
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePractical stability of Au25(SR)18−1/0/+1†
C. B. Collins a,
M. A. Tofanellib,
M. F. Crookc,
B. D. Phillipsa and
C. J. Ackerson*a
a,
M. A. Tofanellib,
M. F. Crookc,
B. D. Phillipsa and
C. J. Ackerson*a
aDepartment of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA. E-mail: Ackerson@colostate.edu
bDepartment of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA
cDepartment of Chemistry, Binghamton University, Binghamton, New York 13902, USA
First published on 21st September 2017
Abstract
Superatom electron shell and/or geometric shell filling underlies the thermodynamic stability of coinage and alkali metal clusters in both theoretical and experimental results. Factors beyond simple shell filling contribute substantially to the lifetime of ligated clusters in solution. Such factors include the nature of the solvent, the atmosphere and the steric size of the ligand shell. Here we systematically lay out a ‘practical’ stability model for ligated metal clusters, which includes both shell-closing aspects and colloidal stability aspects. Cluster decomposition may follow either fusion or fission pathways. Solvent polarity can be determinative of the decomposition pathway.
Introduction
Gas-phase and solution-phase (ligated) metal clusters comprised of alkali or coinage metals form in discrete, thermodynamically stable sizes for which a possible explanation of is found in superatom complex theory.1 This describes molecule-like, discrete electronic states for the delocalized core metal electrons due to quantum confinement of gold nanoclusters with approximately 150 atoms or fewer. According to the superatom model, clusters with closed electronic shells are more stable than those with open electronic shells. Experimentally, we previously showed that superatomic electron configuration predicts the thermal stability of the clusters in differential scanning calorimetry experiments.2 In this work, we observed that Au25(PET)18−1 (1S21P6) was more thermally stable than Au25(PET)180 (1S21P5) or Au25(PET)18+1 (1S21P4).While superatom electron theory offers explanations for the discrete sizes3–5 and thermal stability of the clusters, little investigation has been done into the practical, solution stability of the monolayer protected clusters.6–8 This is an area that has been well studied for larger colloidal particles,9–12 but it is not clear whether the same principles apply to the smaller particles, and how colloidal descriptions of stability and superatomic descriptions of stability interact.
When a metal cluster or particle ‘decomposes’, that decomposition can be by fission/etching (breaking into smaller component pieces) or fusion (agglomerative growth of particles). Fission is an established pathway of decay for thiolate protected silver clusters.7 Etching represents a type of fission that is widely used in synthesis of metal nanostructures and is highly dependent on oxygen in the case of thiol protected gold nanoparticles.13–16 Instability through agglomerative growth pathways for nanoparticles is well established for water-soluble colloids17 and also for organo-soluble colloids.18,19 Theoretically, colloidal stability is attributed to a combination of van der Waals attraction, electrostatic double layer repulsion, and steric terms, described classically in DLVO and extended DLVO theory.17,20 For water soluble nanoclusters, manipulation of ligand shell charge has been shown to improve solution stability.8,21
In this work, we endeavour to establish the “practical stability” of thiolate protected metal clusters, using Au25(SR)18 as a well-studied model system in which different oxidation states and ligand shells are easily accessible.21–28 Such practical stability is important to establish as these clusters find their way into catalytic and imaging applications.29,30 We find that practical stability appears to depend both on superatom stability concepts and on colloidal stability concepts.
