
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polypyridyl substituted BODIPY derivatives; water switchable imaging probes that exhibit halogen substituent dependent localisation in live cells†
Ciarán Dolanab,
Aisling Byrneab,
Conor Longa,
Krzysztof Czamaracd,
Agnieszka Kaczorcd,
Malgorzata Baranskacd and
Tia E. Keyes *ab
*ab
aSchool of Chemical Sciences, Dublin City University, Dublin 9, Ireland. E-mail: tia.keyes@dcu.ie; Tel: +353 1 7008185
bNational Centre for Sensor Research, Dublin City University, Dublin 9, Ireland
cFaculty of Chemistry, Jagiellonian University, Ingardena 3, 30-060 Krakow, Poland
dJagiellonian Centre of Experimental Therapeutics (JCET), Jagiellonian University, Bobrzynskiego 14, 30-348 Krakow, Poland
First published on 11th September 2017
Abstract
The synthesis and photophysical properties of water responsive 1,10-phenanthrolyl and 2,2′-bipyridyl substituted BODIPY derivatives prepared as lipid probes for cell imaging are reported. These compounds exhibit intense emission in non-aqueous media that is reversibly extinguished in aqueous media. Halogen substitution at the BODIPY indacene core decreases the emission quantum yields and causes red spectral shifts of emission maxima of the order H > Br > I. The emission was quenched on binding of the phenanthrolyl or bipyridyl to cations Fe2+, Cu2+ and Zn2+. The origin of the water switching effect and the impact of halogen substitution was investigated by modelling the electronic structure of the fluorophore using DFT methods. All compounds showed excellent permeability to live cells and were found, under imaging conditions, to generally exhibit low cytotoxicity. The absence of emission in the aqueous environment facilitated the collection of high contrast images from membranous regions and lipid droplets in live cells. The staining pattern in HeLa cells was found to depend on halogen substitution. Across both bpy and phen derivatives the halogenated probes showed the strongest targeting of lipid droplets within cells whilst the parent unsubstituted compounds were more widely dispersed in the cytoplasm. Resonance Raman imaging was used to map the distribution of probes within the cell and confirmed that the compounds showed strong co-localisation with lipid rich regions of the cell.
1 Introduction
Fluorescent dyes based on the 4,4′-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) core have been the focus of intensive research because of their useful photophysical properties. Important photophysical characteristics include high fluorescence quantum yields, narrow absorption and emission bands, good solubility across a range of solvents, high (photo)chemical stability and relatively good biological uptake.1–6 In addition, the synthetic versatility of BODIPY derivatives allows their visible absorption and emission characteristics to be varied through relatively simple structural modifications to the pyrrole core.7–10 Thus, BODIPY-based fluorophores can be adapted for use as labels for reagents and biomarkers, in optoelectronic switching applications, as singlet oxygen sensors, and as therapeutic agents.3,11–13Structural modification to the BODIPY core alters the photophysics of the fluorophore. In particular, the extension of the degree of π conjugation through the central indacene unit can have a significant impact (Fig. 1).11,14 Unfortunately, these ‘extended-BODIPY dyes’ tend to be more susceptible to photodegradation.15 Adding donor or acceptor groups to the BODIPY core, particularly groups capable of photoinduced electron transfer (PET), has led to the development of BODIPY-based pH or chemosensors.1,11,16–20 Varying the substituent at the 8 position (meso-position) can affect the spectroscopic and photophysical properties of BODIPY compounds, however, while the impact of such changes can be difficult to predict, the effects of such substitution are generally weaker than substitution at the indacene units.9,21
 | ||
| Fig. 1 Numbering system used for the central BODIPY indacene unit. | ||
BODIPY dyes have been widely used as cellular imaging contrast agents.5,22–27 Functionalisation,10,18,22,23 nanoparticle encapsulation4,20,28 or conjugation to biomolecules can be used to improve aqueous solubility.19
A number of BODIPY derivatives are non-emissive in aqueous media while emitting strongly from organic solvent. The origin of this effect has been attributed to the formation of non-fluorescent dimers or higher aggregates in water.10 And, introduction of bulky substituents at the 2,6-positions of the BODIPY core which inhibit aggregation results in intense fluorescence, even in the solid state.29 Similarly, it has been shown that the presence of methyl substituents at the 1,7-positions can force aromatic substituents at the 8-position into an orthogonal geometry and disrupting intramolecular π–π interactions.30 The formation of non-luminescent charge-transfer states in BODIPY derivatives with donor/acceptor functionalities has also been attributed to the extinction of BODIPY emission in water.52
This aqueous light-switching behaviour is of value in cell imaging, facilitating high contrast imaging of lipid or proteinaceous structures. However, few studies on water switching BODIPY fluorophores in cellular imaging have been reported to date. Herein, we describe a family of 1,10-phenanthrolyl and 2,2′-bipyridyl meso substituted BODIPY compounds, which showed reduced or no emission when in contact with an aqueous environment, but which showed intense emission when in non-aqueous media. We have developed these materials as contrast agents for resolving lipid droplets or membranous structures in the cell. Association of the probe within sub-cellular membrane/protein or lipid structures turns the emission on. We explore the impact of substitution with halogen atoms at the indacene core has on the emission properties of the compounds as well as their uptake and localisation in live cells. The localisation of the probe within a single cell line varies with halogen substitution. We have monitored the distribution of the phenathrolyl-BODIPY probes within live and fixed cells using Raman microscopy and show that the compounds co-localise strongly with lipid rich cellular regions consistent with fluorescence imaging.
Finally, BODIPY derivatives with appended coordination sites have been widely studied as sensors for transduction of metal ion binding reported through fluorescence.5–7,11,12,19,24,25,31–35,37–46 We have also recently shown that complexation of a ruthenium metal centre to a polypyridyl-based BODIPY fluorophore does not alter BODIPY emission permitting the use of the complex as a ratiometric oxygen sensor.35 As the reported BODIPY derivatives are functionalised at the meso-position with metal coordinating ligands, we report a preliminary investigation into their ability to complex biologically relevant metal ions in non-aqueous media and the effect of metal binding on photophysical properties of the dyes.
2 Experimental
2.1 Apparatus
1H NMR, 13C NMR and 19F NMR spectra were performed on either a Bruker AC 400 MHz or Bruker Avance III 600 MHz spectrometer using DMSO-d6 or CDCl3 as solvents unless otherwise stated. Deuterated solvents were used for homo/hetero nuclear lock and the signals are referenced to the deuterated solvent peaks. Mass spectrometry was carried out at the Chemistry and Chemical Biology Laboratory, University College Dublin using a Bruker Esquire LC_00050 electron spray interface (ESI) with a positive ion polarity. Elemental analysis was carried out by Chemistry and Chemical Biology Laboratory, University College Dublin on an Exador analytical CE440 analyser. UV-vis spectra were recorded on a Jasco V-670 spectrophotometer using a 1 cm quartz cuvette and spectra were corrected for solvent absorbance. Solution based emission spectra were recorded on a Varian Cary Eclipse fluorescence spectrophotometer with an excitation slit width of 2.5 nm and an emission slit width of 2.5 nm, unless otherwise stated. Low temperature (77 K) emission experiments were performed by dissolving the BODIPY compound in a butyronitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :propionitrile (5:4 v/v) glass. Solid state fluorescence was carried out on a HR 1000 Labram confocal Raman micro-spectrometer (Horiba Jobin-Yvon). The spectrometer was equipped with diffraction gratings (600 or 1800 grooves per mm), an Olympus BX40 microscope with a 50× objectives and the slit allowed the spectral resolution to be 2 cm−1. The Raman and fluorescence signals were detected by a Peltier-cooled CCD detector following 473 nm laser excitation. The integration times ranged from 2 to 5 seconds. Quantum yields were obtained by absorbance matching each BODIPY dye with a fluorescein standard (θf = 0.91 in methanol) solution. The corresponding integrated photon flux was then determined and the quantum yields calculated as previously reported.47 Luminescent lifetimes were obtained using a Picoquant Fluorotime 100 TCSPC (time correlated single photon counting) system, exciting at 450 nm (picosecond pulse diode lasers) and the band pass dielectric filter was set for detection at 460 nm and above. 10000 counts were collected for each lifetime measurement and all measurements were performed in triplicate using Nanoharp software to record decay plots. Typical pulse repetition rates of the excitation source were 1 × 105 s−1 with a typical pulse width of 300 ps. FluoroFit software was used to analyse the luminescent decays which fitted well to mono-exponential decay function. The quality of the lifetime fit was determined from visual examination of the residuals and a χ2 value of <1.5. Molecular modelling was performed using B3LYP hybrid density functional with a TZVP basis set as implemented in the Gaussian 09 program suite.48–50
:propionitrile (5:4 v/v) glass. Solid state fluorescence was carried out on a HR 1000 Labram confocal Raman micro-spectrometer (Horiba Jobin-Yvon). The spectrometer was equipped with diffraction gratings (600 or 1800 grooves per mm), an Olympus BX40 microscope with a 50× objectives and the slit allowed the spectral resolution to be 2 cm−1. The Raman and fluorescence signals were detected by a Peltier-cooled CCD detector following 473 nm laser excitation. The integration times ranged from 2 to 5 seconds. Quantum yields were obtained by absorbance matching each BODIPY dye with a fluorescein standard (θf = 0.91 in methanol) solution. The corresponding integrated photon flux was then determined and the quantum yields calculated as previously reported.47 Luminescent lifetimes were obtained using a Picoquant Fluorotime 100 TCSPC (time correlated single photon counting) system, exciting at 450 nm (picosecond pulse diode lasers) and the band pass dielectric filter was set for detection at 460 nm and above. 10000 counts were collected for each lifetime measurement and all measurements were performed in triplicate using Nanoharp software to record decay plots. Typical pulse repetition rates of the excitation source were 1 × 105 s−1 with a typical pulse width of 300 ps. FluoroFit software was used to analyse the luminescent decays which fitted well to mono-exponential decay function. The quality of the lifetime fit was determined from visual examination of the residuals and a χ2 value of <1.5. Molecular modelling was performed using B3LYP hybrid density functional with a TZVP basis set as implemented in the Gaussian 09 program suite.48–50
Metal ion complexation studies were performed in acetonitrile at BODIPY concentrations of 10 μM. 3 molar of each metal ion (copper(II) acetate, iron(II) chloride and zinc(II) acetate) were prepared and titrated into the BODIPY solution. Spectra were not corrected for dilution effects as dilution percentage was deliberately kept below 2%. Absorbance, emission and lifetime measurements were performed following each addition of metal ion solution until a BODIPY:metal ion ratio of 1:3 was obtained.
