by 14 Da less of the molecular weight of 2, and the upfield shift of C-25 by 4.42 ppm. Therefore, a hydroxy group instead of an acetoxy unit at C-24 in compound 3 was determined by similar analyses. ROESY associations of H-3/H5, H-16/CH3-18, H-20/CH3-18 and H-16/H-23 both in 2 and 3 suggested the α, β, β, and β orientations of H-3, H-16, H-20, and H-23, respectively. The S configuration of C-24 of 2 and 3 was proposed by the same way as that of 1. Accordingly, the structures of 2 and 3 were established as shown.
Cimifrigine D (4) had the molecular formula C37H59NO10 as determined by HREIMS (m/z 677.4129 [M]+, calcd 677.4139). In the 1H NMR spectrum, resonances for an anomeric proton at δH 4.79 (H-1′, 1H, d, J = 7.1 Hz), and a cyclopropane methylene at δH 0.20 (6H, d, J = 3.9 Hz) and 0.48 (1H, d, J = 3.3 Hz) were observed. These data indicated 4 was a 9,19-cycloartane triterpene substituted with a sugar unit. Analyses of NMR spectroscopic data revealed that, structurally, the aglycone part of 4 was identical to 2. The sugar unit in 4 was located to C-3 on the basis of HMBC correlation between the anomeric proton at δH 4.79 (J = 7.1 Hz) and the methine signal at δC 88.59 (C-3). In addition, by comparing its TLC and specific rotation with a standard after acid hydrolysis, the sugar was determined as L-arabinose. ROESY correlations of H-16/CH3-18, H-20/CH3-18 and H-16/H-23 suggested an α-orientation of the substituents at C-16, C-20 and C-23, respectively. Whereas, a β-orientation of the substituent at C-3 was established by the cross-peak of H-3/H-5. The configuration of C-24 was proposed as S by the same way as that of 1. Therefore, the structure of 4 was elucidated as shown.
On the basis of the HREIMS peak at m/z 719.4281 [M]+ (calcd 719.4245), the molecular formula of cimifrigine E (5) was determined as C39H61NO11. Comparison of NMR data of compounds 5 and 4 revealed these two compounds were structurally identical except for the sugar unit. The molecular weight difference between 5 and 4 was 42 Da, consistent with an acetyl unit. Besides, H-2′ signal of 5 was shifted to downfield at δH 5.95, which together with the HMBC correlation between the acetoxy methyl group (δH 2.13) and C-2′ (δC 74.83) located an acetoxy group at C-2′ of 5. The sugar was identified as L-arabinose using the same way as that of 4. The orientations of H-3, H-16, H-20, and H-23 were determined as α, β, β, and β, respectively, by analyses of ROESY correlations. In addition, the configuration of C-24 was elucidated as S by comparison of coupling constant of H-24 of 5 with those of known isodahurinyl-type triteroids.11,13,35,36 Thus, the structure of 5 was determined as shown.
The spectroscopic features of cimifrigines F (6) and G (7) resembled to each other except for the sugar unit. The molecular formulas of 6 and 7 were determined as C35H55NO8 and C37H57NO9, respectively, by HREIMS ([M]+ m/z 617.3943, calcd 617.3928, and [M]+ m/z 659.4053, calcd 659.40337, respectively). Same to that of compounds 4 and 5, the molecular weight difference between 6 and 7 was 42 Da, which equivalent to an acetyl moiety. In addition, the acetoxy group in 7 was located to C-2′ based on the HMBC correlation of the acetoxy methyl group (δH 2.11) and C-2′ (δC 74.37). Structurally, the aglycone part of 6 and 7 was similar to that of 2 except that a terminal double bond was formed between C-25 and C-26 in 6 and 7. HMBC correlations from the olefinic protons at δH 5.30 (for 6) and 5.27 (for 7) and δH 5.04 (for 6) and 5.01 (for 7) to C-24 (δC 78.74 for 6 and 78.82 for 7), C-25 (δC 146.53 for 6 and 146.57 for 7), and CH3-27 (δC 18.55 for 6 and 18.59 for 7) further supported this elucidation. The sugar obtained after acid hydrolysis was identified as L-arabinose by comparing its TLC and specific rotation with a standard. The α-orientation of the substituents at C-16, C-20 and C-23 were determined by ROESY correlations of H-16/CH3-18, H-20/CH3-18 and H-16/H-23. Whereas, correlation of H-3/H-5 indicated the β-orientation of the substituent at C-3. The coupling constant of H-24 for 6 and 7 was 6.3 Hz which consistent with those of dahurinyl-type compounds (6–9 Hz).11,13,35,36 Thus, the stereochemistry of C-24 was elucidated as S and the structure of 6 and 7 were determined as shown.
