
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A high modulus hydrogel obtained from hydrogen bond reconstruction and its application in vibration damper†
Longxiang Zhu,
Jianhui Qiu * and
Eiichi Sakai
* and
Eiichi Sakai
Department of Machine Intelligence and Systems Engineering, Faculty of Systems Engineering, Akita Prefectural University, Akita, 015-0055, Japan. E-mail: qiu@akita-pu.ac.jp
First published on 12th September 2017
Abstract
Hydrogels are wet and soft materials with rubber-like properties. The excellent biocompatibility and stimuli-responsiveness have made hydrogels excellent candidates in the field of materials science. However, most of the hydrogels are extremely soft (modulus of approximately 0.1 MPa) as compared to rubber materials; this greatly limits their application in the field of material engineering. In this study, an Al3+-reinforced carboxymethyl cellulose/polyacrylic acid hydrogel was first synthesized by a facile, visible-light-triggered, one-pot polymerization method. Subsequently, the as-prepared hydrogel was reinforced by evaporation-swelling (E-S) treatment to obtain a hydrogel (HM-Gel) with a 10-fold higher elastic modulus. This hydrogel exhibits a tensile strength of 1.26–1.74 MPa and an elastic modulus of 0.59–1.94 MPa. Moreover, the HM-Gel, with an excellent vibration absorption ability, can find applications in the field of industrial engineering and bioengineering.
Introduction
Hydrogels are engineering materials with a high water content and exhibit a three-dimensional network structure.1,2 Due to their inherently excellent biocompatibility and stimuli-responsiveness, hydrogels have been applied in the field of tissue engineering.3–5 In recent years, hydrogels incorporating effective energy dissipation mechanisms or presenting distinctive structures exhibit remarkable overall mechanical properties (tensile strength of 0.2–10 MPa; fracture elongation of 100–6000%, and toughness of 100–15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 J m−2).6 These hydrogels, which exhibit similar properties to conventional rubbers, are excellent candidates for vibration control materials in tissue engineering.7 The vibration absorption material must have sufficient rigidity (to reduce the amplitude during vibration); therefore, this material must have a high modulus (e.g., cartilage, a natural vibration absorption material, has a modulus greater than 1 MPa).8 However, the modulus of most hydrogels is of the order of 0.1 MPa,9,10 which greatly limits their use as a vibration absorption material.
000 J m−2).6 These hydrogels, which exhibit similar properties to conventional rubbers, are excellent candidates for vibration control materials in tissue engineering.7 The vibration absorption material must have sufficient rigidity (to reduce the amplitude during vibration); therefore, this material must have a high modulus (e.g., cartilage, a natural vibration absorption material, has a modulus greater than 1 MPa).8 However, the modulus of most hydrogels is of the order of 0.1 MPa,9,10 which greatly limits their use as a vibration absorption material.
In recent years, researchers have developed a number of high-modulus hydrogels through novel design concepts and special structures.11 Gong et al. have designed an oppositely charged polyelectrolyte PMPTC/PNaSS hydrogel.12 In the preparation process, a uniformly dispersed polyelectrolyte hydrogel was obtained by a two-step polymerization reaction. The resulting hydrogel was dialyzed to ensure that the ionic bonds in the hydrogel were sufficiently strong. The highest modulus of the PMPTC/PNaSS hydrogel can reach 7.9 MPa. In addition, Lin et al. improved a dual-crosslinked hydrogel consisting of covalent and ionic bonds.13 In this hydrogel, the initial PAM–PAA network was synthesized using a covalent cross-linker, and then, the coordination bonds were formed after immersing the single network hydrogel in a highly concentrated Fe3+ solution. The modulus of the PAM/PAA hydrogel can reach 3.5 MPa. Moreover, optimization of the network structure in the as-prepared hydrogel is an effective way to increase the modulus. For instance, increasing the crystallinity of the polymer chains within the hydrogel by the addition of a salt or base solution at high concentration can enhance the modulus of the hydrogel. Thus, the modulus of the PAM-CS hydrogel can reach 0.3–0.4 MPa through crystallization enhancement; however, the use of a strong base may cause potential problems in applications.14 Therefore, it is a big challenge to produce a hydrogel with high modulus through a convenient, efficient, and green synthesis method. Most hydrogels contain a variety of complex hydrogen bond networks across polymer chains; these hydrogen bond interactions between polymer chains can be leveraged to achieve a reconstruction of the hydrogel network to improve the modulus of the hydrogel.
In our previous study, we prepared a carboxymethyl cellulose/polyacrylic acid hydrogel with excellent mechanical properties (tensile strength of 0.85 MPa, fracture elongation of 700%, and modulus of 0.18 MPa) by a facile, visible-light-triggered, one-pot polymerization method.15,16 Herein, a aluminum ion cross-linked hydrogel (Gel) was synthesized by the same method, and then, the as-prepared Gel was reinforced via the evaporation-swelling method (E-S method) to obtain the high-modulus hydrogels (HM-Gel), with a tensile strength of 1.26–1.74 MPa and an elastic modulus of 0.59–1.94 MPa. In addition, both Gel and HM-Gel exhibited improved damping performance due to the presence of aluminum ion as a reinforcement cross-linking site; thus, the applicability of the hydrogel as a vibration control material is greatly improved. This E-S method provides a way for increasing the modulus of many reported hydrogels and can broaden their application as a vibration control material.
