
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structures and magnetic properties of isoreticular polyrotaxane-type two-dimensional coordination polymers†
In-Hyeok Park ac,
Tun Seng Herngbc,
Huiyeong Jua,
Jun Ding*c,
Shim Sung Lee*a and
Jagadese J. Vittal*c
ac,
Tun Seng Herngbc,
Huiyeong Jua,
Jun Ding*c,
Shim Sung Lee*a and
Jagadese J. Vittal*c
aDepartment of Chemistry and Research Institute of Natural Science, Gyeongsang National University, Jinju 52828, South Korea. E-mail: sslee@gnu.ac.kr
bDepartment of Materials Science and Engineering, National University of Singapore, 119260, Singapore. E-mail: msedingj@nus.edu.sg
cDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: chmjjv@nus.edu.sg
First published on 25th September 2017
Abstract
Two isoreticular polyrotaxane-type two-dimensional coordination polymers (2D CPs) [Co2(bpy)(sdb)2]·2H2O (1) and [Co2(bpe)(sdb)2]·DMA·1.5H2O (2) (where sdb = 4,4′-sulfonyldibenzoate; bpy = 4,4′-bipyridyl and bpe = 4,4′-bipyridyl ethylene) have been synthesized and their solid state structures have been determined by X-ray crystallography. The solid state structures made up of wheels and axles have been synthesized using a bent dicarboxylate (sdb), two linear spacer ligands and Co(II). Interestingly, the sheets are two-fold entangled by [Co2(sdb)2Co2] wheels and bpy or bpe axles. Due to the shorter length of the bpy ligand, the [Co2(sdb)4] moieties in 1 are brought closer than in the sheet formed in 2. The magnetic properties of 1 and 2 have been discussed and fitted according to a spin Hamiltonian. 1 showed predominantly antiferromagnetic interactions with exchange integral of −1.522 cm−1. While 2 exhibited the paramagnetic behaviour below 40 K and antiferromagnetic behaviour at higher temperature.
Introduction
Coordination polymers (CPs) are a class of highly crystalline materials formed by metal ions or metal clusters with exo-dentate spacer ligands.1,2 One, two and three-dimensional CPs can be designed to exhibit high surface area, porosity, optical and magnetic properties.3–7 Among these, polyrotaxane structures can be