Experimental
Clusters were synthesized and purified as previously reported.2 For air free decomposition studies, the dry cluster was suspended in solvent that was rigorously purged with argon and then transferred to a sealed Schlenk cuvette using Schlenk technique to avoid introduction of air. The cuvettes were sealed to avoid contamination by atmosphere and solvent evaporation. The in air samples were also put into the same Schlenk cuvette but balloons were used to ensure there was a constant atmosphere of air in the head space. UV-vis spectra were taken on an ocean optics usb4000 spectrometer using a DT-mini-2-GS deuterium light source at approximately 4 hour intervals throughout the day using the respective solvent in the same type of cuvette as a blank before each measurement. Concentration of the samples were found using the previously reported molar absorptivity at 680 nm of 8800 cm−1 M−1.31 Wavelengths at 680 and 605 nm were monitored so that the trough at 605 could be consistently set to zero and the feature at 680 nm could be compared between measurements. Then the starting concentration was normalized to one and the decrease in concentration was monitored as a decrease in the 680 nm feature. The concentrations were then plotted vs. time to determine the reactant order in Au25 according to the integrated rate laws for zeroth, first and second order reactions. The best fit parameters were then used to determine the rate constant k and the half-life according to the integrated rate law for that given order.Results and discussion
We observe that under some conditions, Au25(SR)18 cluster lifetime is extraordinarily longer than under other conditions, where the clusters degrade rapidly. Fig. 1, left panel illustrates this, showing Au25(SC4H9)180 in rigorously degassed THF, kept solvated under an argon atmosphere in the top set of traces. As judged by the Au25(SC4H9)180 optical absorbance spectrum, the cluster does not substantially decay over the course of 6 days (spectra are offset for clarity). By comparison, the spectrum of the same compound, when solvated in un-stabilized THF (bottom traces), loses the characteristic linear absorption features of Au25(SC4H9)180, evolving to a rather featureless spectrum typical of a heterogeneous mixture within the course of 3 days. Mechanistic information about decay pathways is extracted by examining the decrease in absorbance of the spectral feature at 680 nm, attributed to the HOMO–LUMO gap of the Au25(SR)18 cluster. Fitting the evolution of this peak to integrated kinetic rate laws (Table 1) produces information on the order of decay, rate constant, and half-life of the cluster in different environments. An example of this fitting is shown in Fig. 1, right panel.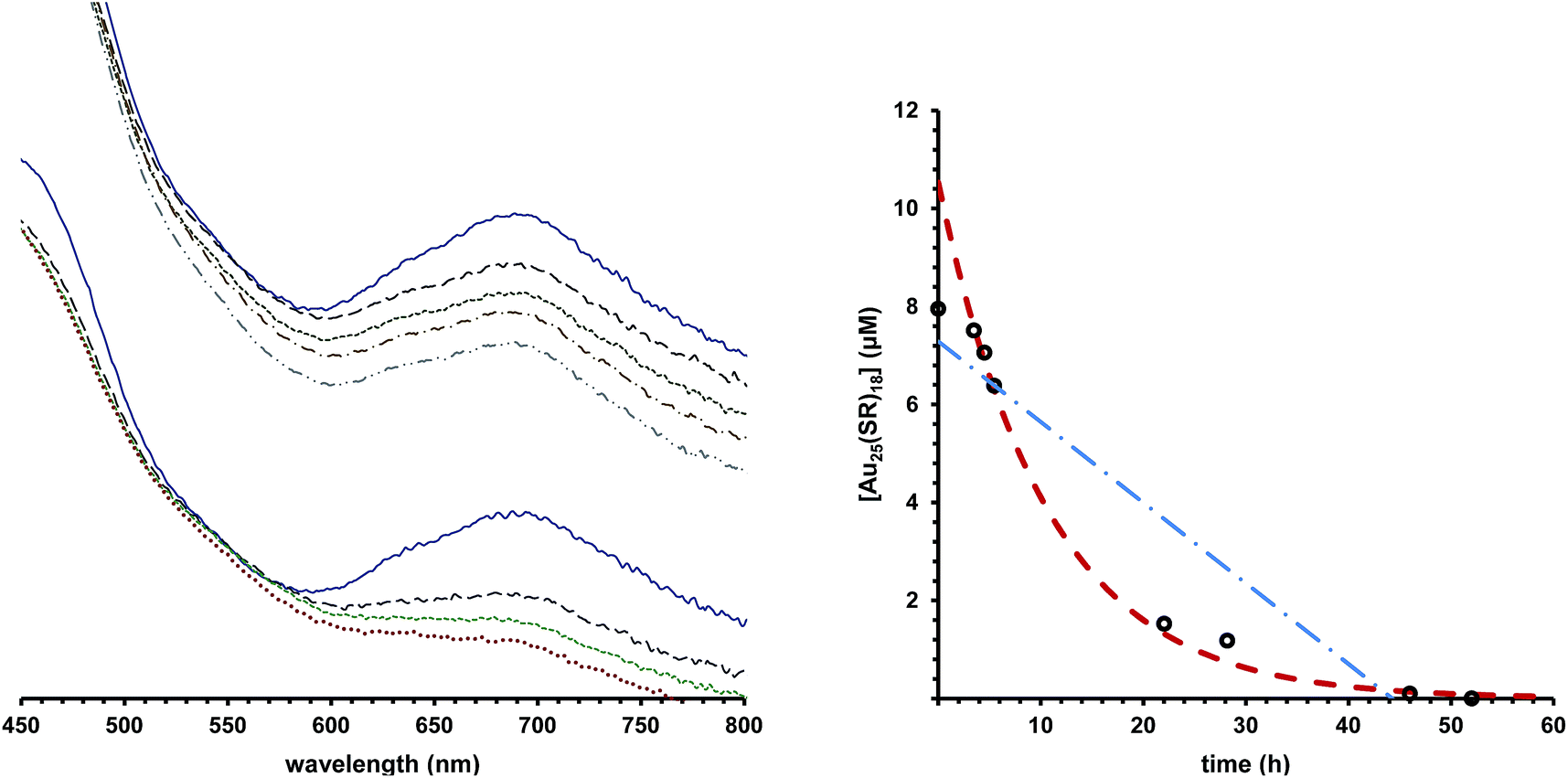 | ||
