
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereoselective one pot five-component reaction toward 4-(tetrazole)-1,3-oxazinanes†
Ajay L. Chandgude a,
Daniele Narduccia,
Katarzyna Kurpiewskab,
Justyna Kalinowska-Tłuścikb and
Alexander Dömling*a
a,
Daniele Narduccia,
Katarzyna Kurpiewskab,
Justyna Kalinowska-Tłuścikb and
Alexander Dömling*a
aDepartment of Drug Design, University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands. E-mail: a.s.s.domling@rug.nl
bJagiellonian University, Faculty of Chemistry, Department of Crystal Chemistry and Crystal Physics Biocrystallography Group, Ingardena 3, 30-060 Krakow, Poland
First published on 26th October 2017
Abstract
A diastereoselective one pot five-component reaction toward the synthesis of 4-(tetrazole)-1,3-oxazinanes has been reported. The sonication-accelerated, catalyst-free, simple, general and highly time efficient, Asinger–Ugi-tetrazole reaction was used for the synthesis of diverse 4-(tetrazole)-1,3-oxazinanes. The reaction exhibit excellent diastereoselectivity and broad substrate scope.
Introduction
The oxazine motif attained significant attention due to their widespread availability in natural products, such as aragupetrosine, bujeine, pagicerine, quimbeline, and upenamide.1 The oxazines scaffold is present in many pharmacologically active agents and drugs, such as pranlukast, dirithromycin, and dolutegravir.2 It is also used as intermediate for the synthesis of drugs like oxacephem antibiotics.2cOn the other hand, the tetrazole is a highly important synthetic scaffold for a wide range of areas and applications. It is extensively used in medicinal and organic chemistry, also in industries such as explosives, agrochemicals, materials, and polymers.3 Their use as a carboxylic acid isostere and cis-amide bond isostere in peptides have many advantages, such as extra lipophilicity, metabolic stability, and hydrogen bonding to increase potency.4 Heterocycles are important in drug design and are present in half of the top 200 drugs.5 Thus, recently the use of heterocycle linked tetrazole scaffolds got major attention as a privileged core structure for the development of drug candidates. This combination is an effective strategy to balance drug-like properties. Owning the importance of heterocycles linked tetrazoles resulted into reports of many examples of bioactive agents, such as pyridine-tetrazole, Akt1 and Akt2 dual inhibitors;6 pyrazole-tetrazole, antileishmanials7 or as cardiotonic agents;8 pyridine-tetrazole, antibacterial;9 piperazines-tetrazole, type 2 diabetes;10 isoxazole-tetrazole, for AMPA receptors;11 and also for ionotropic glutamate receptors.12 Moreover, in non-medical applications, use of cyclic ketimines-tetrazoles as organocatalysts,13 and pyridine-tetrazoles in lanthanide-based applications14 are also well known.
Strategies for the synthesis of heterocycle-tetrazole can be categorized into three types. First, the coupling of heterocycle with tetrazole (Fig. 1A).15 Second, synthesis of cyano-heterocycle followed by the tetrazole formation (Fig. 1B).7 Third, tetrazole synthesis followed by post-condensation reaction toward heterocycle formation (Fig. 1C).16 These methods involve more than two steps, harsh coupling conditions, and also the synthesis of starting material for the coupling can be tedious.