2.2 Materials
All materials were purchased from Sigma-Aldrich (Ireland) or TCI (Europe) and were used without further purification unless otherwise noted. Reaction progression was monitored by TLC. Flash chromatography was performed to purify crude reaction mixtures using an automated flash chromatography system (Analogix-Intelliflash 310) with silica gel pre-packed chromatographic columns (40–63 μm, 60 Å), equipped with a UV-visible detector (200–400 nm) and auto collector. The detection wavelengths used were 254 nm and 365 nm. Anhydrous MgSO4 was used to dry all organic extracts. 5-Bromo-(1,10-phenanthrolyl) (1),51 4-bromo-(2,2′-bipyridyl) (2),52 4-(1,10-phenanthrolyl) benzaldehyde (3) and 1,3,5,7-tetramethyl-5-phenanthrolyl-4,4′-difluoroboradiazaindacene (5) and were synthesised as previously reported.35 All aqueous samples were prepared by dissolving the sample in a solvent mixture containing acetonitrile:water (2:98 v/v).
2.3 Synthesis
:MeOH (9:1 v/v) as mobile phase. The product was then recrystallised from hot CHCl3 with cold pentane to yield 4 as a brown solid (189 mg, 85%). 1H NMR (400 MHz; CDCl3) δ (ppm): 10.09 (s, 1H), 8.79 (dd, J = 5.2 & 0.8 Hz, 1H), 8.71 (m, 2H), 8.46 (dt, 1H), 8.02 (dd, J = 8.4 & 1.6 Hz, 2H), 7.93 (dd, J = 6.4 & 1.6 Hz, 2H), 7.86 (td, 1H), 7.57 (dd, J = 5.2 & 2.0 Hz, 1H), 7.35 (ddd, J = 4.8 & 1.6 Hz, 1H). 13C NMR (CDCl3) δ (ppm): 191.8, 157.0, 155.8, 150.0, 149.2, 148.0, 144.2, 137.1, 136.5, 130.4, 127.9, 124.1, 121.7, 121.3, 119.2, 31.0.:MeOH, 95:5 v/v) to leave the orange/brown coloured product 8 (67 mg, 24%). 1H NMR (400 MHz; CDCl3) δ (ppm): 8.78 (dd, J = 8.4 & 1.6 Hz, 1H), 8.75–8.70 (m, 2H), 8.47 (d, J = 4.2 Hz, 1H), 7.93 (d, J = 4.0 Hz, 2H), 7.87 (td, 1H), 7.61 (dd, J = 8.0 & 4.0 Hz, 1H), 7.44 (d, J = 1.6 Hz, 2H), 7.35 (td, 1H), 6.00 (s, 2H), 2.56 (s, 6H), 1.43 (s, 6H). 13C NMR (CDCl3) δ (ppm): 156.8, 155.9, 155.8, 149.9, 149.2, 148.3, 143.1, 140.8, 139.0, 137.2, 135.9, 131.3, 128.9, 127.9, 124.1, 121.6, 121.4, 119.0, 14.7. 19F NMR (CDCl3) δ (ppm): −146.1 (d), −146.3 (d). HRMS (ToF MS ES+): m/z, calculated for C29H26BN4F2: 479.2219, found 479.2234.:MeOH (95:5 v/v) as mobile phase to yield the dark purple product.2.4 Cellular uptake studies
HeLa cells were seeded at 1.5 × 105 cells in 2 mL culture media on poly-L-lysine coated, 16 mm coverslips in a 12-well plate and left for 24 h at 37 °C with 5% CO2. The BODIPY compounds were added to the wells to give a final concentration of 10 μM and were incubated for 24 h at 37 °C with 5% CO2. Prior to imaging, the cells were washed once with PBS supplemented with 1.1 mM MgCl2 and 0.9 mM CaCl2. The cells were imaged live using a Leica TSP DMi8 confocal microscope using a 100× oil immersion objective lens. For BODIPY dyes 5 and 8, 498 nm was isolated from a white light laser and used to excite the compounds, and a 505–540 nm band pass filter was used to collect the emission. For BODIPY dye 6 and 9, a 530 nm white light laser was used to excite the compounds, and a 540–570 nm band pass filter was used to collect the emission. For BODIPY dye 7 and 10, a 530 nm white light laser was used to excite the compound and a 540–570 nm band pass filter was used to collect the emission.2.5 Raman cellular studies
Raman measurements for both live and fixed cell samples were performed on a WITec Alpha 300 system equipped with a solid state laser (532 nm excitation), CCD detector and 63× magnification immersive objective (numerical aperture 1.0). Human Microvascular Endothelial Cells (HMEC-1, passage 7) were seeded onto CaF2 slides with a concentration of 160000 cells per well and incubated for 24 h in the BODIPY-containing medium (10 μM final dye concentration). After incubation, cells were washed twice with PBS solution, fixed with 2.5% glutaraldehyde solution for 6 min and then rinsed twice with PBS solution. The samples were then stored in PBS solution until ready for measurement. Raman imaging of cells were performed in PBS solution with a maximum laser power (ca. 30 mW), sampling step of 0.3 μm and integration time of 0.5 s for fixed cells and with 0.4 μm and 0.7 s for live cells, respectively. Single spectra were acquired with the integration time 0.5 s for 10 accumulations. The laser power was ca. 9 mW at the sample surface. At least six good-quality data sets were acquired for each sample.