| Fig. 1 Left panel shows the optical spectrum of Au25(SC4H9)18 under argon as synthesized (solid blue trace), after approximately 1 day (purple-dashed trace), 2 days (green small-dash trace), 3 days (red dot-trace), 5 days (brown dash-dot trace), and 6 days (grey dash-dot-dot trace). The top set of traces show the evolution of the Au25(SC4H9)18−1 spectrum in stabilized THF, while the bottom set of traces shows the same experiment in unstabilized THF. The right panel plots the decay of Au25(PET)180 in acetone THF measuring concentration with 8800 cm−1 M−1 extinction coefficient at 680 nm, black circles. Fits to this data by zeroth order decay and first order decay are shown as blue and red traces, respectively. | ||
| Reactant order | Integrated rate law fit | R2 value |
|---|---|---|
| Zero (red) | [A] = [A]0 − kt | 0.895 |
| First (blue) | [A] = [A]0e−kt | 0.972 |
| Second (not shown) | 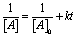 |
0.0156 |
Many prior reports have noted informally that ligand steric size and/or net charge impacts the practical stability of thiolate protected gold clusters, suggesting that aspects of colloidal stability are important in overall practical stability of thiolate protected clusters.32,33 The extent of the influence of ligand size on cluster stability, to our knowledge, has not previously been systematically investigated. To systematically establish the influence of ligand steric size on practical stability of Au25(SR)18, we investigated both the thermal and temporal stability of the compound with R groups ranging in size from (SCH2CH3, ethanethiol) to (S(CH2)11CH3, dodecanethiol), as well as (SCH2CH2C6H5, phenylethanethiol, PET). To determine temporal stability of Au25(SR)18, optical spectra of the clusters were acquired at approximately 4 hour intervals over the length of the experiment. The temporal stability in air, shown as half-life of the cluster, for Au25(SR)180 in dichloromethane solvation.where SR = ethanethiol, hexanethiol, octanethiol, dodecanethiol and phenylethanethiol is shown in Fig. 2, right panel. The thermal stability of a similar set of clusters was determined by differential scanning calorimetry (DSC). We previously used DSC to investigate the stability of Au25(PET)18 clusters as a function of oxidation state.2 The raw DSC traces (ESI p S17†) show a large endotherm that we attribute to a decomposition temperature. The decomposition temperatures of clusters protected by ethanethiol, butanethiol, phenylethane thiol and hexanethiol are shown in the right panel of Fig. 2.