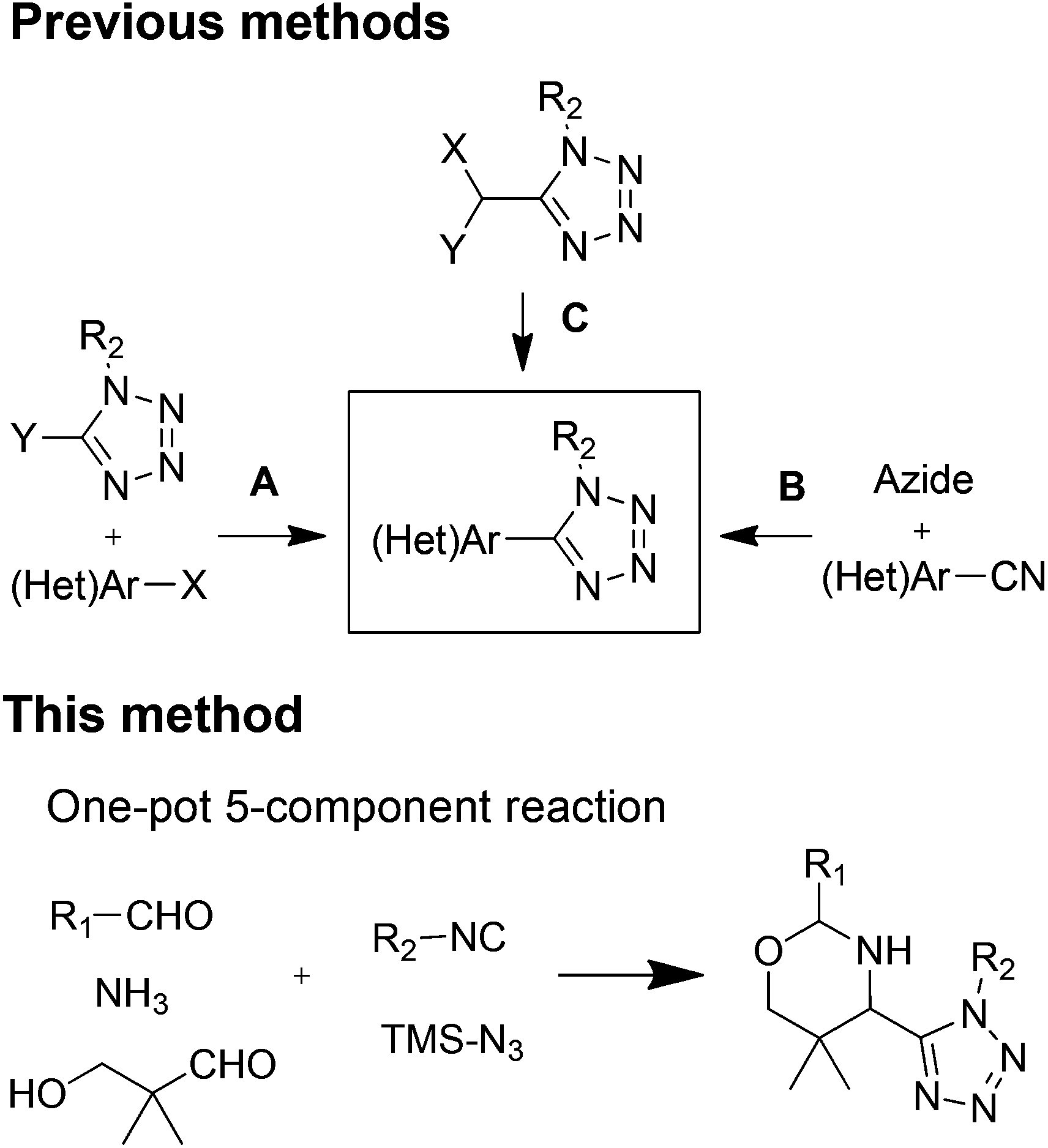 | ||
| Fig. 1 In situ bis-heterocycle synthesis. | ||
Here we are reporting the first example of an in situ oxazine-tetrazole motif synthesis by using a one-pot five-component reaction. The oxazine-tetrazole scaffold is accessible in one pot, time efficiently with high diastereoselectivity and diversity.
Results and discussion
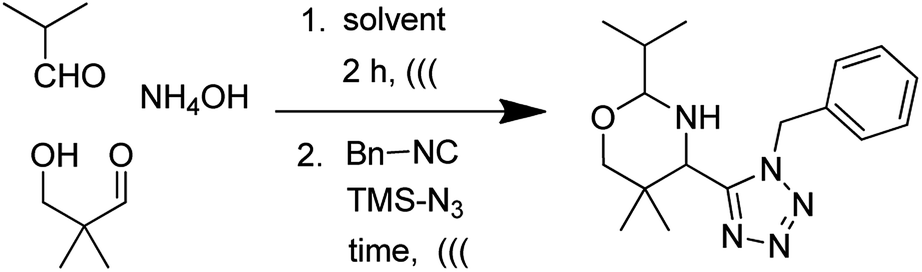
We envisioned the use of Asinger–Ugi-tetrazole union for the first time to synthesize an oxazines-tetrazole scaffold. We start our optimization by using isobutyraldehyde, ammonium hydroxide, 3-hydroxypivalaldehyde, benzyl isocyanide and TMSN3. The reaction in methanol at room temperature resulted in only trace product formation (Table 1, entry 1). Union of an Asinger reaction with other MCRs is known to be low yielding.17 Therefore we move our attention towards the use of sonication in MCR which can be highly effective.18 Further optimization was carried out with sonication at room temperature.