Materials and methods
Materials
Carboxymethyl cellulose sodium salt (CMC, EP), acrylic acid (AA, EP), ceric ammonium nitrate ((NH4)2Ce(NO3)4, CAN, GR), and aluminium(III) chloride hexahydrate (AlCl3·6H2O, GR) were purchased from Nacalai Tesque, Inc. All chemical reagents were used as received. Distilled water was used in all experiments.Sample preparation
The HM-Gels were prepared via two steps consisting of visible-light-triggered polymerization and evaporation-swelling. Briefly, 40 mg of CAN, 3.60 g of AA, and 10.0 mL of distilled water were added to a vial stepwise and mixed under magnetic stirring in an ice bath. Subsequently, 0.40 g of CMC was slowly added to the mixture under vigorous magnetic stirring. The precursor hydrogel was stably dispersed for 40 min under vigorous stirring. A range of concentrations of AlCl3·6H2O (0–1.5 mol%, Al3+ to AA) was added to a series of vials, and the mixtures were stirred for 20 min. The viscous precursor hydrogels were transferred to transparent glass molds and covered with glass plates. Finally, the glass molds were immersed in water at 20 °C and placed 20 cm below a visible light source (LS-M210, Sumita) for 5 min. The prepared carboxymethyl cellulose/polyacrylic acid hydrogels were named Gel-X, where X denoted the Al3+ concentration (mol%) relative to that of monomer (AA). The obtained Gels were placed in a dry oven and evaporated at various temperatures for 24 h (for 25 °C, the hydrogel was evaporated at room temperature for 3 days). The dried Gels were immersed in distilled water for 50 h to obtain a swelling equilibrium. The swollen hydrogels at equilibrium with a high modulus were named HM-Gel. Unless otherwise specified, all hydrogels were evaporated in air at 60 °C.Characterization
The phase transition time of the hydrogels was determined by dynamic mechanical analysis (DMA, RSA-G2, TA Instrument, New Castle, DE, USA). The improved experimental set-up is shown below: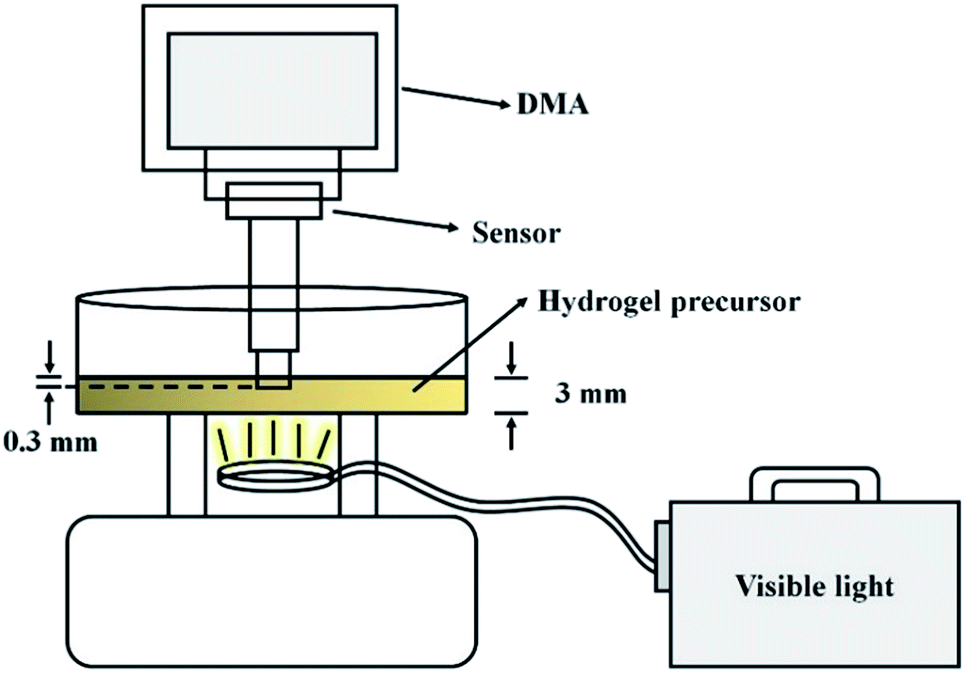
We used a stress relaxation modulus program for the hydrogel gelation time tests using a disc model at 25 °C, 1 Hz, and 1% strain. The program and light were turned on at the same time.
The uniaxial elongation properties were measured using the JIS K6251 (a Japanese industrial rubber) as a standard via a universal testing machine (Instron 3300 series, 50 N load cell, Instron Co., Ltd., Canton, America); the procedure for the measurements was as follows: hydrogels were cut in a dumbbell shape with a 75 mm length (l), 25 mm gauge length (lo), 4 mm width (w), and 2 mm height (h), and tests were conducted at a cross-head speed of 500 mm min−1. The compression properties were measured using the same universal testing machine at a cross-head speed of 1 mm min−1. Hydrogels were cut in a disc shape with 9 mm diameter and 2 mm height.
The storage modulus and loss modulus of the hydrogel specimens were measured as a function of frequency using dynamic mechanical analysis (DMA, RSA-G2, TA Instrument). Hydrogels were cut into a disc shape with 9 mm diameter and 2 mm height. Before measurements, an axial force of 0.981 N was applied on the specimens. The frequency sweeping experiments were performed under a constant strain amplitude (1%) in the frequency range from 100 to 0.1 Hz.