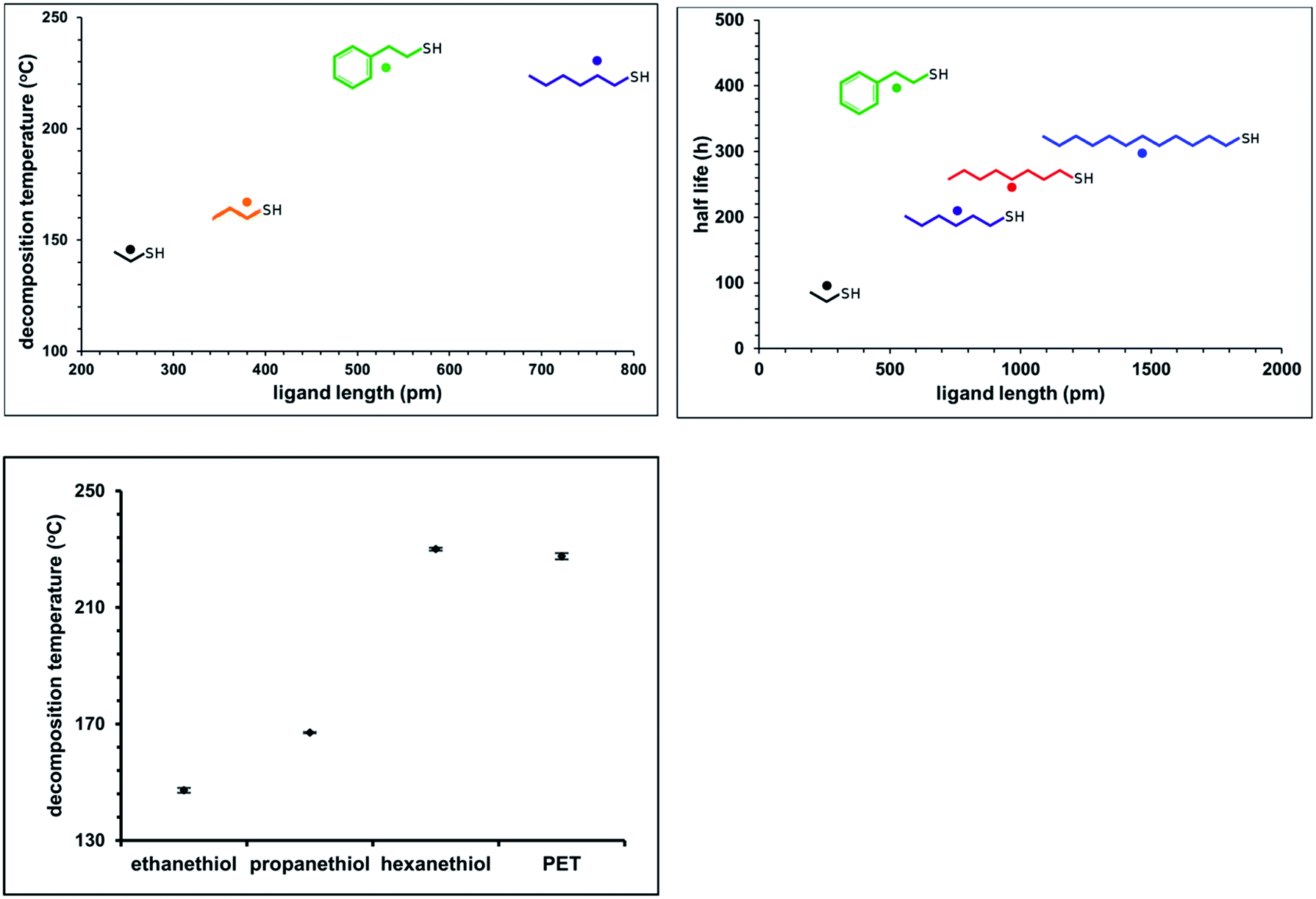 | ||
| Fig. 2 Left-hand panel shows the thermal stability of the clusters as it related to steric size of the protecting ligand. Right-hand panel shows the temporal stability of Au25(SR)180 as it relates to the steric size of the protecting ligand, air free in DCM. | ||
Overall, we observe a strong correlation with sterically larger ligands conferring greater thermal and temporal stability. The exception to this trend, phenylethanethiol, is easily rationalized as a more sterically demanding ligand which, by virtue of its branched nature, provides a larger ‘cluster cone angle’34 relative to its length, when compared to straight chain alkanethiols. We expect that branched alkanethiols may also show increased stability relative to absolute chain length.