|
|||
|---|---|---|---|
| Entry | Solvent | Time (h) | Yieldb (%) |
| a The reaction was carried out with isobutyraldehyde (1 mmol), ammonium hydroxide (1.5 mmol), 3-hydroxypivalaldehyde (1 mmol), benzyl isocyanide (1.2 mmol) and TMSN3 (1.2 mmol) in 0.5 ml solvent.b Yield of isolated product.c Without sonication at room temperature. nd-not determined. nr-no reaction. | |||
| 1c | MeOH | 12 | Trace |
| 2 | MeOH | 2 | 51 |
| 3 | MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (3:1) :H2O (3:1) |
4 | 21 |
| 4 | MeOH:H2O (1:1) |
4 | 17 |
| 5 | MeOH:H2O (1:3) |
6 | 15 |
| 6 | EtOH | 2 | nd |
| 7 | H2O | 7 | nr |
| 8 | Dioxane | 2 | 32 |
| 9 | THF | 2 | 29 |
| 10 | TFE | 3 | 16 |
| 11 | DCM | 3 | 17 |
| 12 | MeCN | 3 | 33 |
| 13 | Toluene | 2 | 60 |
| 14 | Toluene:H2O (1:1) |
4 | 11 |
| 15 | Toluene:H2O (3:1) |
3 | 19 |
| 16 | Toluene:H2O (4:1) |
2 | 25 |
| 17 | p-Xylene | 4 | 15 |
First, we optimized the ammonia source. We screened different ammonia sources, like NH4OH, NH4Cl, and NH4OAc. NH4OH in 1.5 equivalent was found to be the best. When the reaction was performed in MeOH, a promising 51% yield was obtained (Table 1, entry 2). Next, we move our attention towards solvent screening. Use of MeOH:H2O solvent systems, such as 3:1, 1:1 or 1:3 resulted in less product formation, like 21%, 17%, and 15% respectively (Table 1, entries 3–5). However, EtOH as solvent gave the desired product only in trace amounts. When water was used as a solvent, the reaction did not proceed further probably due to the water insolubility of the reactants (Table 1, entry 7). Use of dioxane and THF provided a similar yield of ∼30% (Table 1, entries 8–9). TFE and DCM gave lower yields. Toluene turned out to be the best solvent with 60% yield (Table 1, entry 13). However, an attempt to make the protocol greener by using toluene:water solvent system resulted in a lowering to 25% yield (Table 1, entries 14–16); while xylene did not ameliorate the reaction yield.