Cimifrigines A–G (1–7) were evaluated against human HL-60, SMMC-7721, A549, MCF-7, and SW-480 cell lines for their cytotoxicities. All compounds (1–7) showed different levels of activities with the glycoside derivatives (4–7) stronger than the aglycone analogues (1–3) (Fig. 4, Table S1†). Compounds 5 and 7 exhibited as potent activities as positive control cisplatin (IC50: 0.5 to 5.4 μM) against all cell lines with IC50 values ranging from 0.8 to 6.3 μM. However, 4 and 6 indicated broad spectrum and moderate activities with IC50 values around 8.9 to 14.3 μM. Structurally, the C-2′ position was substituted by an acetoxy unit in 5 and 7. While, a hydroxy group was located at this position in 4 and 6. Therefore, based on the aforementioned data, it may gave the preliminary conclusion that sugar unit is critical to the cytotoxicities of this new type of CTs. Besides, hydrophobic groups like acetoxy at the sugar unit could enhance the activities as much as one order of magnitude.
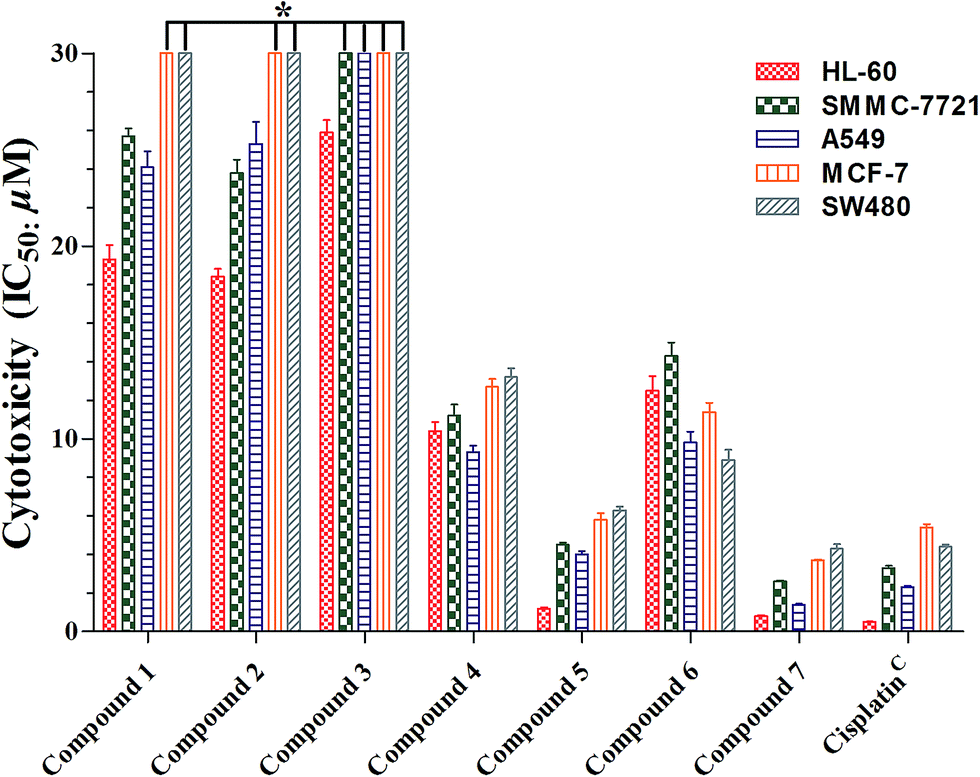 | ||
| Fig. 4 Cytotoxicity of compounds isolated from the flowers of C. frigida. Results are expressed as the average (n = 3) of IC50 values (μM). 0.1% DMSO as the solvent control and did not show any cytotoxicity to those cell lines. CUsed as a positive control substance for the cytotoxicity assay. *The IC50 values are >30 μM. | ||
Conclusions
As mentioned in the Introduction besides the roots of Cimicifuga spp., nontraditionally used part, such as fruits, also contained novel active CTs. Therefore, we expanded the research target to the flowers of C. frigida, a species of which the chemical constituents have not been reported yet. As a result, seven new dahurinol-type analogues, cimifrigines A–G (1–7), with an oxime group at C-15, were isolated and identified. Significantly, all of the compounds had cytotoxic effect and two of them showed comparable activity as that of cisplatin, the positive control, in the in vitro cytotoxicity assay. Besides, preliminary structure–activity relationship also discussed in the study which afforded potential informations for further chemical modifications. In summary, to the best of our knowledge, this is the first example of naturally occured oxime group bearing cytotoxic CTs. In addition, on the basis of the delightful discoveries from the fruits and flowers of Cimicifuga spp., we assume that the more sophisticated parts of this genus, such as pollen and vegetative organ, may also contain novel active chemical constituents and deserved further investigations.Experimental section
General experimental procedures