We also examined the effect of cluster oxidation state on stability. Previously, we suggested that ‘closed shell’ superatom electron configurations are associated with greater thermal stability.2 Here we expand this investigation to examine how the thermal stability of different oxidation states correlates to the steric size of the protecting ligand. Fig. 3 shows the observed decomposition temperatures for Au25 clusters protected by ethanethiol (orange), propanethiol (purple), hexanethiol (blue), and phenylethanethiol (red) in the −1, and 0 oxidation states. As we observed before, increasing the oxidation state of the clusters, deviating from a full superatomic electron shell generally decreases their thermal stability. Fig. 3 shows that when varied simultaneously, the steric size of the ligand provides a larger contribution than oxidation state to practical thermal stability.
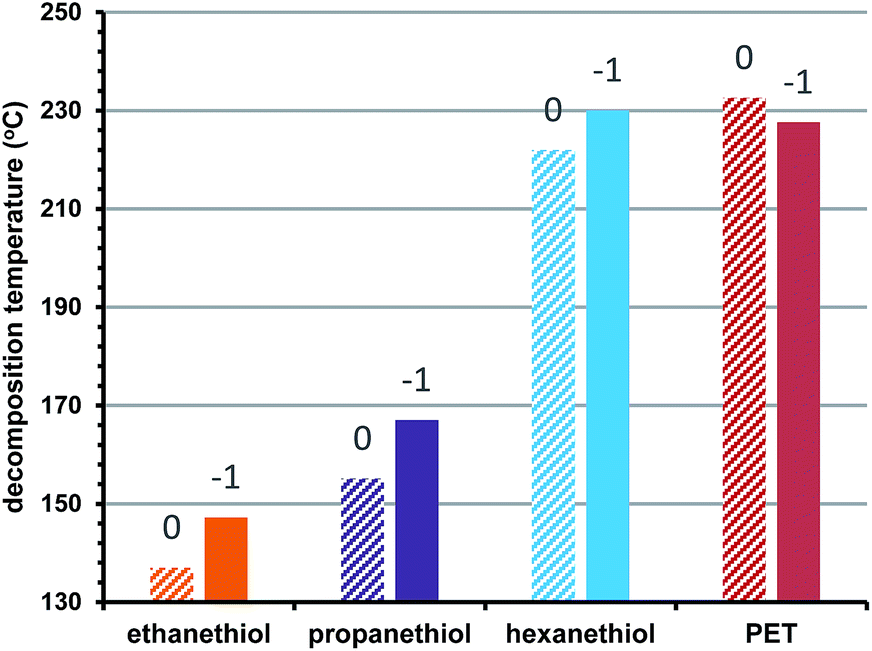 | ||
| Fig. 3 Oxidation state vs. thermal decomposition temperature. Charge states are shown above each column. | ||
In contrast to the influence of oxidation state on thermal stability of desolvated clusters, we observe a weaker correlation between oxidation state and lifetime in solution for solvated clusters. Instead, we observe that the nature of the solvent appears to dominate the lifetime of the clusters in solution (Fig. 4 and 5).