3 Results and discussion
3.1 Synthesis
Scheme 1 shows the synthetic steps toward preparation of the 1,10-phenanthrolyl and 2,2′-bipyridyl-based BODIPY fluorescent probes. Bromo-functionalised precursors 1 and 2 were synthesised according to literature procedures.51,52 Suzuki–Miyaura protocol was then employed to synthesise the benzaldehyde-containing ligands 3 and 4 in good yield of 79% and 85%, respectively. The BODIPY derivatives, 5 and 8, were synthesised via a standard one pot trifluoroacetic acid catalysed condensation reaction with 2,4-dimethylpyrrole in a solution containing benzaldehyde precursor in nitrogen purged CH2Cl2. The mixture was stirred at room temperature for 5 h under a nitrogen atmosphere. Tetrachorobenzoquinone was then added and the reaction stirred for a further 30 min followed by the addition of Et3N and BF3OEt2 with stirring overnight under nitrogen. The crude products were then isolated by silica gel chromatography to give BODIPY dyes 5 and 8 in yield of 30% and 24%, respectively.35 Heavy atom substitution at the 2,6 positions of the indacene core was achieved using either N-bromosuccinimide or N-iodosuccinimide in hexafluoropropanol to give the fluorophores 6, 7, 9 and 10 (Scheme 1) in excellent yields (>86%) following isolation by silica gel chromatography.2 The structure and purity of the novel BODIPY derivatives were confirmed by NMR spectroscopy (1H, 13C and 19F) and high resolution electrospray ionisation mass spectrometry. Notably, as expected, upon bromo- or iodo-functionalisation, the 1H NMR spectra confirmed loss of the protons located at the 2,6-position of the BODIPY core unit at approximately δH 6 ppm.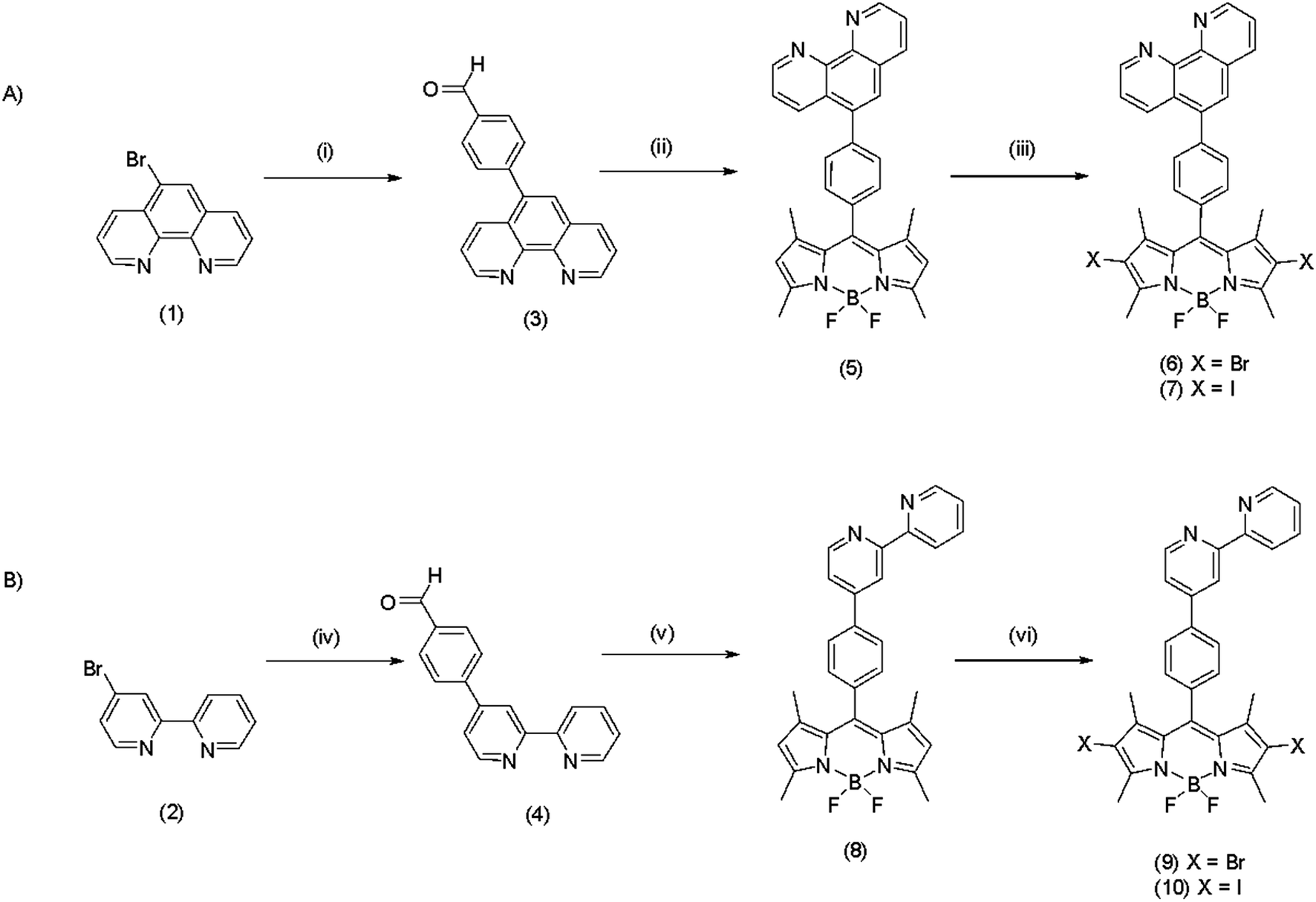 | ||
| Scheme 1 The synthesis of (A) 1,3,5,7-tetramethyl-(phenanthrolyl)-BODIPY and (B) 1,3,5,7-tetramethyl-(bipyridyl)-BODIPY derivatives. (i) and (iv) 4-Formylphenylboronic acid, Pd(dfpp)2Cl2·CH2Cl2, K2CO3, dioxane:H2O (4:1 v/v), reflux 6 hours; (ii) and (v) 2,4-dimethyl-1H-pyrrole, TFA, CH2Cl2, stirring 4 hours; tetrachlorobenzoquinone, Et3N, BF3OEt2, 12 hours; (iii) and (vi) NBS or NIS, hexafluoropropanol, stirring 20 minutes. | ||
3.2 Optical and photophysical properties
The absorbance and emission spectra for the BODIPY derivatives 5–10 in acetonitrile are shown in Fig. 2. There is relatively little variation in the visible absorbance spectra of the phenanthrolyl and bipyridyl-based BODIPY analogues. Their visible absorbance is characterised by an intense feature at ca. 496 nm. Halogenation at the 2,6 positions of compounds 5 and 8, induces a red shift in this absorbance maximum to 523 nm for the brominated derivatives 6 and 9, and to 529 nm for the iodinated dyes 7 and 10. These transitions are assigned to spin-allowed π–π* transitions involving pyrromethene-BF2 fluorophores30 and weaker broad bands observed ca. 370 nm are attributed to the S0 → S2 (π–π*) transitions for each compound.18 Strong absorbance bands in the UV region (ca. 270 nm) originate from π–π* transitions of the polypyridyl moieties.30 However, it is noted that the absorbance spectrum of exhibits 5 an uncharacteristic UV spectrum which is attributed to variation in electronic behaviour between the two orientations of the phenanthrolyl unit; orthogonal and parallel to the BODIPY core, similar UV spectra have been recorded for a related BODIPY compound.7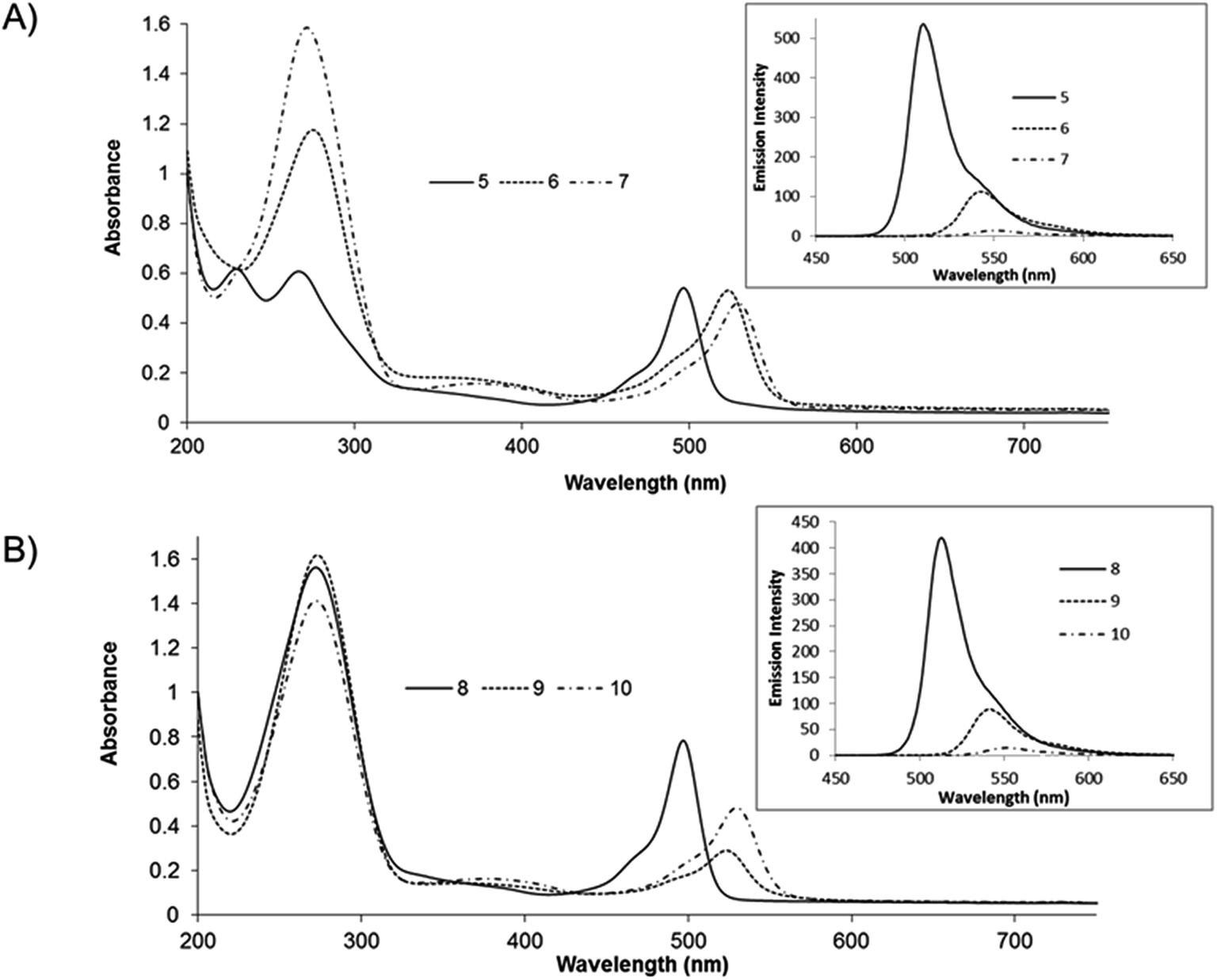 | ||
| Fig. 2 Absorbance and emission (inset) spectra of (A) compounds 5–7 and (B) compounds 8–10, measured in degassed acetonitrile at room temperature. Emission spectra were recorded by exciting each sample at its maximum visible absorbance and slit widths were kept constant at 2.5 nm. All samples were of 10 μM concentration. | ||
Excitation into the absorbance maxima of compounds 5 and 8 leads to an intense fluorescence centred around 510 nm with quantum yields and lifetimes consistent with other BODIPY compounds.18 The emission (λmax) is shifted to the red on halogenation, by comparison with the parent (unhalogenated) compound Table 1. The red shifts followed the order Br > I and were accompanied by dramatic reductions in fluorescence quantum yield and lifetime. The quantum yield of the brominated BODIPY is less than half that of the unhalogenated compound and decreases by approximately an order of magnitude on iodo functionalisation. The sequential red-shift in absorbance energy and quenching of emission intensity is attributed to increasingly efficient intersystem crossing induced by the heavy atom effect of the auxochromes yielding dark triplet states consistent with previous reports on halogenated BODIPY fluorophores10,18 The fluorescence maxima were not strongly affected by low temperature (77 K) and there was no evidence for phosphorescence from any of the complexes at this temperature (ESI material Fig. S29 & S30†).