Column chromatography (CC) was run on silica gel (200–300 mesh, Qingdao Marine Chemical, Inc.), and Lichroprep RP-18 (40–63 μm, Merck). Semipreparative HPLC was carried out on an Agilent 1100 liquid chromatography system using an YMC-Pack 10 mm × 250 mm column (Pro C18 RS). Precoated TLC plates (200–250 μm thickness, silica gel 60 F254, Qingdao Marine Chemical, Inc.) were used for thin-layer chromatography. The spots in TLC were visualized by heating after spraying with 10% aq. H2SO4. 1D and 2D NMR spectra were performed on Bruker DRX-500 and Avance III-600 MHz spectrometers (Bruker, Zűrich, Switzerland) with solvent signal as internal reference. ESIMS and HRESIMS were run on a Shimadzu LCMS-IT-TOF mass spectrometer (Shimadzu, Kyoto, Japan) or an Agilent G6230 TOF MS (Agilent Technologies, Palo Alto, USA). Infrared spectra were tested on a Shimadzu IR-450 instrument with KBr pellets. A JASCO P-1020 digital polarimeter was applied to record optical rotations, using MeOH as solvent. X-ray diffraction was realized on a Bruker SMART APEX CCD crystallography system.Plant material
The flowers of Cimicifuga frigida (1.0 kg) were collected from Litang County, Sichuan Province, China, in September 2012. Prof. Wang Zongyu, Kunming Institute of Botany, Chinese Academy of Sciences, identified the species. A voucher specimen (KUN no. 201209003) has been deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, PR China.Extraction and isolation
MeOH (4 L, 3 times, 7 days each) was used to extract the dried and milled flowers of Cimicifuga frigida (1.0 kg) at room temperature. MeOH was evaporated under vacuum at 50 °C to afford the extract (87.4 g). The extract gave fractions A (10.3 g), B (18.1 g), C (13.4 g), and D (12.3 g) by silica gel CC (2.5 kg, 10 × 150 cm) eluted with CHCl3–MeOH [100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 (4 L), 50:1 (8 L), 10:1 (7 L), 0:100 (4 L)]. Another six sub-fractions (B.1–B.6) were obtained through RP-18 CC (500 g, 6 × 50 cm), gradiently eluted with MeOH–H2O from 60:40 to 100:0. Fraction B.3 (2.7 g) yielded compounds 1 (3.8 mg), 2 (3.2 mg), and 3 (1.9 mg) by silica gel CC (40 g, 3 × 60 cm) eluting with CHCl3–Me2CO from 30:1 gradient to 10:1 and semipreparative HPLC (eluted with CH3CN–H2O, gradient from 65:35 to 85:15). Sub-fractions (C.1–C.4) were obtained by RP-18 CC (eluted with MeOH–H2O, gradient from 50:40 to 85:15) on fraction C. Consequently, 5 (3.3 mg), and 7 (2.8 mg) were purified from fraction C.2 (1.8 g) by silica gel CC (40 g, 3 × 40 cm) eluted with CHCl3–Me2CO (gradient from 20:1 to 10:1), and then repeated semipreparative HPLC (eluted with CH3CN–H2O, gradient from 50:50 to 65:35). Fraction C.3 gave compounds 4 (2.5 mg) and 6 (2.7 mg) by successively silica gel CC (40 g, 3 × 40 cm, eluted with CHCl3–Me2CO 10:1, 13 L), and semipreparative HPLC (eluted with CH3CN–H2O, gradient from 50:50 to 65:35).