For the cyclic tensile tests, loading–unloading measurements were performed using the same universal testing machine at a constant velocity of 100 mm min−1. The Gels were cut into pieces with the following size: 60 mm length (l), 10 mm width (w), 2 mm height (h), and 30 mm gauge length (lo). The samples were covered with oil to prevent water evaporation. Tensile stress (σ) was calculated as σ = F/wh, where F is the load.
Swelling experiments were performed by immersing the Gels and dried-Gels in conical flasks filled with distilled water. The flasks were placed in a temperature-controlled bath at 25 °C for 50 hours. The swelling ratio (Qm) was calculated by the following equation:
| Qm = Ws/Wa |
The changes of the CMC crystalline network structure in Gel and HM-Gel were investigated using an optical microscope (Eclipse model ME 600D, Nikon, Japan). The thin hydrogel films were obtained by shock-freezing using a microtome (RM2145, Leica Microsystems, Japan). The length, width, and thickness of the hydrogel films were 15 mm, 2 mm, and 200 μm, respectively.
Differential thermal analysis (DTA) of the samples was performed using a TG instrument (DTG-60, Shimadzu Co., Ltd., Kyoto, Japan) at a scan rate of 10 °C min−1 from room temperature to 400 °C under a N2 atmosphere (flow rate of 50 mL min−1). Samples of 4–6 mg were placed in aluminum crucibles using an empty aluminum crucible as a reference.
To fully understand the changes in the internal network, we estimated the effective network chain density (N) based on the cyclic stress–strain curves of HM-Gel (dried in air at 60 °C), according to the following equation reported in the literature:
| τ = NRT[α − (1/α)2] |
For the experiments evaluating the damping performance, a commercially available test tube mixer was used as the vibration source; the vibration machine was equipped with a hydrogel vibration damper, a support plate, and a force sensor. The sampling frequency was set at 20 points per second.
All the hydrogel performance tests were performed at room temperature.
Results and discussion
Visible-light intensity for hydrogel formation
Light as an efficient and easily acquired trigger has been extensively used for radical-initiated polymerization.17 Visible light, which is safe, low cost, and easy to use, is favored in synthetic polymerization applications.18 In our previous study, visible light was found to be an efficient trigger for the synthesis of cellulose-containing hydrogels.15 In this polymerization, ceric ions attack the glucopyranose units to form a CMC–cerium complex. Then, Ce4+ ions are reduced to Ce3+ within the complex, and a free radical and aldehyde are obtained by the rupture of the carbon 2 and 3 bonds of the glucopyranose unit.19 Finally, the free radical initiates the monomer polymerization and a network is formed through end-group termination of the active polymer chains (Scheme 1). This polymerization can even be performed under sunlight, and the gelation process is completed within only 1 minute (Fig. 1, inset). To study the effect of light intensity on hydrogel performance, a metal halide lamp (LS-M210, Sumita) was selected as a stable visible light source. The luminous emittance is proportional to the output energy. Fig. S1† shows that the luminous emittance was in the range of 58–215 kLux depending on the output energy (from 25% to 100%). Since the generated radicals are highly dependent on the induction of visible light, the gelation time is closely related to the visible light intensity. The pseudo relaxation modulus showed that the hydrogel precursors from the viscous state to the just-formed solid state require 68 s, 30 s, 20 s, and 22 s at the visible light output energy of 25%, 50%, 75%, and 100%, respectively (Fig. 1). This suggests that high light intensity helps to shorten the gelation time, but higher light intensity has a limited effect on the gelation time. Moreover, the visible light intensity greatly affects the mechanical properties of the hydrogel. At a lower output energy (25%), the tensile strength, fracture elongation, and elastic modulus of Gel-1.0 were 0.59 MPa, 640%, and 0.096 MPa, respectively. When a higher output energy of 50%, 75%, and 100% was applied, the tensile strength and elastic modulus of Gel-1.0 clearly improved (Fig. S2†). However, the fracture elongation slightly decreased with the increasing output energy. | ||
| Scheme 1 Proposed mechanism for the formation of the hydrogel under visible light and the reinforced process by the evaporation-swelling method. | ||
 | ||