| Compound | Absorbance | Emission | Lifetimesc | Quantum yieldb | |
|---|---|---|---|---|---|
| λabs (nm) (logεmax) |
λem (nm) R.T. | λema (nm) 77 K | τ (ns) ± 1.5% | θf | |
| a Measured in butyronitrile:propionitrile (5:4 v/v).b Calculated using fluorescein (θf = 0.91 in methanol) as standard.47c lifetimes were carried out under aerated conditions, but did not change on deaeration. |
|||||
| 5 | 496 (4.73) | 510 | 509 | 3.314 | 0.23 |
| 6 | 523 (4.72) | 542 | 538 | 1.291 | 0.09 |
| 7 | 529 (4.68) | 552 | 548 | 0.159 | 0.02 |
| 8 | 498 (4.89) | 513 | 511 | 2.405 | 0.21 |
| 9 | 522 (4.46) | 541 | 537 | 1.277 | 0.06 |
| 10 | 530 (4.68) | 552 | 546 | 0.426 | 0.02 |
3.3 Water switchable luminescence
All of the compounds reported here were found to be non-emissive in water. A representative water switching response is shown for compound 5 in Fig. 3. At approximately 80% v/v water, a sharp decrease in emission intensity is observed and the emission is completely extinguished in 98% v/v water (solution also contained 2% v/v acetonitrile to maintain dissolution of the compound). It is important to note however, that emission intensity is recovered once the compound is extracted and re-dissolved in acetonitrile solution.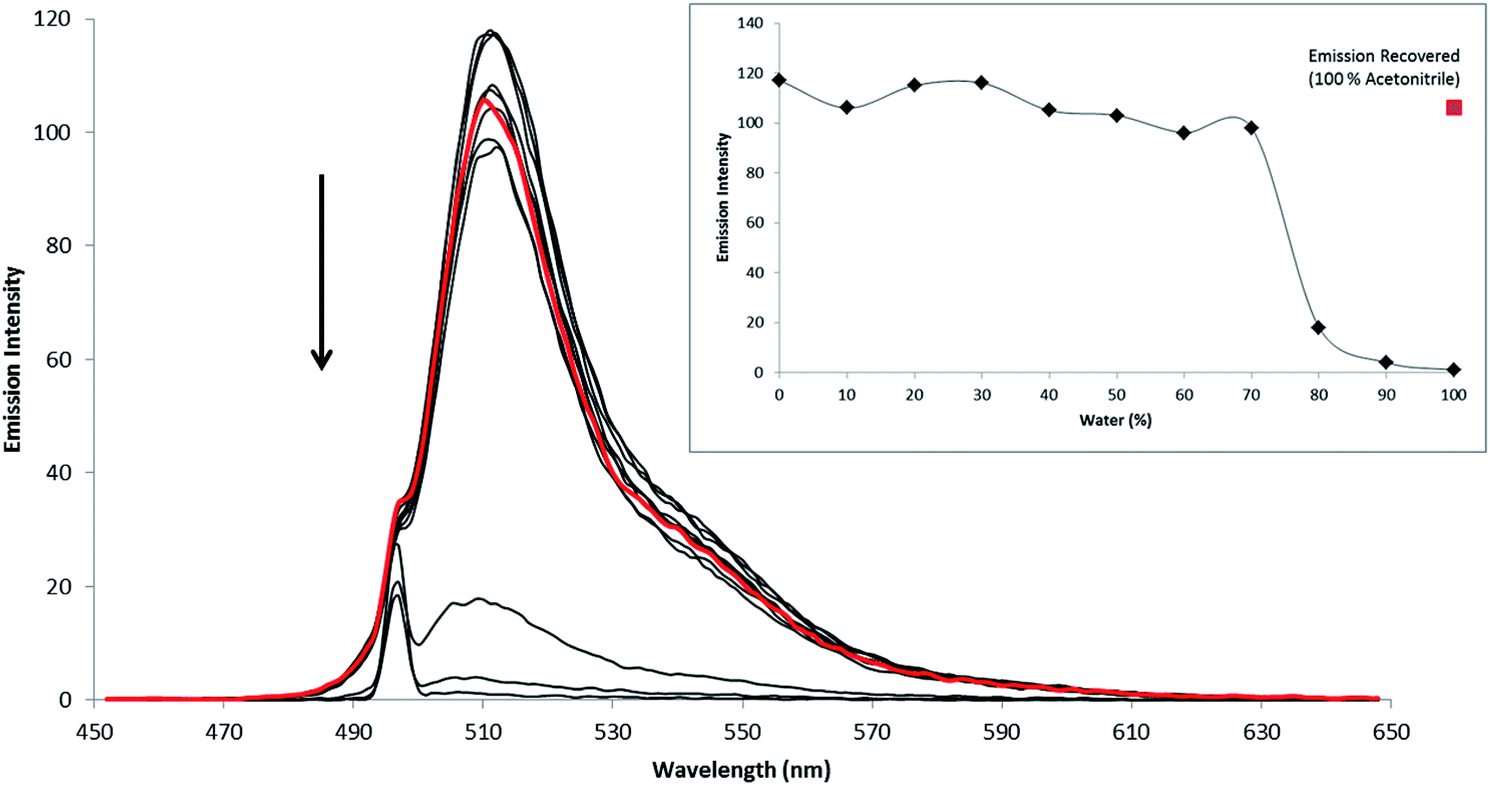 | ||
| Fig. 3 Quenching of the fluorescent emission of compound 5 (10 μM) in acetonitrile–water solutions. The water ratio was varied from 0% to 98%. The red plot signifies the emission recovery of the BODIPY compound. (Inset) shows the changes to peak fluorescence intensity upon dilution by water and the corresponding emission recovery (red data point). The shoulder at 496 nm is artefactual due to the excitation source, 2.5 nm slit widths were used throughout. | ||
While many BODIPY derivatives are emissive both in organic and aqueous solvents, there are a number of reports of BODIPY derivatives that are similarly weakly or non-emissive in aqueous environments.10,20,53 This effect has been attributed in a number of compounds to dye aggregation induced by poor solubility in aqueous media. Although alternative mechanisms of solvent induced quenching have been reported for BODIPY including, switchable PET fluorescence quenching, and charge transfer mechanisms.52
The behaviour observed here is comparable to behaviour of aza-BODIPY fluorophores reported by Hu et al.; where fluorescence is extinguished in solutions ranging from 50–70% water content.4 The sharp threshold of emission extinction in water was explained by hydrophobic aggregation leading to non-emissive clusters.4,10
Interestingly, for the compounds reported here, we observed that the water:acetonitrile ratio at which emission is quenched is independent of the BODIPY concentration. If aggregation was responsible for the emission extinction this should occur at lower water ratios for higher concentrations of dye. Moreover, all the BODIPY derivatives remain luminescent in the solid state (ESI material, Fig. S31†). If aggregation were responsible for emission extinction, the solid would not be expected to emit. Furthermore, in previous reports of aggregation induced quenching, the absorbance bands of the fluorophore are broadened in aqueous media, whereas as shown here for compounds 5–10 (ESI Fig. S32 & S33†) the absorbance bands remain sharp albeit with solvent induced blue shifts. Together this data may suggest that aggregation induced quenching is not responsible for the emission quenching of 5–10. Furthermore, in a ruthenium BODIPY dyad reported by us recently, containing pendant BODIPY 5, we observed water induced extinction of the BODIPY (but not the ruthenium) emission. This complex would not be expected to aggregate as it has good solubility in water and the charge on the complex would also mitigate against aggregation.