:0 (4 L), 50:1 (8 L), 10:1 (7 L), 0:100 (4 L)]. Another six sub-fractions (B.1–B.6) were obtained through RP-18 CC (500 g, 6 × 50 cm), gradiently eluted with MeOH–H2O from 60:40 to 100:0. Fraction B.3 (2.7 g) yielded compounds 1 (3.8 mg), 2 (3.2 mg), and 3 (1.9 mg) by silica gel CC (40 g, 3 × 60 cm) eluting with CHCl3–Me2CO from 30:1 gradient to 10:1 and semipreparative HPLC (eluted with CH3CN–H2O, gradient from 65:35 to 85:15). Sub-fractions (C.1–C.4) were obtained by RP-18 CC (eluted with MeOH–H2O, gradient from 50:40 to 85:15) on fraction C. Consequently, 5 (3.3 mg), and 7 (2.8 mg) were purified from fraction C.2 (1.8 g) by silica gel CC (40 g, 3 × 40 cm) eluted with CHCl3–Me2CO (gradient from 20:1 to 10:1), and then repeated semipreparative HPLC (eluted with CH3CN–H2O, gradient from 50:50 to 65:35). Fraction C.3 gave compounds 4 (2.5 mg) and 6 (2.7 mg) by successively silica gel CC (40 g, 3 × 40 cm, eluted with CHCl3–Me2CO 10:1, 13 L), and semipreparative HPLC (eluted with CH3CN–H2O, gradient from 50:50 to 65:35).
X-ray crystal structure analysis
A Bruker APEX DUO diffractometer equipped with an APEX II CCD was used to obtain the intensity data at 100 K, using Cu Kα radiation. Bruker SAINT was applied for cell refinement and data reduction. The structure was determined by direct methods using SHELXS-97.38 Refinements were performed with SHELXL-97, using full-matrix least-squares, with anisotropic displacement parameters for all the non-hydrogen atoms. The H atoms were placed in calculated positions and refined using a riding model. Molecular graphics were calculated with PLATON.X-ray crystallography of compound 1
Colorless crystals of compound 1 (CCDC: 1545247†) was obtained by methanol extract of Cimicifuga frigida. The X-ray crystallographic data of 1: C33H53NO6, monoclinic, pace group P21, a = 9.5634(4) Å, b = 11.2799(5) Å, c = 14.9190(6) Å, α = 90.00°, β = 103.4570(10)°, γ = 90.00°, V = 1565.19(11) Å3, Z = 2, Dcalcd = 1.188 g cm−3. The final R1 values were 0.1056 (I > 2σ(I)). The final wR(F2) values were 0.2898 (I > 2σ(I)). The goodness of fit on F2 was 1.492. Crystal size, 0.54 × 0.40 × 0.10 mm3. Flack parameter = 0.2(3). The Hooft parameter is 0.13(8) for 1536 Bijvoet pairs.Hydrolysis and identification of the sugar units in compounds 4–7
The MeOH solution (3 mL) of each compound (1.5 mg) was refluxed with 0.5 N HCl (2 mL) for 2 h. CHCl3 (3 × 10 mL) was used to extract the reaction mixture after diluting with H2O. A monosaccharide was given by neutralizing each aqueous layer with Ag2CO3 and filtering the precipitate. The monosaccharide from compounds 4–7 had an Rf (EtOAc–CHCl3–MeOH–H2O, 3:2:2:1) and specific rotation of [α]20D +63.4 (c0.06, MeOH) corresponding to those of L-arabinose (Sigma-Aldrich).
Cytotoxicity bioassay
The human tumor cell lines HL-60, SMMC-7721, A549, MCF-7, and SW-480 were used in the cytotoxic assay. These cell lines were obtained from ATCC (Manassas, VA, USA). DMEM medium (Hyclone, USA), supplemented with 10% fetal bovine serum (Hyclone, USA), was used to culture cells in 5% CO2 at 37 °C. The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) method was applied to evaluate the cytotoxicity.39,40 Briefly, cells were seeded into each well of a 96-well cell culture plate. After 12 h of incubation at 37 °C, the test compound was added. Each tumor cell line was exposed to compounds 1, 2, and 3 at concentrations of 3, 10, 20, 50, and 100 μM triplicates for 48 h, to compounds 4 and 6 at concentrations of 1, 3, 10, 30, and 50 μM triplicates for 48 h, and to compounds 5 and 7 at concentrations of 0.1, 0.3, 1, 5, 10, and 20 μM triplicates for 48 h, respectively. Cisplatin (Sigma, USA) was used as a positive control. After compound treatment, cell viability was detected and a cell growth curve was graphed. IC50 values were calculated by Reed and Muench's method.41 (All compounds were dissolved in DMSO as 100 mM stock and 0.1% DMSO was used as the solvent control).Acknowledgements
This project was supported by the National Natural Science Foundation of China (81302670 and U1132604) and the Foundation of State Key Laboratory of Phytochemistry and Plant Resources in West China (P2015-KF03).Notes and references
- E. Liske and P. Wustenberg, Menopause, 1998, 5, 250–255 CrossRef.