With optimized conditions in hand, next, we tested the scope and limitations of this reaction by reacting various aldehydes and isocyanides (Table 2). Different linear and branched aliphatic aldehydes such as isobutyraldehyde, propanal, butyraldehyde, and valeraldehyde provide moderate to good yields of 21% to 60% (Table 2, entries 2–7). Good to excellent yield were obtained with aliphatic-aromatic aldehydes like benzyl and phenylacetaldehyde. Benzaldehyde and 2-chloro benzaldehyde are valid substrates in this reaction with providing moderate yields of 35% and 45% respectively (Table 2, entries 12 and 13). However, the reaction with ketone resulted in only trace product formation. It is important to mention that, the preformation of imine from aldehyde and ammonium hydroxide is needed to get high yield which normally requires 30 minutes to 1 hour preincubation. The slow addition of 3-hydroxypivalaldehyde over 30 min also helped to get a clean reaction. After the addition of isocyanide and TMSN3, the reaction completes within 2–4 hours.
|
||||
|---|---|---|---|---|
| Entry | R1-CHO | R2-NC | Yieldb (%) | drc |
| a The reaction was carried out with isobutyraldehyde (1 mmol), ammonium hydroxide (1.5 mmol), 3-hydroxypivalaldehyde (1 mmol), benzyl isocyanide (1.2 mmol) and TMSN3 (1.2 mmol) in 0.5 ml solvent.b Yield of isolated product.c dr ratio determined by NMR analysis. | ||||
| 1 | 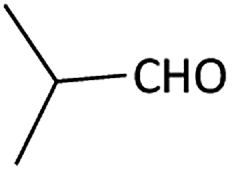 |
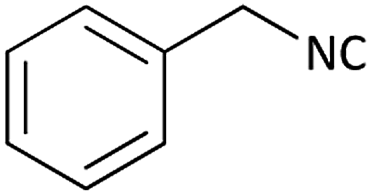 |
(1a) 60 | 78:22 |
| 2 | 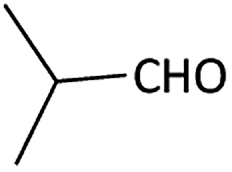 |
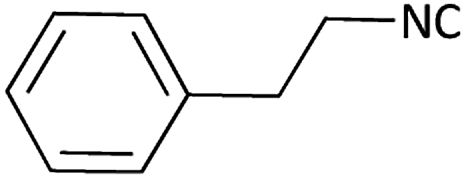 |
(1b) 51 | 91:09 |
| 3 | 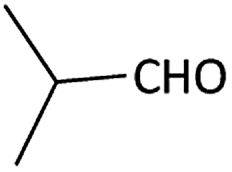 |
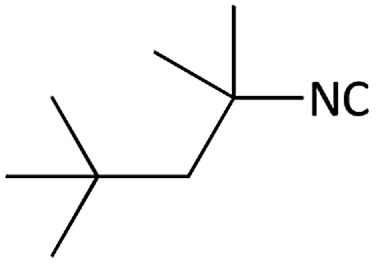 |
(1c) 56 | 90:10 |
| 4 | 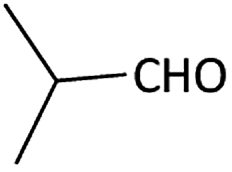 |
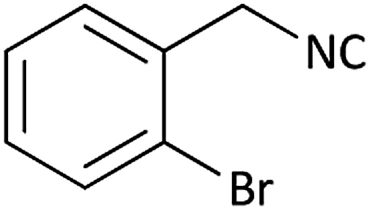 |
(1d) 47 | 88:12 |
| 5 | 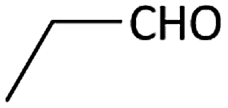 |
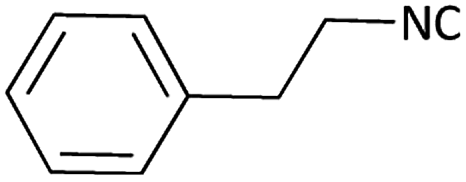 |
(1e) 38 | 90:10 |
| 6 | 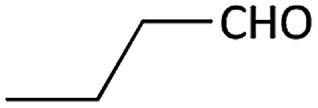 |
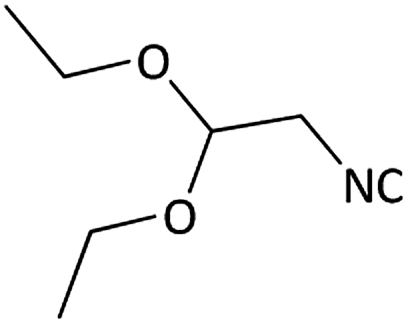 |