designed from wheels formed by two bent spacer ligands with two metal ions and axles from linear spacer co-ligands.8–18 For instance, the two-dimensional (2D) CPs containing polyrotaxane structures can be designed and constructed from bent dipyridyl ligands and linear dicarboxylates or vice versa.19–25 Recently, we reported several supramolecular isomers in polyrotaxane-type CPs of Zn(II) and Co(II) using a bent ligand, 4,4′-sulfonyldibenzoate (sdb, Scheme 1) and a long linear spacer ligand, 1,4-bis[2-(4′-pyridyl)ethenyl]benzene (bpeb).26,27 The polyrotaxane-type Co(II) CPs show typically antiferromagnetic behaviour due to the long spacer ligand bpeb which prevents the neighbouring paddlewheel Co(II) coming closer in the solid state structures. Hence we have employed shorter bipyridyl spacer ligands namely 4,4′-bipyridyl (bpy) and 4,4′-bipyridyl ethylene (bpe) to synthesize similar polyrotaxane CPs and investigated their magnetic behaviour (Scheme 1). The details are given below.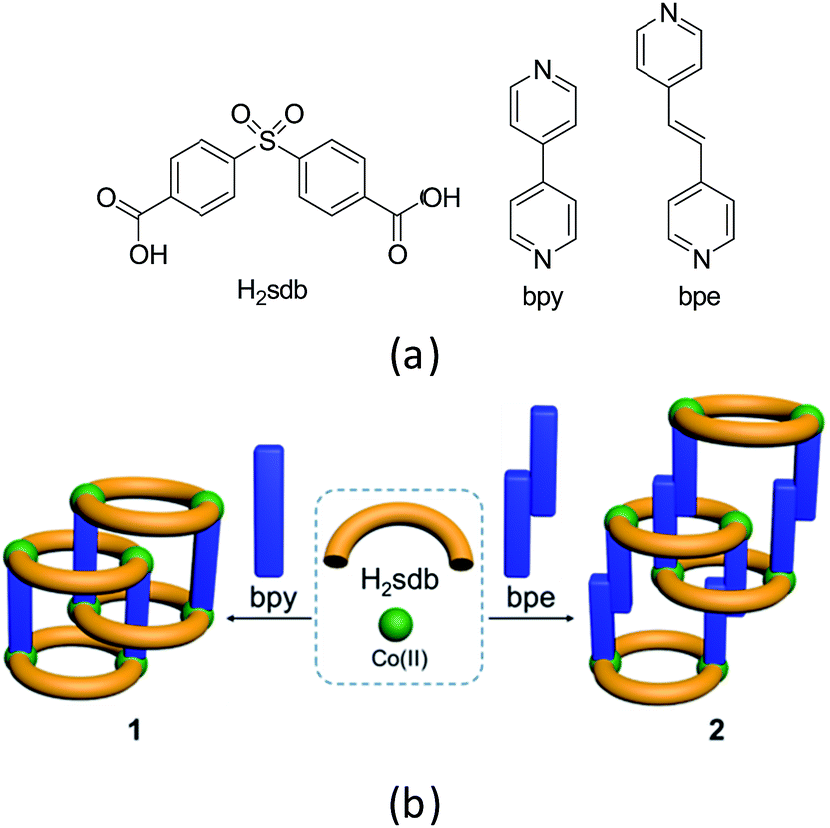 | ||
| Scheme 1 (a) Structural diagrams of H2sdb, bpy and bpe. (b) Polyrotaxane structures (1 and 2) formed from these two ligands with Co(II). | ||
Results and discussion
The single crystals of [Co2(bpy)(sdb)2]·2H2O (1) and [Co2(bpe)(sdb)2]·DMA·1.5H2O (2) were synthesized by the solvothermal reactions of Co(NO3)2·6H2O and H2sdb with bpy or bpe, respectively, in the equimolar ratio in the mixtures of dimethylformamide (DMF) or dimethylacetamide (DMA)/dimethylsulfoxide (DMSO)/water at 120 °C. The two isoreticular polyrotaxane-type 2D CPs (1 and 2) are constructed from the paddlewheel repeating unit [Co2(O2C–C)4]. The adjacent paddlewheel units are bridged by two sdb ligands to form a ring, [Co2(sdb)2Co2] thus generates a one-dimensional (1D) structure. These chains are further connected by the bpy or bpe spacer ligands in the axial positions of the paddlewheel units yielding 2D CPs. Interestingly, the [Co2(sdb)2Co2] rings and bpy (1) or bpe (2) axles from the neighbouring 2D structures produced polyrotaxane structures. The bulk homogeneity of the both products was confirmed by PXRD patterns (Fig. S1 and S2†). The details of the individual structures are described below.[Co2(bpy)(sdb)2]·2H2O (1)