| Fig. 1 Effect of various light intensities on the time of hydrogel state transformation: pseudo relaxation modulus–time curves of Gel-1.0. The inset is Gel-1.0 before and after polymerization under sun light. | ||
Improvement of mechanical properties
To improve the mechanical performance of hydrogels, researchers have intensively investigated different reinforced networks including double networks,3 topological networks,20 polyampholyte based networks,21 hydrogen bond cross-linked networks,22 or ionic cross-linked networks.23 Among them, the ionic cross-linking is one of the most deeply investigated. Metal cations play an important role in hydrogels containing anionic polymer chains as they can form coordination bonds with the negatively charged polymer chains to increase the cross-linking density of hydrogels.24,25 When an external loading is applied, these coordination bonds act as reversible sacrificial bonds and rupture to dissipate the applied energy. An improvement in tensile strength, elastic modulus, and fracture elongation was observed by adding 0.1 mol% Al3+ (Fig. 2). However, the mechanical properties of the hydrogels tend to remain stable with the addition of more than 0.5 mol% Al3+; in particular, within the 500% elongation, the stress–strain curves of Gel-0.5, Gel-1.0, and Gel-1.5 were almost coinciding. This phenomenon indicates that an excess of Al3+ concentration has little effect on the mechanical properties of the hydrogel since most of the carboxyl groups in the PAA chain are maintain in the acid form (–COOH).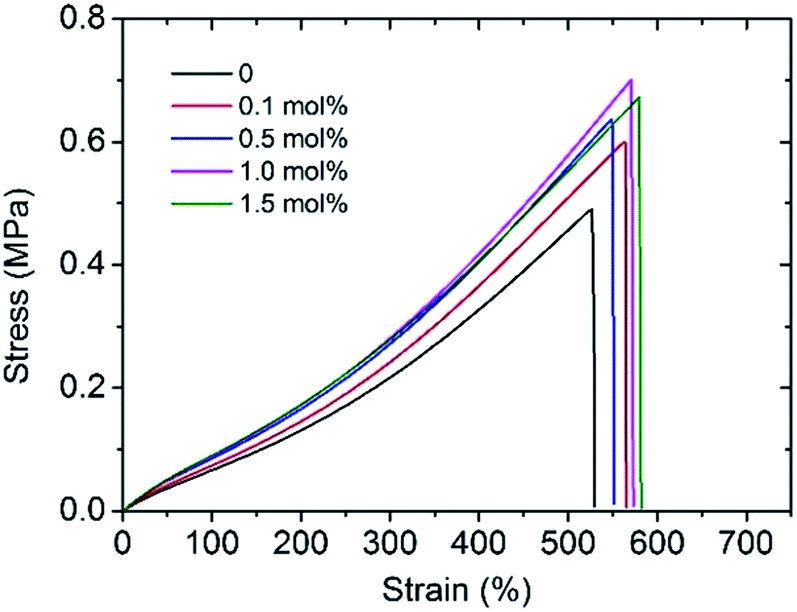 | ||
| Fig. 2 Stress–strain curves of Gels with various Al3+ contents. | ||
The hydrogels prepared by visible-light-triggered polymerization possessed remarkable mechanical properties (e.g., Gel-0.5, tensile strength of 0.66 MPa, strain of 550%, and elastic modulus of 0.106 MPa). However, Gel-0.5, as well as many reported hydrogels,26,27 showed a lower elastic modulus as compared to some synthetic hydrogels (e.g., polyelectrolyte hydrogels)28 or natural tissues (e.g., cartilage).29,30 In this study, we applied the evaporation-swelling (E-S) method on the as-prepared Gel to achieve an approximately 10-fold increase in the elastic modulus of the hydrogel (Scheme 1). The mechanical properties of the as-prepared Gel underwent fundamental changes as a result of the restructuring of the hydrogel internal network. Fig. 3a shows that the E-S method-treated Gel (HM-Gel-0.5, air) reaches a high tensile strength and elastic modulus of 1.50 MPa and 0.96 MPa, respectively, which are 2.27 and 9 times higher than those of Gel-0.5. When the evaporation operation was conducted in vacuum, the HM-Gel-0.5 (vacuum) exhibited tensile strength (1.78 MPa) and elastic modulus (1.29 MPa) that were 2.70 and 12.2 times higher than those of Gel-0.5, respectively (Fig. 3a). Apparently, the HM-Gels dried in vacuum displayed a higher tensile strength and elastic modulus than the HM-Gels dried in air, whereas a lower fracture elongation was found in the HM-Gels dried in vacuum. (Table S1†).
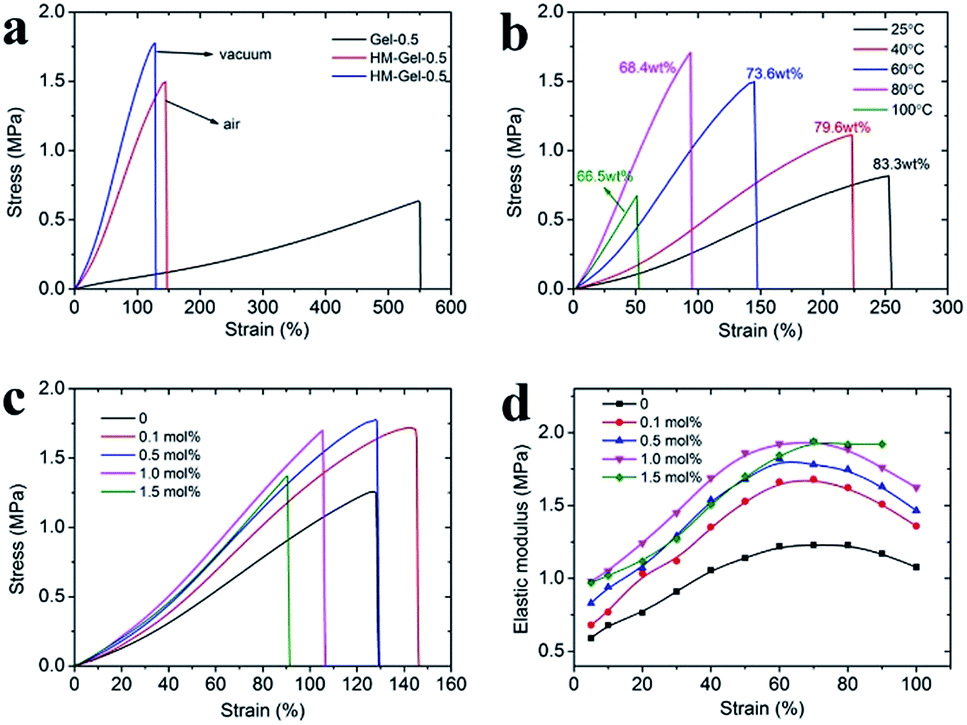 | ||
| Fig. 3 (a) Stress–strain curves of Gel-0.5, HM-Gel-0.5, (dried in air at 60 °C, red line; and evaporated in vacuum at 60 °C, blue line); (b) stress–strain curves of HM-Gel-0.5 obtained at various evaporation temperatures, the water content of the swollen hydrogel at equilibrium is as indicated; (c) stress–strain curves of HM-Gels (dried in vacuum) with various Al3+ contents; (d) elastic modulus of the HM-Gels calculated from the derivative of the stress–strain curves in (c). | ||
As shown in Fig. 3b, the evaporation temperature is a decisive factor for the mechanical performance of HM-Gels. When the evaporation proceeds at room temperature, the tensile strength and elastic modulus of HM-Gel-0.5 were only 1.24-fold and 1.83-fold, respectively, as compared to those of Gel-0.5. The tensile strength and elastic modulus of HM-Gel-0.5 dramatically increased with the increasing evaporation temperature, whereas the fracture elongation gradually decreased. This indicates that the evaporation temperature during the evaporation-swelling process is critical for the reconstruction of an internal network of the hydrogel. However, when Gel-0.5 was evaporated at 100 °C for 24 h, the degeneration of the CMC network resulted in a substantial impairment of the mechanical properties. In addition, the evaporation temperature in the E-S method has a substantial effect on the water content of the hydrogel after the swelling equilibrium is reached. The water content of the hydrogel decreased from 83.3 wt% to 66.5 wt% as the evaporation temperature increased (Fig. 3b, inset labels).