(1f) 55 | 91:09 |
| 7 | 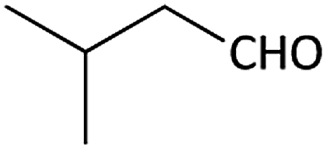 |
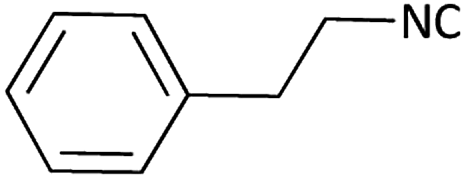 |
(1g) 25 | 90:10 |
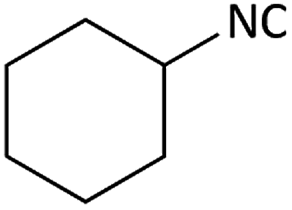
| 8 | 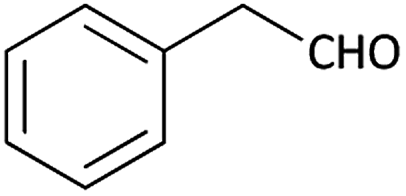 |
 |
(1h) 34 | 96:04 |
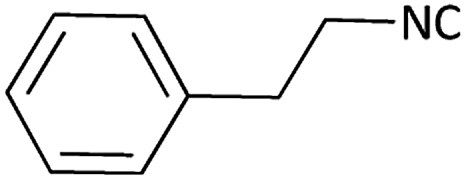
| 9 | 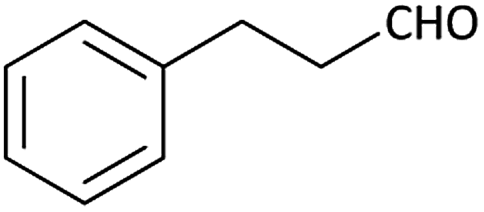 |
 |
(1i) 48 | 94:06 |
| 10 | 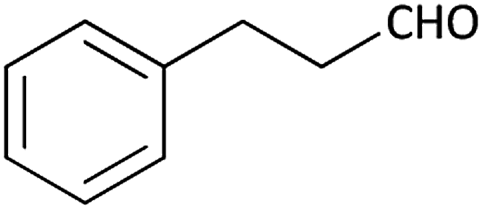 |
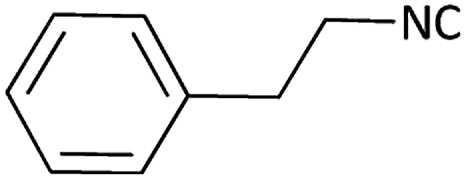 |
(1j) 50 | 90:10 |
| 11 | 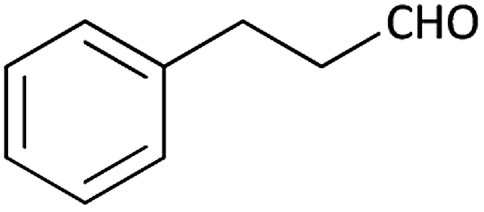 |
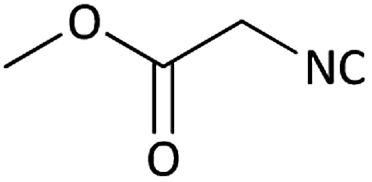 |
(1k) 83 | 91:09 |
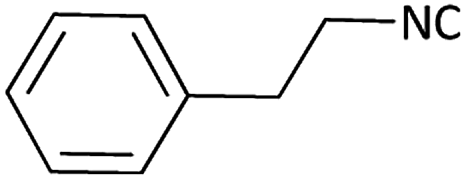
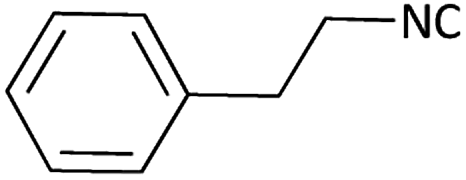
| 12 | 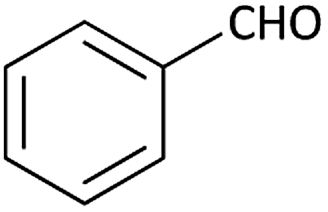 |
 |
(1l) 35 | 94:06 |
| 13 | 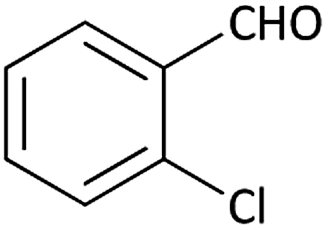 |
 |
(1m) 45 | 92:08 |
| 14 | 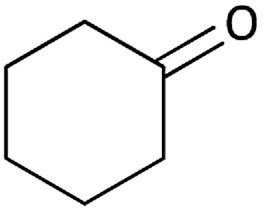 |
 |
(1n) Trace | — |
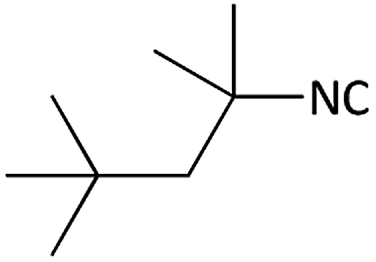
Further, we screened different isocyanides. Aliphatic isocyanides like tert-octyl isocyanide and cyclohexyl isocyanide worked well (Table 2, entries 3 and 9). Aromatic isocyanides like benzyl and phenylethyl isocyanide with different aldehydes, product yields were good. The glycine isocyanide provided the excellent yield of 83% (Table 2, entry 11). The functional group protected isocyanide, diethoxy-acetaldehyde was also compatible in this reaction, which is interesting for further postmodification condensation or for unions with other MCR (Table 2, entry 6). Also, a tolerance of a 2-bromo benzyl isocyanide is interesting for potential postmodification reactions (Table 2, entry 4).
In all examples a higher diastereoselectivity was observed. Aliphatic, aromatic aldehydes and also all isocyanides show more than 90:10 diastereoselectivity. However with benzyl isocyanide and 2-bromo benzylisocyanides low diastereoselectivity was observed.
The structures have been confirmed by NMR, MS (low and high resolution) and also by X-ray crystallography (Fig. 2).