3.4 Computational studies
The molecular and electronic structures of fluorophores 5–7 were modelled by DFT methods and the nature of singlet excited states were calculated using TD-DFT methods. Estimating the excitation energies of BODIPY compounds using TD-DFT methods is prone to significant errors. These are thought to arise because of difficulties with the differential electron correlation, contributions of multi-reference character, and double excitations which cannot be modelled accurately by the single determinant description used in TD-DFT methods. Mean absolute errors of up to 0.5 eV are common for the vertical excitation energies calculated using a variety of pure and hybrid functionals such as BLYP, B3LYP or cam-B3LYP (see ESI† materials for a comparison for BLYP, B3LYP or cam-B3LYP for the phenBODIPY derivatives). Empirical correction factors are required to bring the calculated excitation energies close to the experimental values obtained experimentally. However, in this work, rather than their absolute energies, our interest is focussed on understanding the relative energies of accessible singlet excited states as these can provide useful insights into the photophysics of these compounds and help explain the light switch behaviour in different media.9,54,55 The hybrid density functional B3LYP was used here because the BLYP functional tended to underestimate the relative energies of charge-transfer states, while cam-B3LYP appears to overestimate them.61 The B3LYP/TD-DFT method was used to estimate the relative energies of the singlet excited states of 5–10. Electron density difference maps were constructed for the excited states provide a picture of the electron drift compared to the ground state.22 These electron density difference maps are presented for 5 to 7 in Fig. 4 and for 8 to 10 in ESI materials.† The regions with reduced electron density relative to the ground state are coloured blue, whereas those regions with a higher electron density in the excited state are coloured red. For all compounds in this study, the lowest excited singlet (S1) state (at the Franck–Condon region) is localised at the BODIPY core and transition to this excited state is responsible for the sharp, low-energy absorption feature in the UV/vis spectrum. This state is emissive and results in the sharp luminescence feature in the emission spectrum. In contrast, charge-transfer states involving the BODIPY core and the aromatic substituent on the meso-position are non-luminescent. Chemical and/or environmental changes which affect the nature of the lowest energy excited state, for instance changing from BODIPY-based to charge-transfer, will change the luminescent properties of the compound. Consequently we have calculated the relative energies of the low lying singlet states (S1 to S4) of 5, 6, and 7 to see if charge-transfer states exist close (in energy terms) to the lowest energy BODIPY-centred emissive state Fig. 4. | ||
| Fig. 4 Singlet excited state energies for 5, 6, and 7 (H2, Br2 and I2 respectively) and the electron density difference maps for 5 (left) and 7 (right); levels indicated in red are phenanthrolyl to BODIPY charge-transfer in character while those in blue are BODIPY to phenanthrolyl, black indicates BODIP-based states. | ||
The energy of the S1 state is reduced upon substitution of the BODIPY core with a halogen atom. This is consistent with the observed shift in the low energy absorption feature, but the calculations indicate that the nature of the state as BODIPY-based is unchanged. For the non-halogenated compound 5, the S2 and S3 states are charge-transfer in character. S2 can be described as a phen to BODIPY, while S3 is a BODIPY to phen charge-transfer state. The S2 and S3 states have similar energies although the small oscillator strengths indicate that they cannot be efficiently populated by excitation of the ground-state (Fig. 4). They may well be thermally populated from the S1 state, or their energies my drop below that of the BODIPY centres state as the excited state structure evolves from the Franck–Condon region. In contrast however, in the bromo 6 and iodo 7 compounds the S2 state has phenanthrolyl to BODIPY charge-transfer character whereas S3 is BODIPY localised and energetically well-resolved from S2 (Fig. 4). Based on these calculations we propose that competitive population of a non-radiative charge-transfer excited state is responsible for quenching of the luminescence in aqueous environments.
For the bipyridyl compounds the lowest excited states for 8, 9 and 10 are, as expected, BODIPY localised. However whereas for 8 the S2 state is a charge transfer state for 9 and 10 S2 are BODIPY localised. Although, charge transfer states lie close in energy within a cluster of states above the S2 for these compounds.
3.5 The impact of metal ion binding at the polypyridyl site on BODIPY photophysics
BODIPY based ion sensors containing amine metal binding sites have been fairly widely reported.5,11 In such probes emission quantum yield has been observed to increase upon metal ion binding because the amine behaves as an electron donor and undergoes photoinduced electron transfer to the BODIPY excited state, quenching its emission. Such quenching is reduced by the presence of the ion, thus the sensors “light up” on metal ion coordination.5,11In the present examples, the well-known metal binding ligands, bpy and Phen are substituted at the BODIPY meso position so we were interested to ascertain the impact of metal ion binding on the photophysics of these materials. Three biologically relevant cations were selected for study: Cu2+, Fe2+ and Zn2+. Fig. S36† shows the changes in the emission intensity of 8 in acetonitrile over the addition of 3 molar equivalents of each cation. Fig. 7 (inset) also show the corresponding UV-vis absorbance measurements upon metal binding. The BODIPY compounds are strongly emissive in acetonitrile and emission is quenched when metal ion is added to solution. Addition of Cu2+ or Fe2+ induced significant quenching of emission intensity of compound 8, whereas Zn2+ had relatively little effect. This observation contrasts with the reported quenching of luminescence by Zn2+ binding to terpyridine-modified BODIPY compounds.32,56 Nevertheless, this trend was consistent across all compounds 5–10 analysed, the relative fluorescence intensities changes across each compound are shown in Fig. 5. In particular, dyes 5 and 8 exhibited an overall emission intensity decrease of >80% on saturation coordination of Cu2+ and Fe2+ ions compared with only 13% for Zn2+.
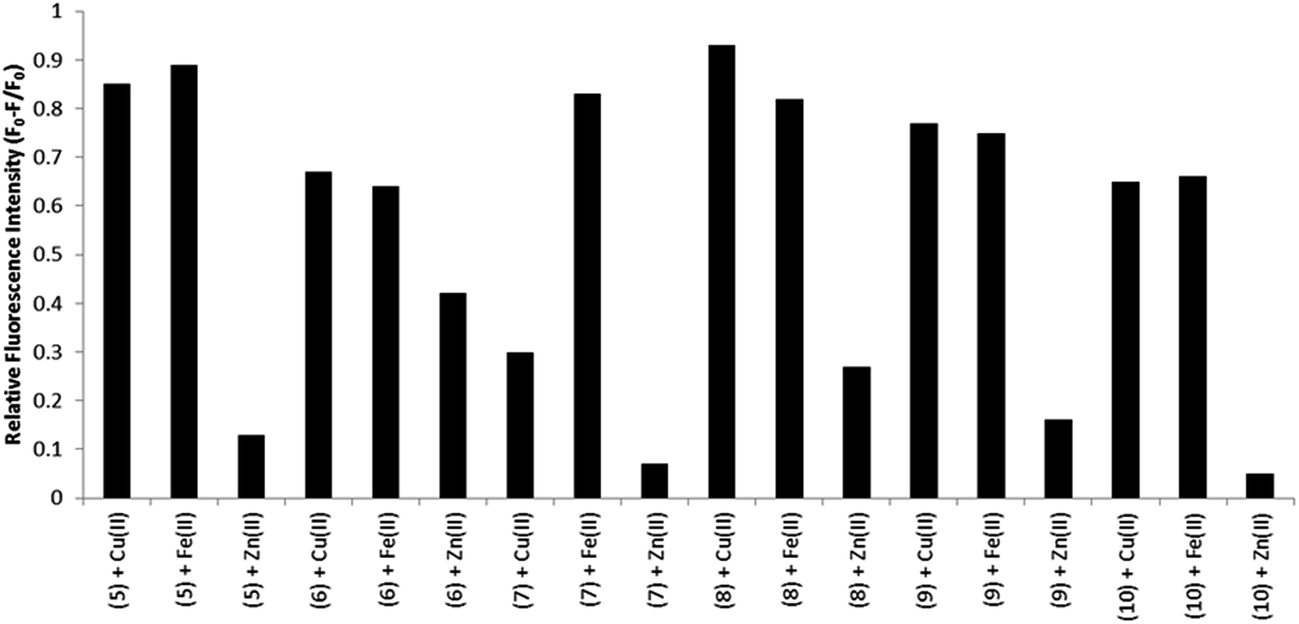 | ||
| Fig. 5 Relative fluorescence emission intensity of BODIPY compounds 5–10 (1 equiv., 10 μM) in the presence of various metal cations (3 equiv.) in acetonitrile solution. The excitation wavelength varied depending on the maximum visible absorbance for 5–10. The corresponding emission data was recorded as the maximum peak emission for each individual experiment. | ||
The molar extinction coefficients and maximum absorbance wavelength in the visible region remained unchanged following metal ion binding. Thus the changes to the emission intensity do not originate from alterations to absorption cross section at excitation, although changes to the UV absorbance do occur with appearance of new bands appear ca. 310 nm attributed to perturbation of the n–π* and π–π* polypyridyl transitions on metal binding.32 Emission quenching data for all compounds are presented in the ESI (Fig. S36–S41†). Applying the mole ratio (Job) method (e.g. Fig. S42†) to the emission data for each of the BODIPY derivative with Cu2+ or Fe2+ the stoichiometric ratio of M:BODIPY was obtained as 1:2 for Cu(II) and 1:3 for Fe(II) which is as expected for bpy or phen based ligands for these two metals and is consistent with bipyridyl-based BODIPY sensors.19,56 Finally, it is important to note that the fluorescence lifetimes of the BODIPY compounds remained unchanged with metal complexation. This observation indicates that the quenching mechanism results from the formation of a non-emissive metal bound complex, rather than dynamic quenching through photoinduced electron transfer to/from the BODIPY centre. This precludes the use of such compounds in fluorescence lifetime imaging to monitor metal distribution but conversely means that metals are not a source of interference in such imaging.