Blue blocks of single crystals of [Co2(bpy)(sdb)2]·2H2O (1) crystallized in orthorhombic space group Cmcm with Z = 4 and the asymmetric unit contains quarter of the formula unit. In the crystal structure of 1 (Fig. 1), the atoms Co1, Co2, N1, C3, C4 and N2 have crystallographic m2m symmetry and the other C atoms in the bpy ligand occupy crystallographic m plane. The paddlewheel is produced by applying this m2m operation (Fig. 1a). Two sdb ligands are bonded to two neighbouring paddlewheels along a-axis forming Co2(sdb)2 ring (Fig. 1b). The Co–Co diagonal distance in this ring, 12.737 Å corresponds to the length a of the unit cell. The axial positions are occupied by bpy ligands which are along the crystallographic b-axis. The distance of the Co–bpy–Co⋯Co repeating unit, 13.765 Å is the length b of unit cell. The connectivity of Co2(bpy)(sdb)2 produced a 2D structure in ab-plane with S1, O3 and O4 atoms sit on crystallographic m plane. Two-fold entanglement of these rings formed by Co2(sdb)2 with dimension, 8.59 Å × 8.59 Å (the distances between centres of Co(II) and S⋯S distances in the ring) by the axle bpy ligands from the neighbouring 2D structure generates a polyrotaxane structure (Fig. 1c–f). This polyrotaxane entanglement has been observed in several 2D structures before.8–27 By virtue of X-ray crystallographic symmetry, bpy ligand is at the centre of this square cavity. The total potential solvent accessible area volume as calculated by PLATON in 1 is 288.3 Å3 which is 7.6% of the unit cell volume 3809.5 Å3 despite two-fold interpenetration.28–30 | ||
| Fig. 1 2D grid-type polyrotaxane of [Co2(bpy)(sdb)2]·2H2O (1). (a) Coordination environment of Co(II) atoms. Symmetry codes: (A) 1 − x, y, z; (B) 1 − x, y, 1/2 − z; (C) x, y, 1/2 − z; (D) x, −1 + y, z. (b) Basic building unit. (c) Top view. (d) The 2-fold entanglement showing the 2D square-type grids. (e) and (f) Rotaxane units. | ||
[Co2(bpe)(sdb)2]·DMA·1.5H2O (2)