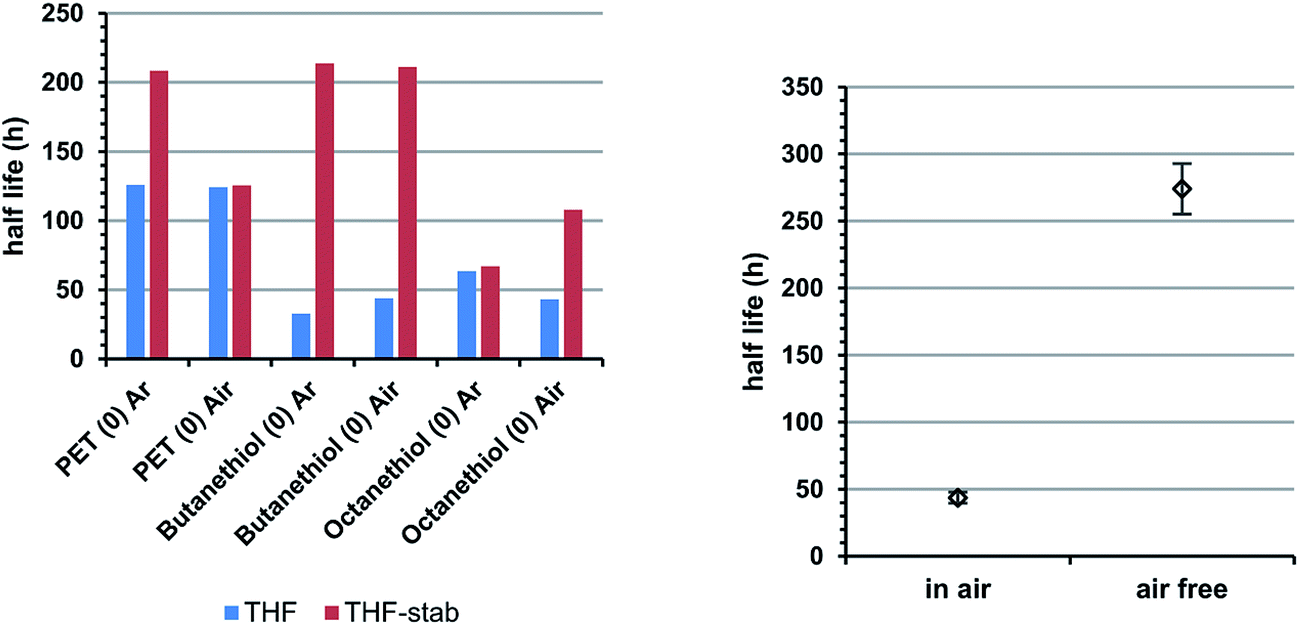 | ||
| Fig. 4 Left panel shows lifetimes of Au25(SR)180 in THF and stabilized THF. Generally, the inclusion of BHT increases observed half-life. The right panel shows inclusion vs. exclusion of ambient atmosphere for Au25(PET)18−1 in dichloromethane, showing the lifetime of the cluster is substantially decreased by exposure to atmosphere. | ||
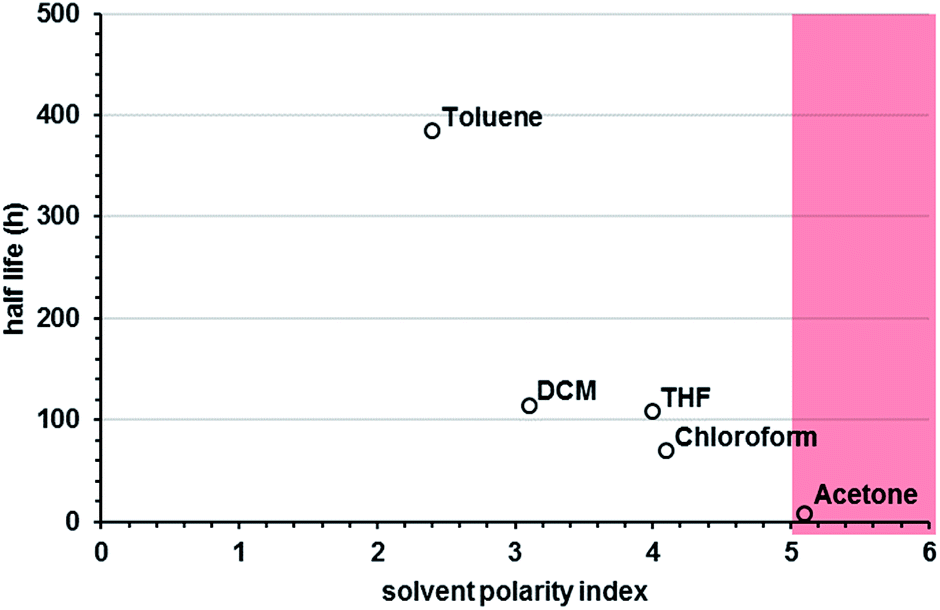 | ||
| Fig. 5 Au25(PET)180 half lives in toluene, dichloromethane (DCM), tetrahydrofuran, chloroform and acetone in air. The red-overlay represents regions of polarity solvent polarity where fusion mechanisms are (increasingly) observed. | ||
We also examined the effect storage of Au25(SR)18 in solution under ambient atmosphere compared to in solution under argon. In general, we note that storage under argon preserves cluster integrity for longer periods of time than storage under atmosphere. Fig. 4, right panel shows the lifetime of Au25(PET)18 in DCM under air and under argon. Here, storage under ambient atmosphere notably decreases the half life of the cluster. In the case of THF solvation, however, stabilized THF, which includes a radical inhibitor butylated hydroxytoluene (BHT) allows much longer half lives, independent of the storage atmosphere. That argon atmospheres and BHT specifically both tend to enhance cluster lifetimes is suggestive of dissolved O2 playing a role in the mechanism of cluster decomposition, potentially through an oxygen dependent etching process.13
In our preliminary observations, we noted that the nature of the solvent appeared to play a substantial role in the lifetime of solvated Au25(SR)18. We therefore made a systematic investigation of the effect of solvent on the lifetime of the clusters in solution. In testing several solvents used for organo-soluble clusters, we observed a strong correlation between solvent polarity and the lifetime of the cluster in solution, with more polar solvents promoting more rapid decomposition. Fig. 5 shows the half lives of Au25(PET)180 in a selection of solvents arranged according to their polarity index.35 The polarity index is a relative measure of the degree of interaction with a polar test solute, from Burdick & Jackson solvents.35 In general we find that clusters are markedly more stable in toluene than many of the other solvents screened. Some combinations of ligand and solvent, such as butanethiol protected particles in toluene (ESI p S15†) resulted in no measurable decay of the particles over 20 days.