- Y. Nian, H. Zhu, W. R. Tang, Y. Luo, J. Du and M. H. Qiu, J. Nat. Prod., 2013, 76, 896–902 CrossRef CAS PubMed.
- Y. Nian, J. Yang, T. Y. Liu, Y. Luo, J. H. Zhang and M. H. Qiu, Sci. Rep., 2015, 5, 9026–9031 CrossRef CAS PubMed.
- D. J. McKenna, K. Jones, S. Humphrey and K. Hughes, Altern. Ther. Health Med., 2001, 7, 93–100 CAS.
- S. Nobuko, K. Mutsuo, T. Harukuni, M. Teruo, E. Fumio, N. Hoyoku, N. Masahiro, S. Yohiro and H. L. Kuo, Bioorg. Med. Chem., 2005, 13, 1403–1408 CrossRef PubMed.
- Chinese Pharmacopoeia Commission, The Pharmacopoeia of Chinese People's Republic, ed. Y. Li, The Chemical Industry Publishing House, Beijing, China, 2010, vol. 1, pp. 68–69 Search PubMed.
- J. X. Li and Z. Y. Yu, Curr. Med. Chem., 2006, 13, 2927–2951 CrossRef CAS PubMed.
- N. M. Bao, Y. Nian, W. H. Wang, X. L. Liu, Z. T. Ding and M. H. Qiu, Phytochem. Lett., 2015, 12, 200–202 CrossRef CAS.
- W. H. Wang, Y. Nian, Y. J. He, L. S. Wan, N. M. Bao, G. L. Zhu, F. Wang and M. H. Qiu, Tetrahedron, 2015, 71, 8018–8025 CrossRef CAS.
- N. M. Bao, Y. Nian, G. L. Zhu, W. H. Wang, L. Zhou and M. H. Qiu, Fitoterapia, 2014, 99, 191–197 CrossRef CAS PubMed.
- Y. Nian, H. Y. Wang, L. Zhou, J. Su, Y. Li and M. H. Qiu, Planta Med., 2013, 79, 60–69 CAS.
- Y. Nian, H. Y. Wang, J. Su, L. Zhou, G. Feng, Y. Li and M. H. Qiu, Tetrahedron, 2012, 68, 6521–6527 CrossRef CAS.
- Y. Nian, X. M. Zhang, Y. Li, Y. Y. Wang, J. C. Chen, L. Lu, L. Zhou and M. H. Qiu, Phytochemistry, 2011, 72, 1473–1481 CrossRef CAS PubMed.
- Y. Nian, Y. L. Zhang, J. C. Chen, L. Lu, C. Qing and M. H. Qiu, J. Nat. Prod., 2010, 73, 93–98 CrossRef CAS PubMed.
- Y. Nian, J. C. Chen, L. Lu, X. M. Zhang and M. H. Qiu, Helv. Chim. Acta, 2009, 92, 112–120 CrossRef CAS.
- L. Lu, J. C. Chen, Y. Li, C. Qing, Y. Y. Wang, Y. Nian and M. H. Qiu, Chem. Pharm. Bull., 2012, 60, 571–577 CrossRef CAS PubMed.
- H. Y. Wang, Y. Nian, C. Y. Ma, Y. B. Song, L. Zhou and M. H. Qiu, Chin. J. Chem., 2012, 30, 1265–1268 CrossRef CAS.
- D. S. Li, Y. Nian, Y. Sun and M. H. Qiu, Helv. Chim. Acta, 2011, 94, 632–638 CrossRef CAS.
- L. R. Sun, J. Yan, L. Zhou, Z. R. Li and M. H. Qiu, Molecules, 2011, 16, 5701–5708 CrossRef CAS PubMed.