Although the concentration of Al3+ has a limited effect on the mechanical properties of the as-prepared hydrogels, it is noted that the mechanical properties of HM-Gels can be greatly changed via the introduction of Al3+. The tensile strength increased to about 1.70 MPa upon increasing the Al3+ content from 0.1 mol% to 1.0 mol%, whereas the fracture elongation gradually decreased. However, a higher Al3+ content (1.5 mol%) could reduce the comprehensive mechanical properties of the HM-Gel (Fig. 3c). The elastic modulus was calculated from the derivative of the stress–strain curves of the HM-Gels. At a strain of 30%, the elastic modulus of HM-Gels was in the range of 1.11–1.45 MPa, depending on the Al3+ content, which was higher than 0.91 MPa of HM-Gel without the reinforced cross-linking site (Al3+). The elastic modulus of HM-Gel-1.0 was as high as 1.92 MPa at a strain of 60% (Fig. 3d). The results indicate that an increase in Al3+ content can improve the tensile strength and elastic modulus of the hydrogels, whereas the fracture elongation first increased and then decreased. However, the addition of excess Al3+ reduces the mechanical properties of the hydrogel.
Both Gel and HM-Gel, like most tough hydrogels, are capable of withstanding large compressive stress.31 Fig. 4 shows that the compressive stress of Gel-0.5 and HM-Gel-0.5 can reach 17.1 MPa and 28.8 MPa, respectively, at a compressive strain of 90%. HM-Gel-0.5 exhibited a higher compressive stress than other reported hydrogels under the same strain condition.26,32 In the present study, the compressive modulus of HM-Gel-0.5 was always several times that of Gel-0.5, which should also be attributed to the recombination of the internal network structure of the hydrogel during the evaporation-swelling process.
 | ||
| Fig. 4 Compressive stress–strain curves of Gel-0.5 and HM-Gel-0.5 (the calculated compressive modulus is shown in the inset in the figure). | ||
Internal structure evolution
The conventional method for studying a hydrogel structure is to freeze-dry the hydrogel and then observe the pore structure of the hydrogel by an SEM.33 Although the basic skeleton structure of the hydrogel after freeze-drying is retained, its fine structure changes (e.g. the hydrogel changes from transparent to white). To investigate the actual changes occurring in the internal network of the hydrogel during the evaporation-swelling process, thin films (200 μm) of these two kinds of hydrogel (Gel and HM-Gel) were obtained by shock-frozen section and observed using a polarized optical microscope in the wet state. In this hydrogel, some of the CMC are present in the aggregated state (crystalline cellulose) rather than completely in the form of molecules. Therefore, although the hydrogel is transparent, the internal network is heterogeneous. Due to the polarization property of crystalline cellulose,34 we observed that a large amount of CMC was present in the form of crystals in the hydrogel (Fig. 5). In the as-prepared Gel (Fig. 5a), in addition to undissolved CMC chains exhibiting polarization property (black regions), the isolated CMC molecules involved in the polymerization reaction were not polarized (light-colored regions); this indicated that this CMC fraction was present in the non-crystalline form in the hydrogel. After the as-prepared Gel was treated through the E-S method (Fig. 5b), the edges of the crystalline CMC became irregular and many shadows (crystalline) filled in the light-colored regions as compared to the image shown in Fig. 5a. This indicated that the CMC crystal morphology and crystallinity had undergone tremendous changes. The images show that the E-S method greatly changes the hydrogel internal network of the hydrogel and increases the network's cross-linking density. The FT-IR spectra (Fig. S3†) showed that the absorption peaks of –COO–H in HM-Gel-0.5 were much stronger than those of PAA at 3000–3600 cm−1, indicating both PAA chains and CMC chains were involved in this reinforcement crystallization. In addition, the attenuation of the absorption peak of 1240 cm−1 (–CO–OH) suggested that part of the carboxyl group and aluminum ion form more stable coordination compounds during this treatment process. | ||
| Fig. 5 (a and b) Images of Gel-0.5 and HM-Gel-0.5 films in the wet state under polarized light. The images are in negative mode for easy viewing; (c) effect of Al3+ content on the DTA curves of HM-Gels. | ||
On the other hand, although the coordination bond could enhance the mechanical properties of the HM-Gel, the crystallinity of the HM-Gel was reduced. For example, an excessively high content of aluminum ions (as in HM-Gel-1.5) resulted in a very low crystallinity (the secondary crystallization region at only 75–130 °C), which led to a decrease in mechanical properties (Fig. 5c). The calculated effective network chain density (N) definitely confirmed that with the increase of Al3+ from 0 to 1 mol%, the N increased from 82 mol m−3 to 130 mol m−3, whereas the excessively high amount of Al3+ led to the decrease of the N (Fig. S4†).35 Therefore, by adjusting the content of aluminum ions to control the crystallinity of the HM-Gel, the required mechanical properties can be achieved.