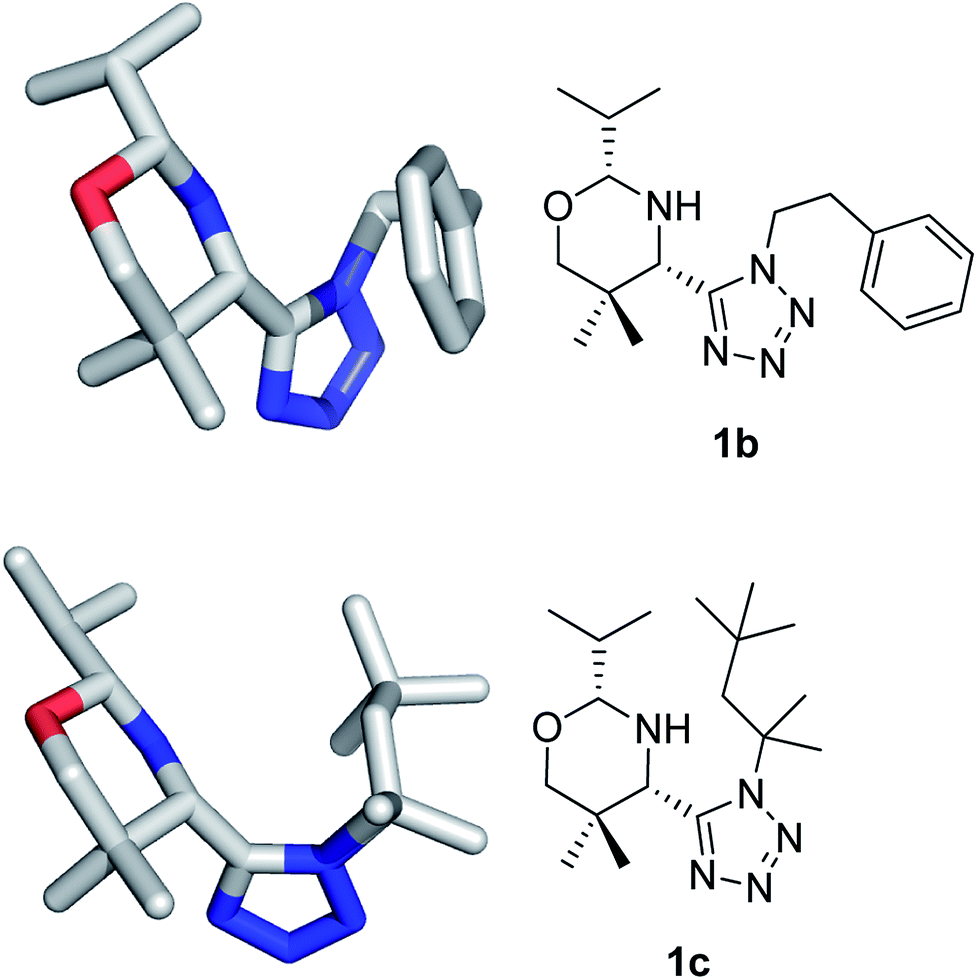 | ||
| Fig. 2 X-ray structures of 1b and 1c. | ||
Proposed mechanism
Based on the previous reports we proposed the following mechanism and which could also explain the high diastereoselectivity of this reaction. In this reaction, first aldehyde, ammonia and 3-hydroxypivalaldehyde react together to form the asymmetrically substituted Asinger reaction product, 5,6-dihydro-2H-1,3-oxazines. With the reference of reported articles and study we assume that out of the two possible half-chair conformations with minimal energy I and II, the energetically preferred half-chair conformation conformer I is strongly favoured.17,19 As the strong steric interactions between one of the methylene protons in 6-position and substitution on the position 2 unflavored the conformer II. However, in conformer I this steric repulsion is reduced by position 6 (Scheme 1).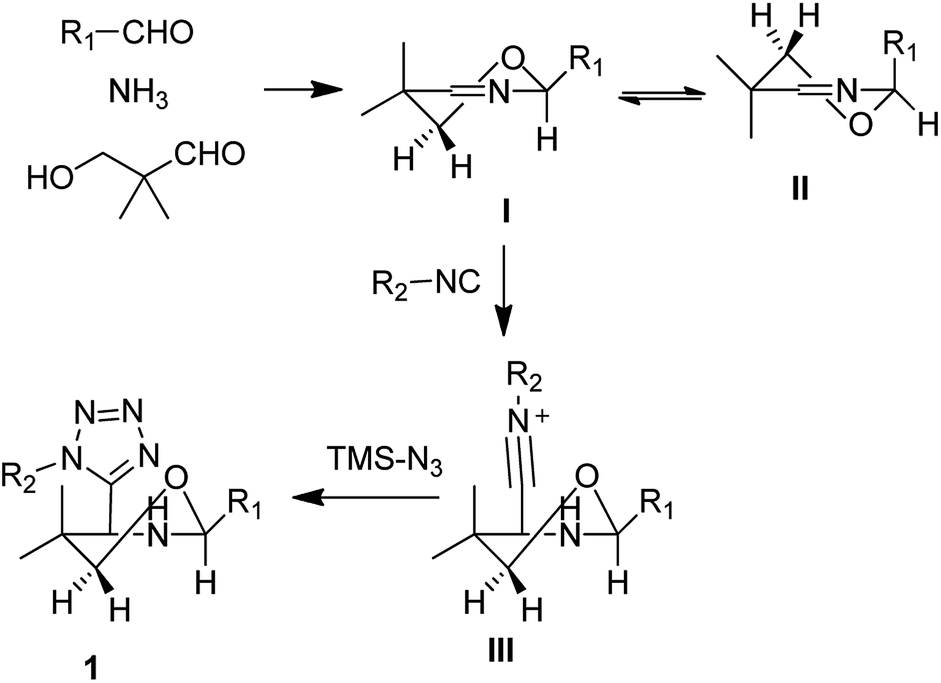 | ||
| Scheme 1 Proposed mechanism of highly diastereoselective Asinger–Ugi tetrazole reaction. | ||
Next, as per previously published research,20 isocyanide will preferentially attack axially on the six membered, 5,6-dihydro-2H-1,3-oxazines to reduce the steric stain to form the intermediate III. Followed by azide attack on this intermediate III formed the final product in high diastereoselectivity as per above mentioned reasons like preferable conformers of 5,6-dihydro-2H-1,3-oxazines and preferred axial attack of isocyanides.