- L. Lu, J. C. Chen, H. J. Song, Y. Li, Y. Nian and M. H. Qiu, Chem. Pharm. Bull., 2010, 58, 729–733 CrossRef CAS PubMed.
- L. Lu, J. C. Chen, Y. Nian, Y. Sun and M. H. Qiu, Molecules, 2009, 14, 1578–1584 CrossRef CAS PubMed.
- L. R. Sun, J. Yan, Y. Nian, L. Zhou, H. J. Zhang and M. H. Qiu, Molecules, 2008, 13, 1712–1721 CrossRef CAS PubMed.
- L. R. Sun, C. Qing, Y. L. Zhang, S. Y. Ji, Z. R. Li, S. J. Pei, M. H. Qiu, M. L. Gross and S. X. Qiu, Beilstein J. Org. Chem., 2007, 3, 1–6 CrossRef PubMed.
- L. R. Sun, J. Yan, L. Lu, S. J. Pei, Z. R. Li, L. Zhou, X. M. Zhang and M. H. Qiu, Helv. Chim. Acta, 2007, 90, 1313–1318 CrossRef CAS.
- Z. Ali, S. L. Khan, R. S. Pawar, D. Ferreira and I. K. Khan, J. Nat. Prod., 2007, 70, 107–110 CrossRef CAS PubMed.
- C. Dan, Y. Zhou, Y. Deng, S. L. Peng, L. S. Ding, M. L. Gross and S. X. Qiu, Org. Lett., 2007, 9, 1813–1816 CrossRef CAS PubMed.
- J. C. Gao, F. Huang, J. C. Zhang, G. Y. Zhu, M. S. Yang and P. G. Xiao, J. Nat. Prod., 2006, 69, 1500–1502 CrossRef CAS PubMed.
- J. X. Li, J. Liu, C. C. He, Z. Y. Yu, Y. Du, S. Kadota and H. Seto, Maturitas, 2007, 58, 59–69 CrossRef CAS PubMed.
- N. Sakurai, J. H. Wu, Y. Sashida, Y. Mimaki, T. Nikaido, K. Koike, H. Itokawa and H. Lee, Bioorg. Med. Chem. Lett., 2004, 14, 1329–1332 CrossRef CAS PubMed.
- M. A. Findeis, F. Schroeder, T. D. McKee, D. Yager, P. C. Fraering, S. P. Creaser, W. F. Austin, J. Clardy, R. Wang, D. Selkoe and C. B. Eckman, ACS Chem. Neurosci., 2012, 3, 941–951 CrossRef CAS PubMed.
- J. H. Lee, T. D. Cuong, S. J. Kwack, J. H. Seok, J. K. Lee, J. Y. Jeong, M. H. Woo, J. S. Choi, H. K. Lee and B. S. Min, Planta Med., 2012, 78, 1391–1394 CrossRef CAS PubMed.
- J. A. Compton, Plantsman, 1992, vol. 14, pp. 99–115 Search PubMed.
- Editorial Committee of Chinese flora, Flora of China, ed. W. C. Wang, China Science Press, Beijing, China, 2004, vol. 27, pp. 93–103 Search PubMed.
- C. Ren, Q. Yuan and Q. E. Yang, Nord. J. Bot., 2012, 30, 585–595 CrossRef.
- Y. Shao, A. Harris, M. F. Wang, H. J. Zhang, G. A. Cordell, M. Bowman and E. Lemmo, J. Nat. Prod., 2000, 63, 905–910 CrossRef CAS.
- G. Kusano, Y. Murakami, N. Sakurai and T. Takemoto, Yakugaku Zasshi, 1976, 96, 82–85 CrossRef CAS.
- R. W. W. Hooft, L. H. Straver and A. L. Spek, J. Appl. Crystallogr., 2008, 41, 96–103 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL97, University of Göttingen, Germany, 1997 Search PubMed.
- T. Mosmman, Immunol. Methods, 1983, 65, 55–63 CrossRef.
- M. C. Alley, D. A. Scudiero, A. Monks, M. L. Hursey, M. J. Czerwinski and D. L. Fine, Cancer Res., 1988, 48, 589–601 CAS.
- L. J. Reed and H. Muench, Am. J. Hyg., 1938, 27, 493–497 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR spectra, HRESIMS spectra of new compounds 1–7, and X-ray crystallographic data of 1. CCDC 1545247. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra07275a |
| This journal is © The Royal Society of Chemistry 2017 |