Swelling property
Hydrogels, as soft and wet materials, consist of a cross-linked network and plenty of water (50–90%) and have been commonly used in aqueous environments.36,37 The swelling behavior of hydrogels is very important for their practical applications.20 The swelling ratio (Qm) of Gels decreased from 6.37 to 2.57 with the increase of Al3+ content. In sharp contrast, the Qm of HM-Gels slightly changed from 1.22 to 0.97 in the same composition (Fig. 6). This large difference in the swelling ratios indicates that the crystallization induced by the E-S method severely hinders the swelling of the hydrophilic polymer network in distilled water. | ||
| Fig. 6 Equilibrium swelling ratio (Qm) of the Gels and HM-Gels with various Al3+ contents. | ||
Viscoelasticity of the hydrogels
In Fig. 7, the storage modulus E′ and tanδ are shown for the as-prepared Gels and HM-Gels as functions of frequency at a strain of 1.0%. The E′ of all hydrogels increased as the frequency increased. Gel-0.5 had a higher E′ than Gel-0 in the experimental frequency range; this suggested that the newly formed cross-linking site could increase the cross-linking network density by introducing Al3+.38 The enhanced effect of Al3+ also occurred in HM-Gel, and the ΔE′ (E′Gel-0.5 − E′Gel-0, and E′HM-Gel-0.5 − E′HM-Gel-0) increased from 100 kPa of Gel to more than 200 kPa of HM-Gel at the frequency of 0.1–100 Hz. These results confirm that the ions generated more coordination compounds, which increased the density of the cross-linked network during the evaporation-swelling process. The E′ of the hydrogels with the same components (i.e., Gel-0 and HM-Gel-0 and Gel-0.5 and HM-Gel-0.5) were also different before and after the evaporation-swelling treatment. The increased E′ indicates that the hydrogen bond recombination inside the hydrogel not only changes the structure of the cross-linked network, but also increases the cross-linking density of the network. The damping factor (tanδ) is determined by the storage modulus and loss modulus ratio.39 Unlike the storage modulus, the tanδ value of hydrogels with the same composition was reduced before and after treatment via the E-S method; this may be because the increased cross-linking density limited the movement of partial polymer chains. The tanδ value of all hydrogels showed a clear critical frequency at approximately 25 Hz (Fig. 7b and S5†) and increased rapidly as the frequency increased; this indicated that the energy dissipation caused by intermolecular friction was proportional to the frequency. For the hydrogel without Al3+, the tanδ value of HM-Gel-0 was significantly lower than that of Gel-0 after evaporation-swelling treatment. However, with the introduction of a certain amount of Al3+ into the hydrogel, the decrease in tanδ value of the hydrogel containing Al3+ before and after the evaporation-swelling treatment was significantly smaller than that of the aluminum-free hydrogel. Interestingly, the tan δ of Gel-0.5 and HM-Gel-0.5 had another critical frequency (approximately 1 Hz); this phenomenon could also be observed in the hydrogel with a higher Al3+ content (over 0.5 mol% Al3+, Fig. S5†). This change in the frequency response may be due to the rupture of ionic bonds within the hydrogel containing Al3+. Therefore, the damping characteristic of hydrogels contributes to their potential application in the field of vibration absorption.