Conclusions
In conclusion, we have developed a diastereoselective one-pot five component reaction for the oxazinane-tetrazoles synthesis. This sonication-assisted, novel, and general reaction has many advances, such as high time efficiency, catalyst-free, diverse scope, and excellent diastereoselectivity. Moreover, due to diverse substrate compatibility, this reaction has a significant potential for postcondensation reactions to get more complex and diverse oxazine-tetrazole structures. Studies towards this area are in progress and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Groningen. The Erasmus Mundus Scholarship “Svaagata” is acknowledged for a fellowship to A. Chandgude. The work was financially supported by the NIH (2R01GM097082-05) and by Innovative Medicines Initiative (grant agreement No. 115489). Funding has also been received from the European Union's Horizon 2020 research and innovation programme under MSC ITN “Accelerated Early stage drug dIScovery” (AEGIS), grant agreement No. 675555. The research (K. K., J. K.-T.) was carried out with the equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (Contract No. POIG.02.01.00-12-023/08).Notes and references
- (a) M. Kobayashi, K. Kawazoe and I. Kitagawa, Tetrahedron Lett., 1989, 30, 4149–4152 CrossRef CAS; (b) X. J. Hu, H. P. He, H. Zhou, Y. T. Di, X. W. Yang, X. J. Hao and L. Y. Kong, Helv. Chim. Acta, 2006, 89, 1344–1349 CrossRef CAS; (c) E. Bombardelli, A. Bonati, B. Danielli, B. Gambetta, E. M. Martinelli and G. Mustich, Experientia, 1975, 31, 139–140 CrossRef CAS; (d) J. I. Jimenez, G. Goetz, C. M. S. Mau, W. Y. Yoshida, P. J. Scheuer, R. T. Williamson and M. Kelly, J. Org. Chem., 2000, 65, 8465–8469 CrossRef CAS PubMed.
- (a) R. Kim, V. Bihud, K. bin Mohamad, K. Leong, J. bin Mohamad, F. bin Ahmad, H. Hazni, N. Kasim, S. Halim and K. Awang, Molecules, 2012, 18, 128–139 CrossRef PubMed; (b) G. Ramachandran, K. I. Sathiyanarayanan, M. Sathishkumar, R. S. Rathore and P. Giridharan, Synth. Commun., 2015, 45, 2227–2239 CrossRef CAS; (c) C. Borel, L. S. Hegedus, J. Krebs and Y. Satoh, J. Am. Chem. Soc., 1987, 109, 1101–1105 CrossRef CAS.
- (a) L. M. T. Frija, A. Ismael and M. L. S. Cristiano, Molecules, 2010, 15, 3757–3774 CrossRef CAS PubMed; (b) A. L. Chandgude and A. Domling, Eur. J. Org. Chem., 2016, 2383–2387 CrossRef CAS; (c) V. Y. Zubarev and V. A. Ostrovskii, Chem. Heterocycl. Compd., 2000, 36, 759–774 CrossRef CAS; (d) S. J. Wittenberger, Org. Prep. Proced. Int., 1994, 26, 499–531 CrossRef CAS; (e) A. Sarvary and A. Maleki, Mol. Diversity, 2015, 19, 189–212 CrossRef CAS PubMed.
- (a) C. X. Wei, M. Bian and G. H. Gong, Molecules, 2015, 20, 5528–5553 CrossRef CAS PubMed; (b) J. Roh, K. Vavrova and A. Hrabalek, Eur. J. Org. Chem., 2012, 6101–6118 CrossRef CAS; (c) L. V. Myznikov, A. Hrabalek and G. I. Koldobskii, Chem. Heterocycl. Compd., 2007, 43, 1–9 CrossRef CAS; (d) R. J. Herr, Bioorg. Med. Chem., 2002, 10, 3379–3393 CrossRef CAS PubMed.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- Z. Zhao, W. H. Leister, R. G. Robinson, S. F. Barnett, D. Defeo-Jones, R. E. Jones, G. D. Hartman, J. R. Huff, H. E. Huber, M. E. Duggan and C. W. Lindsley, Bioorg. Med. Chem. Lett., 2005, 15, 905–909 CrossRef CAS PubMed.