The dark violet block-shaped single crystals of [Co2(bpe)(sdb)2]·DMA·1.5H2O (2) crystallized in monoclinic space group P21/c with Z = 4. Similar to 1, the structure of 2 also adopts a doubly-entangled 2D structure (Fig. 2). The asymmetric unit contains a formula unit. The coordination mode at the Co(II) centre is square-pyramid with four oxygen atoms from four sdb ligands in the square-plane and one nitrogen atom of the bpe ligand at the apical position. The paddlewheel repeating units are connected to four sdb ligands and propagate along the c-axis via Co2(sdb)2 ring to form a 1D polymeric structure (Fig. 2a–c). The square cavity has dimension of 8.58 Å × 8.61 Å (the distances between centres of Co(II) and S⋯S distances in the ring) producing a 1D structure with square cavities due to the use of bent sdb ligand. The bpe ligand also acts as a pillar to this 1D structure to produce the layered structure (Fig. 2d) with crystallographic glide plane and inversion centre.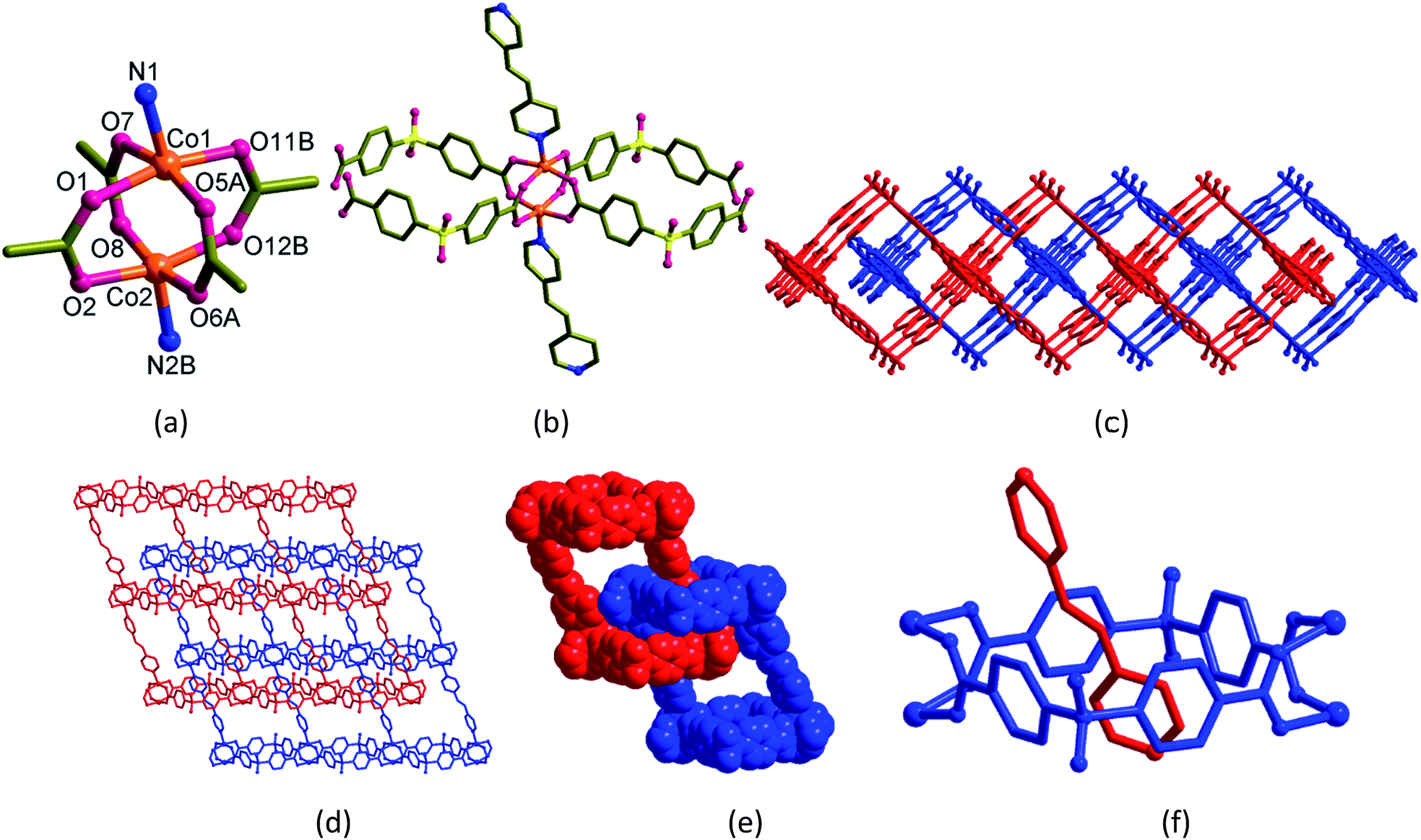 | ||
| Fig. 2 2D polyrotaxane structure of [Co2(bpe)(sdb)2]·DMA·1.5H2O (2). (a) A view showing the environment of Co(II) atoms. Symmetry codes: (A) x, y, 1 + z; (B) x − 1, y, z. (b) Basic building unit. (c) Top view. (d) The 2-fold entanglement of the 2D parallelogram grids. (e) and (f) Rotaxane units. | ||
Like 1, the linear bpe ligand also penetrates the [Co2(sdb)2] square ring to form a rotaxane structure in the 2D grid of 2. The pyridyl ring of bpe ligand is occupied in the cavity centre of the square ring. Again, the unusual entanglement of polyrotaxane was generated by rotaxane arrangement in 2. The total potential solvent area volume as calculated by PLATON in 2 is 874.6 Å3 which is 20.2% of the unit cell volume 4327.7 Å3 despite two-fold interpenetration (Fig. 2d–f).28–30 The void volume was partially occupied by one DMF and one and half water molecules. Unlike 1 showing a square-type grid (Fig. 1d), the 2D parallelogram grid in 2 (Fig. 2d) is generated from the distortion of the paddlewheel structure (Fig. 2a) and the longer length of the bpe spacer ligand.