This data provides insight into decay pathways. Decay of a suspended particle may arise from one of two either fusion of two particles or fission of a single particle into smaller components. These processes have distinct kinetics, with fission pathways following 0th order kinetics and fusion pathways following 1st order kinetics. An analysis of the disappearance of characteristic peaks of Au25 and spectral evolution into something suggestive of a mixture suggests that the mechanism of decay is environment dependent.
We notice that solvent polarity appears not only determinative of half-life but also influences the mechanism of decay. Higher polarity solvents favor 0th order decay (fission) pathways and lower polarity solvents favor 1st order decay (fusion). Example fits of 0th and 1st order decay are shown the ESI p S12.† This suggests that the higher polarity solvents may be driving aggregation of particles capped by non-polar ligands. We expect to observe the opposite trend for particles capped by polar ligands.
The trends shown are expected to be similar for other magic number sized monolayer protected noble metal clusters, but it's unknown if these trends will continue as clusters size increases into the range between cluster and colloidal particle.
Conclusions
Overall, this data allows us to suggest practical aspects of storage and handling that may preserve the integrity of Au25(SR)18 and by extension other thiolate protected clusters. In general, steric stabilization of the clusters appears to provide the greatest tunable variable for manipulating cluster half lives. The nature of the solvent also plays a large and unexpected role in the half-lives of the clusters. Less important but still appreciable contributions are made by oxidation state and relatedly the atmosphere under which clusters are stored. In some conditions, especially when dissolved in toluene with larger ligands the clusters appear to be very stable.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Eric Knudson for helpful conversations in formulating this work.Notes and references
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- M. A. Tofanelli and C. J. Ackerson, J. Am. Chem. Soc., 2012, 134, 16937–16940 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- O. Toikkanen, S. Carlsson, A. Dass, G. Rönnholm, N. Kalkkinen and B. M. Quinn, J. Phys. Chem. Lett., 2010, 1, 32–37 CrossRef CAS.
- A. Desireddy, S. Kumar, J. Guo, M. D. Bolan, W. P. Griffith and T. P. Bigioni, Nanoscale, 2013, 5, 2036–2044 RSC.
- X. Yuan, N. Goswami, I. Mathews, Y. Yu and J. Xie, Nano Res., 2015, 8, 3488–3495 CrossRef CAS.