3.6 Cell uptake studies
A key objective of this work is to determine the utility of these compounds as contrast agents for lipid regions in cell imaging. The luminescence behaviour of the materials in aqueous and non-aqueous environments suggests they may be used to distinguish hydrophobic and hydrophilic regions of the cell. Cellular uptake of compounds 5–10 were measured in live HeLa cells, an adherent cervical cancer cell line. In the presence of minimal organic solvent, all compounds were readily permeable to live cells and Fig. 6 shows the luminescent confocal images of HeLa cells following incubation for 24 hours with 10 μM of these compounds. It can be seen that the compounds emit strongly from populated features within the cytoplasm of the live cells but appear to be excluded from the nucleus. Given that the compounds do not emit in water, the fluorescence images from the cell are expected to originate only from non-aqueous regions of the cell-proteinaceous structures, lipid droplets or membranes which is further confirmed by Raman spectroscopy of HMEC-1 cells described in Section 3.7.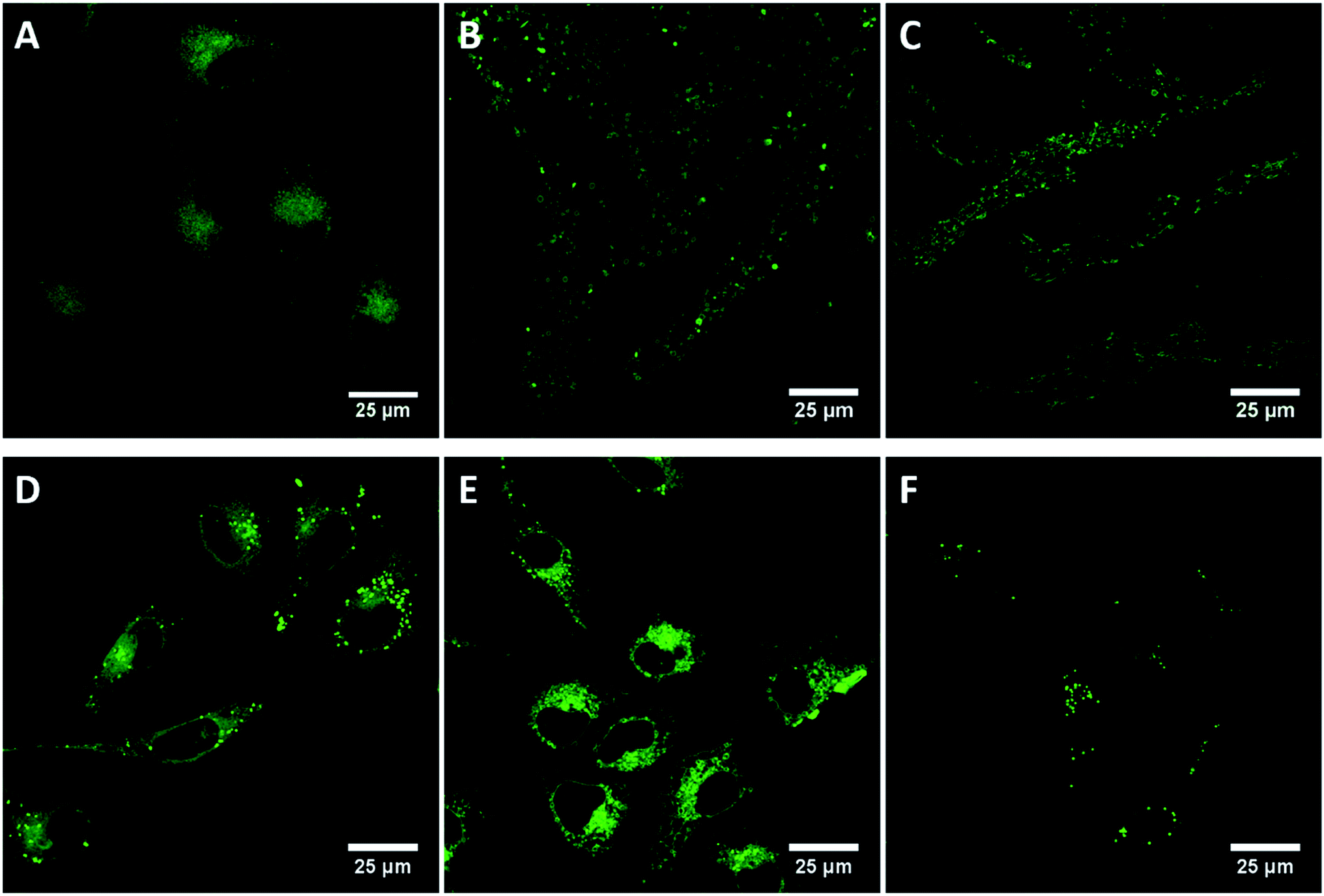 | ||
| Fig. 6 Live cellular uptake studies of BODIPY compounds 5 (A), 6 (B), 7 (C), 8 (D), 9 (E), 10 (F). In HeLa cells. Cells were incubated with 10 μM dye in HeLa cell media for 24 h prior to imaging. For compounds 5 and 8, a 498 nm white light laser was used to excite the compounds and a 505–540 nm band pass filter was used to collect the emission. For 6 and 9, a 530 nm white light laser was used to excite the compound and a 540–570 nm band pass filter was used to collect the emission. For 7 and 10, a 530 nm white light laser was used and the emission was collected using a 535–580 nm band pass filter. | ||
The apparent (as we only see emission when it is hydrophobic environment) distribution of the probes within the cell is interesting as it indicates that halogenation impacts localisation within the living cell. Compound 5, the unfunctionalised phenantroyl-BODIPY parent compound, appears to distribute fairly widely throughout the in the cytoplasmic region in cytoplasmic membranous structures and lysosomes of the HeLa cells, but does not enter any specific organelles and is nuclear excluding, as shown in Fig. 6(A). By contrast, the halogen modified derivatives, compounds 6 and 7, exhibit highly punctuated distribution within the cells, shown in Fig. 6(B and C), yielding high contrast images of what are confirmed via Raman imaging vide infra, to be lipid droplets within the live cells.57,58 Similarly, 8 distributes in the cytoplasm but unlike 5 does show some localisation in lipid droplets (Fig. 6(D)), similar to the distribution of 6 and 7, indicating lipid droplet localisation. Fig. 6(E) shows compound 9 distributed throughout the cytoplasm within membranous regions but does not show strong localisation to lipid droplets, as described below though 9 shows anomalous behaviour compared to the other probes with evidence for cytotoxicity from this probe which is not observed for the other compounds. Like 7, compound 10 is highly localised to lipid droplets in the cytoplasm (Fig. 6(F)).
Unhalogenated BODIPY compounds employed previously as cellular contrast agents have been shown to localise in the lysosomes of HeLa cells25,53 and cytoplasm of a range of other cell lines but have similarly largely been nuclear excluding.5,25,27,33,36,40,59 Far less has been reported on uptake and particularly distribution of halogenated BODIPY derivatives in live cells to date.18
It is important to note that while DMSO was used to solubilise the compounds prior to addition to the cells the final concentration of DMSO in the medium was 0.1% v/v. Such low concentration of solvent would not be expected to affect the viability or promote membrane permeability within the cells,60 and furthermore is not sufficient to switch on emission. Indeed, the viability of the cells on exposure to 5–7 and 8 and 10 over 24 hours was high, with no evidence for cell death indicating the cytotoxicity of the BODIPY compounds is low under imaging conditions employed here. However, as described, after exposure to compound 9 at the same concentrations and incubation conditions, the cells show signs of paraptosis, a type of programmed cell death which is indicated by the presence of the large vacuoles throughout the cytoplasm (Fig. 6(E)).