Magnetic properties
As shown in Fig. 3, 1 and 2 display very contrast χmT behaviour. For 1, χmT increases drastically from 0.27 cm3 K mol−1 at 2 K to 3.08 cm3 K mol−1 at 50 K. Above 50 K, χmT of these samples increases monotonically with increasing temperature. Such χmT reduction at low temperature may associate with the presence of antiferromagnetic exchange coupling between the Co(II) ions and/or to the depopulation of the higher energy Kramers doublets of the Co(II) centres with a 4T1 term as the ground state. At room temperature (300 K), 1 exhibits χmT value of 3.54 cm3 K mol−1 that is higher than expected 1.87 cm3 K mol−1 for spin only high spin Co(II) ions (S = 3/2 and g = 2) and it is closed to the theoretical value (∼3.3 cm3 K mol−1) of spin-orbital coupling of Co(II) ions. Thus, this phenomenon may be attributed to the spin-orbital coupling of Co(II) ions. | ||
| Fig. 3 Temperature dependence of χmT for 1 and 2 measured at 500 Oe from 2 to 350 K. The samples are fitted according to a spin Hamiltonian by the PHI program with g = 1.91 for 1 and 2.02 for 2 (solid lines). | ||
For 2, the value of χmT remains nearly constant from 2 K up to 40 K, suggesting the paramagnetic behaviour. Then the χmT increases monotonically with increasing temperature with χmT of 1.56 cm3 K mol−1 at 300 K. This phenomenon suggests the presence of antiferromagnetic exchange coupling in the CP with increasing temperature.32,33 It is noted that the value of χmT, 1.56 cm3 K mol−1 at 300 K is same order of magnitude as the expected 1.87 cm3 K mol−1 for spin only high spin Co(II) ions (S = 3/2 and g = 2). Thus, magnetic properties of 2 mainly predominated by high spin Co(II) ions.
The magnetic susceptibility data were fitted according to a spin Hamiltonian by the PHI31 program with g = 1.91 for 1 and 2.02 for 2 in the temperature range of 2–350 K. The best fitted data yielded the exchange integral J of −1.522 cm−1 for 1 and −61.281 cm−1 for 2 respectively. Both samples exhibit a temperature independent paramagnetic (TIP) component of 5.0 × 10−4 cm3 K mol−1. These results indicate that antiferromagnetic interaction exists within the Co(II) dimer unit31,32 1 and 2 shows axial zero field splitting (ZFS) parameter (D) of 4.69 and 4.49 cm−1, respectively, with the rhombic ZFS parameter (E) of ±0.056 cm−1. The values of the parameters g, D and E indicate the presence of magnetic anisotropy as expected for the tetrahedrally coordinated Co(II) atom.34
In summary we synthesized two isoreticular pillared-layer 2D CPs (1 and 2) of Co(II) with sdb and two linear bipyridyl spacer ligands with different lengths, bpy and bpe, forming polyrotaxane-type structures. The separation between the [Co2(sdb)2] layers has been varied due to the difference in lengths of the spacer ligands. Hence the variable temperature magnetic properties have been investigated for 1 and 2 which reflects the interplays between structural properties of samples and their magnetic behaviours.