- Y. Moroi, in Micelles, Springer US, 1992, pp. 131–148 Search PubMed.
- J. Zhou, J. Ralston, R. Sedev and D. A. Beattie, J. Colloid Interface Sci., 2009, 331, 251–262 CrossRef CAS PubMed.
- Y. Li and M. A. El-Sayed, J. Phys. Chem. B, 2001, 105, 8938–8943 CrossRef CAS.
- T. Laaksonen, P. Ahonen, C. Johans and K. Kontturi, ChemPhysChem, 2006, 7, 2143–2149 CrossRef CAS PubMed.
- T. A. Dreier and C. J. Ackerson, Angew. Chem., Int. Ed. Engl., 2015, 54, 9249–9252 CrossRef CAS PubMed.
- B. L. V. Prasad, S. I. Stoeva, C. M. Sorensen and K. J. Klabunde, Chem. Mater., 2003, 15, 935–942 CrossRef CAS.
- T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 1999, 103, 9394–9396 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- W. Zhang, in Nanomaterial, ed. D. G. Capco and Y. Chen, Springer Netherlands, 2014, pp. 19–43 Search PubMed.
- M. A. Watzky and R. G. Finke, J. Am. Chem. Soc., 1997, 119, 10382–10400 CrossRef CAS.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- E. M. Hotze, T. Phenrat and G. V. Lowry, J. Environ. Qual., 2010, 39, 1909–1924 CrossRef CAS PubMed.
- Y. Shichibu, Y. Negishi, H. Tsunoyama, M. Kanehara, T. Teranishi and T. Tsukuda, Small, 2007, 3, 835–839 CrossRef CAS PubMed.
- J. Akola, M. Walter, R. L. Whetten, H. Häkkinen and H. Grönbeck, J. Am. Chem. Soc., 2008, 130, 3756–3757 CrossRef CAS PubMed.
- S. Antonello, N. V. Perera, M. Ruzzi, J. A. Gascón and F. Maran, J. Am. Chem. Soc., 2013, 135, 15585–15594 CrossRef CAS PubMed.
- M. A. Tofanelli, K. Salorinne, T. W. Ni, S. Malola, B. Newell, B. Phillips, H. Häkkinen and C. J. Ackerson, Chem. Sci., 2016, 7, 1882–1890 RSC.
- J. F. Parker, C. A. Fields-Zinna and R. W. Murray, Acc. Chem. Res., 2010, 43, 1289–1296 CrossRef CAS PubMed.
- Y. Negishi, N. K. Chaki, Y. Shichibu, R. L. Whetten and T. Tsukuda, J. Am. Chem. Soc., 2007, 129, 11322–11323 CrossRef CAS PubMed.
- A. Venzo, S. Antonello, J. A. Gascón, I. Guryanov, R. D. Leapman, N. V. Perera, A. Sousa, M. Zamuner, A. Zanella and F. Maran, Anal. Chem., 2011, 83, 6355–6362 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- L.-Y. Chen, C.-W. Wang, Z. Yuan and H.-T. Chang, Anal. Chem., 2015, 87, 216–229 CrossRef CAS PubMed.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- H. Kawasaki, S. Kumar, G. Li, C. Zeng, D. R. Kauffman, J. Yoshimoto, Y. Iwasaki and R. Jin, Chem. Mater., 2014, 26, 2777–2788 CrossRef CAS.
- A. C. Templeton, S. Chen, S. M. Gross and R. W. Murray, Langmuir, 1999, 15, 66–76 CrossRef CAS.
- C. J. Ackerson, P. D. Jadzinsky and R. D. Kornberg, J. Am. Chem. Soc., 2005, 127, 6550–6551 CrossRef CAS PubMed.
- D. M. P. Mingos, Inorg. Chem., 1982, 21, 464–466 CrossRef CAS.
- B. & J. Laboratories, Solvent guide, Burdick and Jackson Laboratories, 1984 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Decay spectra and trend fits. See DOI: 10.1039/c7ra07511a |
| This journal is © The Royal Society of Chemistry 2017 |