- J. V. Faria, M. S. dos Santos, A. M. R. Bernardino, K. M. Becker, G. M. C. Machado, R. F. Rodrigues, M. M. Canto-Cavalheiro and L. L. Leon, Bioorg. Med. Chem. Lett., 2013, 23, 6310–6312 CrossRef CAS PubMed.
- L.-M. Duan, H.-Y. Yu, Y.-L. Li and C.-J. Jia, Bioorg. Med. Chem., 2015, 23, 6111–6117 CrossRef CAS PubMed.
- Y. W. Jo, W. B. Im, J. K. Rhee, M. J. Shim, W. B. Kim and E. C. Choi, Bioorg. Med. Chem., 2004, 12, 5909–5915 CrossRef CAS PubMed.
- T. Yoshida, F. Akahoshi, H. Sakashita, H. Kitajima, M. Nakamura, S. Sonda, M. Takeuchi, Y. Tanaka, N. Ueda, S. Sekiguchi, T. Ishige, K. Shima, M. Nabeno, Y. Abe, J. Anabuki, A. Soejima, K. Yoshida, Y. Takashina, S. Ishii, S. Kiuchi, S. Fukuda, R. Tsutsumiuchi, K. Kosaka, T. Murozono, Y. Nakamaru, H. Utsumi, N. Masutomi, H. Kishida, I. Miyaguchi and Y. Hayashi, Bioorg. Med. Chem., 2012, 20, 5705–5719 CrossRef CAS PubMed.
- S. B. Vogensen, R. P. Clausen, J. R. Greenwood, T. N. Johansen, D. S. Pickering, B. Nielsen, B. Ebert and P. Krogsgaard-Larsen, J. Med. Chem., 2005, 48, 3438–3442 CrossRef CAS PubMed.
- A. A. Jensen, T. Christesen, U. Bolcho, J. R. Greenwood, G. Postorino, S. B. Vogensen, T. N. Johansen, J. Egebjerg, H. Brauner-Osborne and R. P. Clausen, J. Med. Chem., 2007, 50, 4177–4185 CrossRef CAS PubMed.
- O. I. Shmatova and V. G. Nenajdenko, J. Org. Chem., 2013, 78, 9214–9222 CrossRef CAS PubMed.
- M. Giraud, E. S. Andreiadis, A. S. Fisyuk, R. Demadrille, D. Imbert and M. Mazzanti, Inorg. Chem., 2008, 47, 3952–3954 CrossRef CAS PubMed.
- (a) Q. Tang and R. Gianatassio, Tetrahedron Lett., 2010, 51, 3473–3476 CrossRef CAS; (b) I. Becker, J. Heterocycl. Chem., 2008, 45, 1005–1022 CrossRef CAS.
- (a) P. Patil, R. Madhavachary, K. Kurpiewska, J. Kalinowska-Tłuścik and A. Dömling, Org. Lett., 2017, 19, 642–645 CrossRef CAS PubMed; (b) S. Gunawan, J. Petit and C. Hulme, ACS Comb. Sci., 2012, 14, 160–163 CrossRef CAS PubMed.
- (a) K. Kehagia, A. Domling and I. Ugi, Tetrahedron, 1995, 51, 139–144 CrossRef CAS; (b) H. Groger, M. Hatam and J. Martens, Tetrahedron, 1995, 51, 7173–7180 CrossRef.
- (a) A. L. Chandgude and A. Dömling, Org. Lett., 2016, 18, 6396–6399 CrossRef CAS PubMed; (b) A. L. Chandgude and A. Dömling, Green Chem., 2016, 18, 3718–3721 RSC.
- A. Dömling and I. K. Ugi, Tetrahedron, 1993, 49, 9495–9500 CrossRef.
- (a) L. Banfi, A. Basso, V. Cerulli, V. Rocca and R. Riva, Beilstein J. Org. Chem., 2011, 7, 976–979 CrossRef CAS PubMed; (b) C. A. Sperger, P. Mayer and K. T. Wanner, Tetrahedron, 2009, 65, 10463–10469 CrossRef CAS; (c) L. El Kaïm, L. Grimaud, J. Oble and S. Wagschal, Tetrahedron Lett., 2009, 50, 1741–1743 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1521456 and 1521499. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra07392e |
| This journal is © The Royal Society of Chemistry 2017 |