Experimental
General
All chemicals were purchased from commercial sources and used as received. All solvents used were of reagent grade. Elemental analyses were carried out on a LECO CHNS-932 elemental analyzer. Thermogravimetric analyses were recorded in a TA Instruments TGA-Q50 thermogravimetric analyzer. Samples were heated at a constant rate of 5 °C min−1 from room temperature to 700 °C and at a continuous flow nitrogen atmosphere. The FT-IR spectra were recorded using Thermo Fisher Scientific Nicolet iS 10 FT-IR spectrometer with KBr pellets. Powder X-ray diffraction (PXRD) patterns were recorded usning Bruker D8 DISCOVER.Preparation of [Co2(bpy)(sdb)2]·2H2O (1)
A mixture of bpy (11.1 mg, 0.071 mmol), H2sdb (22.1 mg, 0.072 mmol) and Co(NO3)2·6H2O (21.0 mg, 0.072 mmol) dissolved in DMF (3 mL), H2O (1 mL) and DMSO (0.5 mL) was placed in a 5 mL glass tube, and then 2–3 drops of 0.1 M NaOH solution was added. The tube was sealed and kept at 120 °C for 48 h, followed by cooling to room temperature over 8 h. Dark blue block-shaped crystals 1 suitable for single crystal X-ray diffraction (SC-XRD) analysis were obtained. Yield: 70%. Anal. calcd for [C38H27Co2N2O13.5S2]: C, 50.18; H, 2.99; N, 3.08; S, 7.05. Found: C, 50.15; H, 2.99; N, 3.20; S, 7.12%. IR (KBr pellet, cm−1) 3432, 3093, 3061, 1629, 1568, 1490, 1409, 1326, 1301, 1217, 1169, 1138, 1101, 1071, 1015, 845, 812, 779, 723, 695, 618 and 584.Preparation of [Co2(bpe)(sdb)2]·DMA·1.5H2O (2)
A mixture of bpe (13.0 mg, 0.071 mmol), H2sdb (21.9 mg, 0.071 mmol) and Co(NO3)2·6H2O (20.8 mg, 0.071 mmol) dissolved in DMA (3 mL), H2O (1 mL) and DMSO (0.5 mL) was placed in a 5 mL glass tube, and then 2–3 drops of 0.1 M NaOH solution was added. The tube was sealed and kept at 120 °C for 48 h, followed by cooling to room temperature over 8 h. Dark blue block-shaped crystals 2 suitable for SC-XRD analysis were obtained. Yield: 80%. Anal. calcd for [C43H35Co2N3O14S2]: C, 51.66; H, 3.53; N, 4.20; S, 6.41. Found: C, 51.82; H, 3.39; N, 4.27; S, 6.51%. IR (KBr pellet, cm−1) 3401, 3095, 3044, 1635, 1612, 1569, 1490, 1409, 1328, 1301, 1169, 1138, 1101, 1070, 1029, 1016, 844, 779, 739, 696, 618 and 551.Crystallographic structure determinations
Crystal data for 1 and 2 at 173 K were collected on a Bruker SMART APEX II ULTRA diffractometer equipped with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) generated by a rotating anode. The cell parameters for the compounds were obtained from a least-squares refinement of the spot (from 36 collected frames). Data collection, data reduction and absorption correction were carried out using the software package of APEX2.35 All of the calculations for the structure determination were carried out using the SHELXTL package.36 Relevant crystal data collection and refinement data for the crystal structures of 1 and 2 are summarized in Table S1.† CCDC 1551657 (1) and 1551656 (2) contain the supplementary crystallographic data for this paper.†Magnetic studies