 | ||
| Fig. 7 Dynamic mechanical analysis of the hydrogels: (a) storage modulus (E′) and (b) damping factor (tanδ) of Gel-0, HM-Gel-0, Gel-0.5, and HM-Gel-0.5. | ||
Cyclic mechanical property
The HM-Gel exhibited excellent tensile and compressive properties during successive cyclic process. Fig. 8a shows that the HM-Gel-0.5 has basic stable stress–time curves. However, it should be noted that the maximum stress for HM-Gel-0.5 slightly decreased from 0.56 MPa at an initial time and then trended to remain unchanged at 0.50 MPa. This may be due to the fact that during the stretching process, the hydrogel network obtained by free radical polymerization generally undergoes rupture and sliding of the polymer chains as well as rupture of ionic and hydrogen bonds, according to a previous study.16 In addition, the slight sliding between the hydrogel and the fixture also contributes to decreasing the maximum stress.40 After the stretched hydrogel was stored at room temperature for 24 hours in a sealed and humid environment, the sample exhibited very similar stress–time curves to the previous curves. The similarity between the curves indicated that the ionic and hydrogen bonds in the Gel were substantially healed.41 The only non-coincident part was the curve of the first loading process, confirming that the irreversible rupture and permanent sliding of the partial polymer chains only occurred during the first stretching process.16 Fig. 8b shows the cyclic compression curves obtained after 10 successive tests at various strains. At a lower strain (less than 25%), the compression loading–unloading curves were always consistent; this indicated that the internal network structure of HM-Gel-0.5 was retained. However, as the compressive strain increases, the network structure of HM-Gel-0.5 changes. At a strain of 50% and 70%, the curves of the second cycle were inconsistent with those of the first cycle; this suggested that a large deformation may cause irreversible damage to the network structure. At a strain of 25%, 50%, and 70% (from the second cycle), the hydrogels displayed a stable hysteresis energy dissipation, which resulted from the rupture of ionic and hydrogen bonds and the friction between polymer chains. Thus, the HM-Gel is a suitable candidate for vibration absorption materials. | ||
| Fig. 8 (a) Cyclic stress–time curves of HM-Gel-0.5 obtained after 10 tests at a strain of 60%, and the same operation was performed after 24 h; (b) cyclic compression curves of HM-Gel-0.5 obtained after 10 tests at various strains. | ||
Vibration absorption of the hydrogel
To verify the actual vibration absorption effect of the hydrogel, we tested the vibrational absorption of the hydrogel (HM-Gel-0.5) using the commercial test tube mixer (Present Mixer 2013, remove the spring bracket, Fig. 9). The test tube mixer was vigorously vibrated on a metal plate and submitted to a large displacement without any cushioning (Movie S1†). When the test tube mixer was placed on the hydrogel, the machine was able to operate stably on the hydrogel after it experienced a partial initial displacement (caused by resonance). Thus, the hydrogel was able to efficiently absorb the vibration and stabilize the test tube mixer (Movie S2†). The detected vibration forces of Gel and HM-Gel were 1.4 N and 0.6 N, respectively, which were lower than the vibration force of the machine without a vibration damper (8.5 N). In a vibration absorption material, stiffness is an important performance index, and a high stiffness can improve the material's vibration absorption performance.8,42 | ||
| Fig. 9 Comparison of the vibration absorption effect obtained with different vibration dampers in free mode. | ||
The stiffness is defined as

where F is the force on the body, δ is the displacement along the force, A is the cross-sectional area, E is the elastic modulus, and l is the length of the element.

For the specific case of an unconstrained uniaxial tension or compression, the elastic modulus can be thought of as a measure of the stiffness of a structure. Thus, the high-modulus hydrogel (HM-Gel-0.5, with an elastic modulus of approximately 1 MPa), obtained by the E-S method, can withstand a greater load under the unit strain than the low-modulus hydrogel (Gel-0.5, with an elastic modulus of approximately 0.1 MPa) and showed a better vibration absorption performance (Fig. 9). Of course, the metal spring bracket displayed the best vibration absorption.
Conclusions
In summary, we first synthesized a hydrogel (Gel) with good mechanical properties via a visible-light-trigger polymerization. To increase the modulus of the hydrogel, such that this can be applied as vibration absorption material, we leveraged the hydrogen bonds in the hydrogel to induce a CMC crystallization to alter the network structure within the hydrogel via the E-S method. The obtained optimal HM-Gel possessed an excellent mechanical properties with tensile strength and elastic modulus of 1.55 MPa and 1.02 MPa, respectively. Furthermore, different evaporation temperatures and the added various aluminum ion concentration have an extremely important effect on the tensile strength, modulus, and swelling ratio of the hydrogel. The coordination bonds formed by aluminum ions and anionic polymers increased the damping in the hydrogel during vibration, thus improving the vibration absorption performance of the hydrogel. In addition, HM-Gel-0.5 exhibited stable mechanical properties in both the cyclic tensile and compression tests. The preliminary application experiment shows that the high-modulus hydrogel has a better vibration absorption effect than the low-modulus hydrogel. We hope that this design concept can enhance the modulus of more hydrogels containing hydrogen bonds, such that these hydrogels can be used not only as machinery vibration absorbers but also as biological vibration absorbers (for example: articular cartilage).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the International Collaboration Project funded by the Shenzhen Government (GJHZ20150316160614843). L. Zhu would like to acknowledge the China Scholarship Council for the Ph.D. program.Notes and references
- E. S. Dragan, Chem. Eng. J., 2014, 243, 572–590 CrossRef CAS.
- X. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- T. Nonoyama, S. Wada, R. Kiyama, N. Kitamura, M. T. Mredha, X. Zhang, T. Kurokawa, T. Nakajima, Y. Takagi, K. Yasuda and J. P. Gong, Adv. Mater., 2016, 28, 6740–6745 CrossRef CAS PubMed.
- A. M. Hopkins, L. De Laporte, F. Tortelli, E. Spedden, C. Staii, T. J. Atherton, J. A. Hubbell and D. L. Kaplan, Adv. Funct. Mater., 2013, 23, 5140–5149 CrossRef CAS.
- W. Huang, A. Tarakanova, N. Dinjaski, Q. Wang, X. Xia, Y. Chen, J. Y. Wong, M. J. Buehler and D. L. Kaplan, Adv. Funct. Mater., 2016, 26, 4113–4123 CrossRef CAS PubMed.
- A. M. S. Costa and J. F. Mano, Eur. Polym. J., 2015, 72, 344–364 CrossRef CAS.
- S. Lv, D. M. Dudek, Y. Cao, M. M. Balamurali, J. Gosline and H. Li, Nature, 2010, 465, 69–73 CrossRef CAS PubMed.