3.7 Cellular imaging using Raman spectroscopy
As emission is only observed from the derivatives reported here when they are in hydrophobic environments, Raman microscopy was exploited to identify if the compounds were localising at hydrophobic regions or were more uniformly dispersed through the cell as this method does not rely on the compound's emission. Raman and fluorescence distribution images of BODIPY complexes 5 and 7 were measured in both live and fixed Human Microvascular Endothelial (HMEC-1) cells using Raman microscopy. The Raman data was collected under 532 nm excitation which is post-resonant with the BODIPY absorbance and hence the Raman spectroscopy is expected to be resonantly enhanced. This resonantly enhanced Raman signal allowed us to map the distribution of the BODIPY compounds within the cells whether or not the compound was emitting thus providing a map of the true distribution of the probes around the cells. Because of the resonance enhancement, the Raman images are dominated by BODIPY signature features rather than cell based components as can be seen in the dual Raman fluorescence images showing BODIPY distribution in cells (ESI Fig. S43 & S44†). Fluorescence and Raman distribution images indicated that both BODIPY dyes 5 and 7 were present inside the cytoplasm, but consistent with confocal fluorescence imaging, shown above, were nuclear excluding.Cluster analysis of the Raman data collected from fixed cells (Fig. 7 and 8) facilitated discrimination of the BODIPY fluorophore contribution from signals from biomaterial within the cellular compartments/organelles.
 | ||
| Fig. 7 Cluster analysis of fixed HMEC-1 cells. (A) Control cell, (B) when exposed to 5 and (C) when exposed 7. Colour codes: red – nucleoli; blue – nucleus; green – endoplasmic reticulum; orange – cytoplasm, brown – class including cell membrane and violet – BODIPY dye. Scale bar is 5 μm. | ||
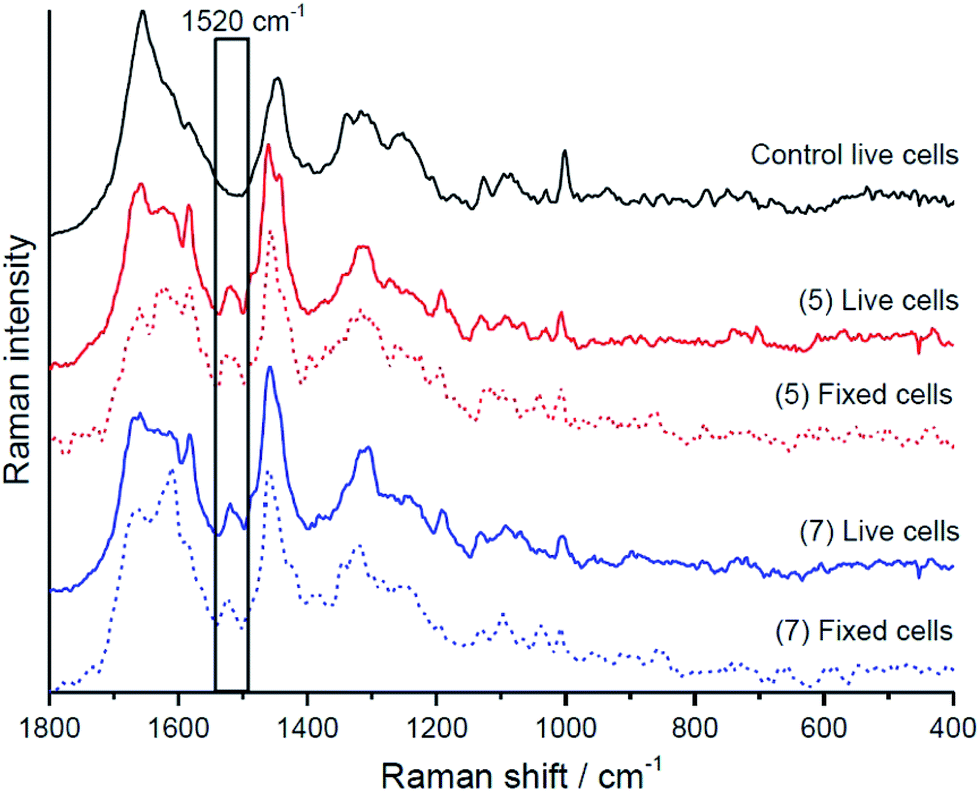 | ||
| Fig. 8 Raman spectra of the class containing 5 and 7 in live and fixed HMEC-1 cells. Cells were incubated with 10 μM of each dye for 24 h and were fixed using 2.5% glutaraldehyde prior to obtaining Raman spectra. A 532 nm laser line was used to excite the compounds. A sampling step of 0.4 μm and integration time of 0.7–1.0 s was used to obtain the Raman spectra. | ||
Raman data indicates that the fluorophores co-localise strongly with lipid in both live and fixed cells for the halogenated sample with association with the endoplasmic reticulum for parent. The lipid distribution is identified and integrated over the band between 3000–3030 cm−1, attributed to the trivalent ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretching vibrations.59 This co-localisation would explain why emission is observed from the BODIPY derivatives in confocal fluorescence imaging as the lipid (i.e.) hydrophobic environment will switch fluorescence on for these compounds. Furthermore, consistent with confocal fluorescence imaging, compound 5 appears to localise in the endoplasmic reticulum and causes the formation of unsaturated lipid bodies. In light of our recent finding, that inflammation results in formation of unsaturated lipids bodies,61 the appearance of such unsaturated lipid bodies in cells incubated with 5 may suggest that in the applied dose it exerts a toxic effect on HMEC cells, whereas 7 does not. Interestingly, based on co-localisation of BODIPY Raman signal with saturated rather than unsaturated lipids 5 was found to preferentially localise at saturated lipids rather than unsaturated lipids in the lipid bodies it evoked.
C–H stretching vibrations.59 This co-localisation would explain why emission is observed from the BODIPY derivatives in confocal fluorescence imaging as the lipid (i.e.) hydrophobic environment will switch fluorescence on for these compounds. Furthermore, consistent with confocal fluorescence imaging, compound 5 appears to localise in the endoplasmic reticulum and causes the formation of unsaturated lipid bodies. In light of our recent finding, that inflammation results in formation of unsaturated lipids bodies,61 the appearance of such unsaturated lipid bodies in cells incubated with 5 may suggest that in the applied dose it exerts a toxic effect on HMEC cells, whereas 7 does not. Interestingly, based on co-localisation of BODIPY Raman signal with saturated rather than unsaturated lipids 5 was found to preferentially localise at saturated lipids rather than unsaturated lipids in the lipid bodies it evoked.
4 Conclusions
A family of 1,10-phenanthrolyl and 2,2′-bipyridyl BODIPY-based fluorophores exhibit emission in non-aqueous solvent but not in water. Heavy atom functionalisation of the BODIPY indacene core was found to modulate emission intensity and wavelength resulting in significant decrease in both fluorescent emission intensity and luminescent lifetimes of the compounds in organic media. This is accompanied by red shifts in emission wavelength but without concomitant appearance of phosphorescence at low temperature. The fluorophores can bind metals at the pendant polypyridyl units. Binding of Cu2+ Fe2+ and Zn2+ leads to quenching of the BODIPY emission intensity in non-aqueous solvent in the order of Cu2+ = Fe2+ ≫ Zn2+ without accompanying changes to emission lifetime. This indicates a static mechanism attributed to switching off the lowest excited state to a weakly or non-emissive BODIPY to polypyridyl charge transfer state on metal binding. The data is consistent with DFT calculations which indicate that charge transfer states resides close in energy to the emissive BODIPY-based LUMO. Observation of both solid-state emission and concentration independent aqueous quenching of the fluorophores suggests that population of such a charge transfer state in water may also be the origin of water switching rather than aggregation induced quenching.Aqueous switchable fluorescent probes are of potential value in cell imaging enabling selective imaging of hydrophobic regions of the cell. Uptake and localisation of the dyes in live was examined using confocal fluorescence and along with Raman microscopy of cells fixed post live uptake. It was found that all dyes explored showed excellent uptake into live HeLa and HMEC-1 cells, and appeared to localise mainly in the cytoplasmic region, excluding the nucleus. Halogenation of the BODIPY had a significant influence on the localisation of the probe in live cells, with evidence for strong localisation of these probes to lipid droplets within the cells. This was confirmed using a resonantly enhanced Raman marker mode for BODIPY at ca. 1520 cm−1 where it was possible to follow the distribution of the BODIPY inside the cell independent of fluorescence to enable understanding of the true distribution of the probe. Through Raman imaging and cluster analysis, the distribution of the BODIPY dyes within HMEC-1 (live and fixed) cells was found to be consistent with confocal fluorescence imaging indicating the probes appeared to localise within lipid droplets within the cells from where fluorescence was switched on. Preliminary evidence indicates that all but one of the BODIPY derivatives show low cytotoxicity and overall it is envisaged that such water switchable BODIPY probes capable of selectively targeting and emitting from lipid droplets may be useful in both imaging and as diagnostic/prognostic markers for cancer cells where such droplets are prevalent compared with healthy cells.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the work of visiting intern student Stefanie Puechner and the financial support of Science Foundation Ireland under grant numbers 15/TIDA/2895 and 14/IA/2488, and the National Science Centre (Poland) under Grant No. DEC-2013/08/A/ST4/00308, in addition to the DJEI/DES/SFI/HEA Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support. Professor R. O'Kennedy, DCU, is gratefully acknowledged for access to cell culture facilities.References
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- L. Wang, J.-W. Wang, A. Cui, X.-X. Cai, Y. Wan, Q. Chen, M.-Y. He and W. Zhang, RSC Adv., 2013, 3, 9219–9222 RSC.
- J.-B. Wang, X.-Q. Fang, X. Pan, S.-Y. Dai and Q.-H. Song, Chem.–Asian J., 2012, 7, 696–700 CrossRef CAS PubMed.
- X. Hu, J. Zhang, Z. Yu, Y. Xie, H. He, J. Qi, X. Dong, Y. Lu, W. Zhao and W. Wu, Nanomed. Nanotech. Biol. Med., 2015, 11, 1939–1948 CrossRef CAS PubMed.
- Y. Wu, X. Peng, B. Guo, J. Fan, Z. Zhang, J. Wang, A. Cui and Y. Gao, Org. Biomol. Chem., 2005, 3, 1387–1392 CAS.