A superconducting quantum interference device (SQUID) magnetometer MPMS was used for the magnetic characterization. Powder sample with a weight of 5–10 mg was sealed in a plastic capsule. Magnetic moment was measured in the temperature range of 2 K to 350 K. The empty plastic capsule exhibited diamagnetism and its magnetic moment was measured for correction. After correction of diamagnetic signal of plastic capsule and sample holder, diamagnetism of monomer and paramagnetic contamination (for example free radical), magnetic susceptibility was fitted using the PHI31 program by means of an isotropic spin Hamiltonian (SH) accounting for the exchange coupling (Heisonberg–Dirac–van Vleck Hamitonian) and zero-field splitting (ZFC).| H = Hex + HZee + HCF |
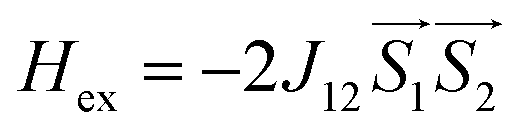

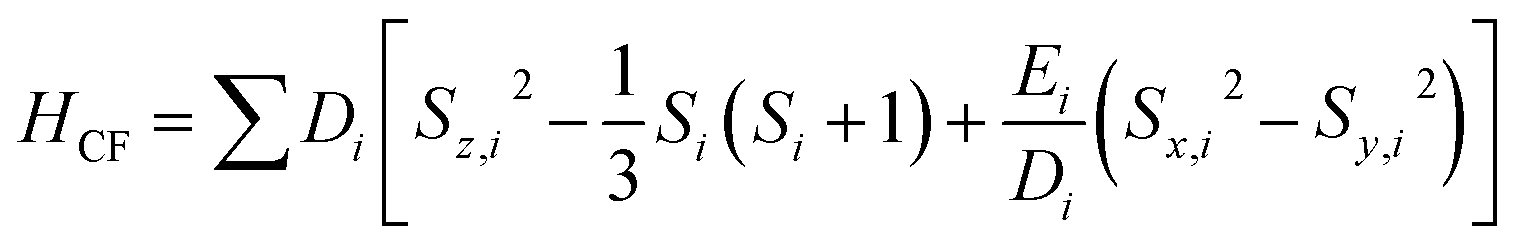
D: axial ZFS parameter; F: rhombic ZFS parameter; ![[S with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0053_20d1.gif) : spin vector;
: spin vector; ![[B with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0042_20d1.gif) : magnetic field vector; g: g-factor; μB: Bohr magneton.
: magnetic field vector; g: g-factor; μB: Bohr magneton.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NRF (2016R1A2A205918799), South Korea and the Ministry of Education, Singapore (Tier 1 FRC Grant No. R-143-000-678-114). S. S. L. acknowledges the support of the GNU Fund for Professors on Sabbatical Leave (2014).References
- S. R. Batten, S. M. Nevillle and D. R. Turner, Coordination Polymers: Design, Analysis and Application, RSC Publishing, London, 2009 Search PubMed.
- Design and Construction of Coordination Polymers, ed. M.-C. Hong and L. Chen, Wiley, 2009 Search PubMed.
- Metal-Organic Frameworks: Design and Application, ed. L. R. MacGillivray, Wiley, Weinheim, 2010 Search PubMed.
- Functional Metal-Organic Frameworks: Gas Storage, Separation and Catalysis, ed. M. Schröder, Topics in Current Chemistry, 2010, vol. 293 Search PubMed.
- Metal-Organic Frameworks, Applications from Catalysis to Gas Storage, ed. D. Farrusseng, Wiley-VCH, Weinheim, 2011 Search PubMed.
- Metal Organic Frameworks as Heterogeneous Catalysts, ed. F. L. i. Xamena and J. Gascon, RSC, 2013 Search PubMed.
- Coordination Polymers and Metal Organic Frameworks: Properties, Types, and Applications (Chemical Engineering Methods and Technology), ed. O. L. Ortiz and L. D. Ramirez, Nova Science Publishers Inc, New York, 2012 Search PubMed.
- H. Ju, E. Lee, S. Kim, I.-H. Park, J.-H. Lee and S. S. Lee, CrystEngComm, 2016, 18, 2621 RSC.
- Y. Bu, F. Jiang, K. Zhou, Y. Gai and M. Hong, CrystEngComm, 2014, 16, 1249 RSC.
- J.-Q. Liu, J. Wu, Y.-Y. Wang, J.-Y. Lin and H. Skiyama, CrystEngComm, 2014, 16, 3103 RSC.
- J. Wu, J.-Q. Liu, Y.-Y. Wang, Z.-P. Gou, D.-H. Xu and H. Skiyama, Inorg. Chem. Commun., 2012, 25, 10 CrossRef CAS.
- J. Yang, J. F. Ma, S. R. Batten and Z. M. Su, Chem. Commun., 2008, 2233 RSC.