- D. D. L. Chung, J. Mater. Sci., 2001, 36, 5733–5737 CrossRef CAS.
- G. Gao, G. Du, Y. Sun and J. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 5029–5037 CAS.
- S. Liu and L. Li, ACS Appl. Mater. Interfaces, 2016, 8, 29749–29758 CAS.
- C. W. Peak, J. J. Wilker and G. Schmidt, Colloid Polym. Sci., 2013, 291, 2031–2047 CAS.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- P. Lin, S. Ma, X. Wang and F. Zhou, Adv. Mater., 2015, 27, 2054–2059 CrossRef CAS PubMed.
- Y. Yang, X. Wang, F. Yang, H. Shen and D. Wu, Adv. Mater., 2016, 28, 7178–7184 CrossRef CAS PubMed.
- L. Zhu, J. Qiu, E. Sakai, L. Zang, Y. Yu, K. Ito, P. Liu and F. Kang, Macromol. Mater. Eng., 2017, 302, 1600509 CrossRef.
- L. Zhu, J. Qiu, E. Sakai and K. Ito, ACS Appl. Mater. Interfaces, 2017, 9, 13593–13601 CAS.
- N. J. Treat, B. P. Fors, J. W. Kramer, M. Christianson, C.-Y. Chiu, J. Read de Alaniz and C. J. Hawker, ACS Macro Lett., 2014, 3, 580–584 CrossRef CAS.
- X. Dai, Y. Zhang, L. Gao, T. Bai, W. Wang, Y. Cui and W. Liu, Adv. Mater., 2015, 27, 3566–3571 CrossRef CAS PubMed.
- L. Fakhru, A. Razi, I. Y. Qudsieh, W. M. Z. W. Yunus, M. B. Ahmad and M. Z. A. Rahman, J. Appl. Polym. Sci., 2001, 82, 1375–1381 CrossRef.
- H. Kamata, Y. Akagi, Y. Kayasuga-Kariya, U. I. Chung and T. Sakai, Science, 2014, 343, 873–875 CrossRef CAS PubMed.
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, Y. Zhao, K. Sato, A. B. Ihsan, X. Li, H. Guo and J. P. Gong, Adv. Mater., 2015, 27, 2722–2727 CrossRef CAS PubMed.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- W. J. Zheng, N. An, J. H. Yang, J. Zhou and Y. M. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 1758–1764 CAS.
- C. H. Yang, M. X. Wang, H. Haider, J. H. Yang, J. Y. Sun, Y. M. Chen, J. Zhou and Z. Suo, ACS Appl. Mater. Interfaces, 2013, 5, 10418–10422 CAS.
- J. Y. Sun, X. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- Q. Chen, L. Zhu, C. Zhao, Q. Wang and J. Zheng, Adv. Mater., 2013, 25, 4171–4176 CrossRef CAS PubMed.
- M. M. Fitzgerald, K. Bootsma, J. A. Berberich and J. L. Sparks, Biomacromolecules, 2015, 16, 1497–1505 CrossRef CAS PubMed.
- J. You, S. Xie, J. Cao, H. Ge, M. Xu, L. Zhang and J. Zhou, Macromolecules, 2016, 49, 1049–1059 CrossRef CAS.
- S. Pal, Design of Artificial Human Joints & Organs, Mechanical Properties of Biological Materials, 2014, ch. 2, pp. 23–40. Search PubMed.
- Z. Wang, F. Tao and Q. Pan, J. Mater. Chem. A, 2016, 4, 17732–17739 CAS.
- G. Du, L. Nie, G. Gao, Y. Sun, R. Hou, H. Zhang, T. Chen and J. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 3003–3008 CAS.
- J. Duan, X. Liang, Y. Cao, S. Wang and L. Zhang, Macromolecules, 2015, 48, 2706–2714 CrossRef CAS.
- J. Fang, A. Mehlich, N. Koga, J. Huang, R. Koga, X. Gao, C. Hu, C. Jin, M. Rief, J. Kast, D. Baker and H. Li, Nat. Commun., 2013, 4, 2974 Search PubMed.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- J. Yang, C. R. Han, J. F. Duan, F. Xu and R. C. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 3199–3207 CAS.
- S. Pina, J. M. Oliveira and R. L. Reis, Adv. Mater., 2015, 27, 1143–1169 CrossRef CAS PubMed.
- J. Kim, A. Conway and A. Chauhan, Biomaterials, 2008, 29, 2259–2269 CrossRef CAS PubMed.
- Z. Cai, D. H. Kwak, D. Punihaole, Z. Hong, S. S. Velankar, X. Liu and S. A. Asher, Angew. Chem., 2015, 54, 13036–13040 CrossRef CAS PubMed.
- D. Shanmugam and M. Thiruchitrambalam, Mater. Des., 2013, 50, 533–542 CrossRef CAS.
- M. Lillie, G. Chalmers and J. Gosline, Connect. Tissue Res., 1994, 31, 23–35 CrossRef CAS PubMed.
- M. Zhong, X. Y. Liu, F. K. Shi, L. Q. Zhang, X. P. Wang, A. G. Cheetham, H. Cui and X. M. Xie, Soft Matter, 2015, 11, 4235–4241 RSC.
- R. Lakes, J. Compos. Mater., 2002, 36, 287–297 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08272j |
| This journal is © The Royal Society of Chemistry 2017 |