- S. Çetindere, S. O. Tümay, A. Kılıç, M. Durmuş and S. Yeşilot, J. Fluoresc., 2016, 26, 1173–1181 CrossRef PubMed.
- S. Karakaya and F. Algi, Tetrahedron Lett., 2014, 55, 5555–5559 CrossRef CAS.
- A. Martin, C. Long, R. J. Forster and T. E. Keyes, Chem. Commun., 2012, 48, 5617–5619 RSC.
- I. K. Petrushenko and K. B. Petrushenko, Spectrochim. Acta, Part A, 2015, 138, 623–627 CrossRef CAS PubMed.
- S. Zhu, J. Zhang, G. Vegesna, F.-T. Luo, S. A. Green and H. Liu, Org. Lett., 2011, 13, 438–441 CrossRef CAS PubMed.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, J. Miao, M. Ren, W. Guo and X. Lv, Sens. Actuators, B, 2016, 226, 364–369 CrossRef CAS.
- M. Galletta, F. Puntoriero, S. Campagna, C. Chiorboli, M. Quesada, S. Goeb and R. Ziessel, J. Phys. Chem. A, 2006, 110, 4348–4358 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- J. K. G. Karlsson and A. Harriman, J. Phys. Chem. A, 2016, 120, 2537–2546 CrossRef CAS PubMed.
- W. Qin, M. Baruah, M. Sliwa, M. Van der Auweraer, W. M. De Borggraeve, D. Beljonne, B. Van Averbeke and N. Boens, J. Phys. Chem. A, 2008, 112, 6104–6114 CrossRef CAS PubMed.
- A. N. Kursunlu, P. Deveci and E. Guler, J. Lumin., 2013, 136, 430–436 CrossRef CAS PubMed.
- G. K. Vegesna, S. R. Sripathi, J. Zhang, S. Zhu, W. He, F.-T. Luo, W. J. Jahng, M. Frost and H. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 4107–4112 Search PubMed.
- L. A. Juárez, A. Barba-Bon, A. M. Costero, R. Martínez-Máñez, F. Sancenón, M. Parra, P. Gaviña, M. C. Terencio and M. J. Alcaraz, Chem.–Eur. J., 2015, 21, 15486–15490 CrossRef PubMed.
- C. Zhao, X. Li, Y. Yang, J. Cao, X. Wang and Y. Zhang, Dyes Pigm., 2014, 101, 130–135 CrossRef CAS.
- H. Son, G. Kang and J. H. Jung, Analyst, 2011, 137, 163–169 RSC.
- A. Martin, R. D. Moriarty, C. Long, R. J. Forster and T. E. Keyes, Asian J. Org. Chem., 2013, 2, 763–778 CrossRef CAS.
- R. D. Moriarty, A. Martin, K. Adamson, E. O’reilly, P. Mollard, R. J. Forster and T. E. Keyes, J. Microsc., 2014, 253, 204–218 CrossRef CAS PubMed.
- S. O. Tümay, E. Okutan, I. F. Sengul, E. Özcan, H. Kandemir, T. Doruk, M. Çetin and B. Çoşut, Polyhedron, 2016, 117, 161–171 CrossRef.
- K. Sreenath, Z. Yuan, J. R. Allen, M. W. Davidson and L. Zhu, Chem.–Eur. J., 2015, 21, 867–874 CrossRef PubMed.
- M. Emrullahoğlu, M. Üçüncü and E. Karakuş, Chem. Commun., 2013, 49, 7836–7838 RSC.
- J. Zhang, B. Zhao, C. Li, X. Zhu and R. Qiao, Sens. Actuators, B, 2014, 196, 117–122 CrossRef CAS.
- C. Grazon, J. Rieger, B. Charleux, G. Clavier and R. Méallet-Renault, J. Phys. Chem. C, 2014, 118, 13945–13952 CAS.
- G.-L. Fu, H. Pan, Y.-H. Zhao and C.-H. Zhao, Org. Biomol. Chem., 2011, 9, 8141–8146 CAS.
- M. Galletta, S. Campagna, M. Quesada, G. Ulrich and R. Ziessel, Chem. Commun., 2005, 4222–4224 RSC.
- J. Rosenthal and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 5536–5537 CrossRef CAS PubMed.
- C. Goze, G. Ulrich, L. Charbonnière, M. Cesario, T. Prangé and R. Ziessel, Chem.–Eur. J., 2003, 9, 3748–3755 CrossRef CAS PubMed.
- N. R. Chereddy, M. V. N. Raju, B. M. Reddy, V. R. Krishnaswamy, P. S. Korrapati, B. J. M. Reddy and V. J. Rao, Sens. Actuators, B, 2016, 237, 605–612 CrossRef CAS.
- A. Harriman, J. P. Rostron, M. Cesario, G. Ulrich and R. Ziessel, J. Phys. Chem. A, 2006, 110, 7994–8002 CrossRef CAS PubMed.
- A. Martin, A. Byrne, C. Dolan, R. J. Forster and T. E. Keyes, Chem. Commun., 2015, 51, 15839–15841 RSC.
- C. Yang, D. Gong, X. Wang, A. Iqbal, M. Deng, Y. Guo, X. Tang, W. Liu and W. Qin, Sens. Actuators, B, 2016, 224, 110–117 CrossRef CAS.
- X. He, J. Zhang, X. Liu, L. Dong, D. Li, H. Qiu and S. Yin, Sens. Actuators, B, 2014, 192, 29–35 CrossRef CAS.
- S. Yin, W. Yuan, J. Huang, D. Xie, B. Liu, K. Jiang and H. Qiu, Spectrochim. Acta, Part A, 2012, 96, 82–88 CrossRef CAS PubMed.
- J. Wang, Y. Xie, Z. Wang and Q. Song, Sens. Actuators, B, 2014, 194, 149–155 CrossRef CAS.
- L. Jiao, J. Li, S. Zhang, C. Wei, E. Hao and M. G. H. Vicente, New J. Chem., 2009, 33, 1888–1893 RSC.
- J. Bañuelos-Prieto, A. R. Agarrabeitia, I. Garcia-Moreno, I. Lopez-Arbeloa, A. Costela, L. Infantes, M. E. Perez-Ojeda, M. Palacios-Cuesta and M. J. Ortiz, Chem.–Eur. J., 2010, 16, 14094–14105 CrossRef PubMed.
- M.-Y. Jia, Y. Wang, Y. Liu, L.-Y. Niu and L. Feng, Biosens. Bioelectron., 2016, 85, 515–521 CrossRef CAS PubMed.
- M. Pamuk and F. Algi, Tetrahedron Lett., 2012, 53, 7010–7012 CrossRef CAS.
- X. Xie and Y. Qin, Sens. Actuators, B, 2011, 156, 213–217 CrossRef CAS.
- D. P. Kennedy, C. M. Kormos and S. C. Burdette, J. Am. Chem. Soc., 2009, 131, 8578–8586 CrossRef CAS PubMed.
- L. Fu, F.-F. Wang, T. Gao, R. Huang, H. He, F.-L. Jiang and Y. Liu, Sens. Actuators, B, 2015, 216, 558–562 CrossRef CAS.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson and others, Gaussian Inc, Wallingford CT.
- M. Hissler, W. B. Connick, D. K. Geiger, J. E. McGarrah, D. Lipa, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2000, 39, 447–457 CrossRef CAS PubMed.
- M. Zalas, B. Gierczyk, M. Cegłowski and G. Schroeder, Chem. Pap., 2012, 66, 733–740 CAS.
- H. Sunahara, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 5597–5604 CrossRef CAS PubMed.
- S. Chibani, A. D. Laurent, B. Le Guennic and D. Jacquemin, J. Chem. Theory Comput., 2014, 10, 4574–4582 CrossRef CAS PubMed.
- A. Charaf-Eddin, B. L. Guennic and D. Jacquemin, RSC Adv., 2014, 4, 49449–49456 RSC.
- B. Turfan and E. U. Akkaya, Org. Lett., 2002, 4, 2857–2859 CrossRef CAS PubMed.
- A. A. R. Mota, P. H. P. R. Carvalho, B. C. Guido, H. C. B. de Oliveira, T. A. Soares, J. R. Corrêa and B. A. D. Neto, Chem. Sci., 2014, 5, 3995 RSC.
- H. Appelqvist, K. Stranius, K. Börjesson, K. P. R. Nilsson and C. Dyrager, Bioconjugate Chem., 2017, 28, 1363–1370 CrossRef CAS PubMed.
- D. E. Ramírez-Ornelas, E. Alvarado-Martínez, J. Bañuelos, I. López Arbeloa, T. Arbeloa, H. M. Mora-Montes, L. A. Pérez-García and E. Peña-Cabrera, J. Org. Chem., 2016, 81, 2888–2898 CrossRef PubMed.
- C. Dolan, R. D. Moriarty, E. Lestini, M. Devocelle, R. J. Forster and T. E. Keyes, J. Inorg. Biochem., 2013, 119, 65–74 CrossRef CAS PubMed.
- K. Czamara, K. Majzner, A. Selmi, M. Baranska, Y. Ozaki and A. Kaczor, Sci. Rep., 2017, 7, 40889 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07493j |
| This journal is © The Royal Society of Chemistry 2017 |