- H. Chen, D. Xiao, J. He, Z. Li, G. Zhang, D. Sun, R. Yuan, E. Wang and Q.-L. Luo, CrystEngComm, 2011, 13, 4988 RSC.
- H. Wu, H. Y. Liu, B. Liu, J. Yang, Y. Y. Liu, J. F. Ma, Y. Y. Liu and H. Y. Bai, CrystEngComm, 2011, 13, 3402 RSC.
- F. Luo, Y. T. Yang, Y. X. Che and J. M. Zheng, CrystEngComm, 2008, 10, 981 RSC.
- Y. Q. Lan, S. L. Li, J. S. Qin, D. Y. Du, X. L. Wang, Z. M. Su and Q. Fu, Inorg. Chem., 2008, 47, 10600 CrossRef CAS PubMed.
- S. R. Batten, CrystEngComm, 2001, 18, 1 Search PubMed.
- S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef.
- X. Guo, Y. Yan, H. Guo, Y. Qi and C. Liu, CrystEngComm, 2016, 18, 2546 RSC.
- M.-D. Zhang, B.-H. Zheng, L. Chen, M.-D. Chen, T. Tao, K. Chen and H. Cao, Dalton Trans., 2016, 45, 3334 RSC.
- L.-L. Liu, C.-X. Yu, F.-J. Ma, Y.-R. Li, J.-J. Han, L. Lin and L.-F. Ma, Dalton Trans., 2015, 44, 1636 RSC.
- Y. Li, X.-Q. Yao, G.-B. Xiao, H.-C. Ma, Y.-X. Yang and J.-C. Liu, J. Mol. Struct., 2015, 1089, 16 CrossRef CAS.
- S.-L. Wang, F.-L. Hu, J.-Y. Zhou, Y. Zhou, Q. Huang and J.-P. Lang, Cryst. Growth Des., 2015, 15, 4087 CAS.
- H.-D. Guo, Z.-A. Fu, X.-M. Guo, X.-L. Li, R.-Z. Chen, Y.-J. Qi and H.-Y. Zou, J. Mol. Struct., 2013, 1038, 8 CrossRef CAS.
- Y. Ma, A.-L. Cheng and E.-Q. Gao, Cryst. Growth Des., 2010, 10, 2832 CAS.
- I.-H. Park, R. Medishetty, J.-Y. Kim, S. S. Lee and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 53, 5591 CrossRef CAS PubMed.
- I.-H. Park, H. Ju, T. S. Herng, Y. Kang, S. S. Lee, J. Ding and J. J. Vittal, Cryst. Growth Des., 2016, 16, 7278 CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164 CrossRef CAS PubMed.
- L. Qin, J.-S. Hu, L.-F. Huang, Y.-Z. Li, Z.-J. Guo and H.-G. Zheng, Cryst. Growth Des., 2010, 10, 4176 CAS.
- N. Benbellat, K. S. Gavrilenko, Y. L. Gal, O. Cador, S. Golhen, A. Gouasmia, J.-M. Fabre and L. Ouahab, Inorg. Chem., 2006, 45, 10440 CrossRef CAS PubMed.
- R. Boča, Coord. Chem. Rev., 2004, 248, 757 CrossRef.
- Bruker, APEX2 Version 2009.1-0 Data Collection and Processing Software, Bruker AXS Inc., Madison, Wisconsin, U.S.A., 2008 Search PubMed.
- Bruker, SHELXTL-PC Version 6.22 Program for Solution and Refinement of Crystal Structures, Bruker AXS Inc., Madison, Wisconsin, U.S.A, 2001 Search PubMed.
- P. de Meester, S. R. Fletcher and A. C. Skapski, J. Chem. Soc., Dalton Trans., 1973, 2575 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns, TGA and crystal data. CCDC 1551657 and 1551656. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra07637a |
| This journal is © The Royal Society of Chemistry 2017 |