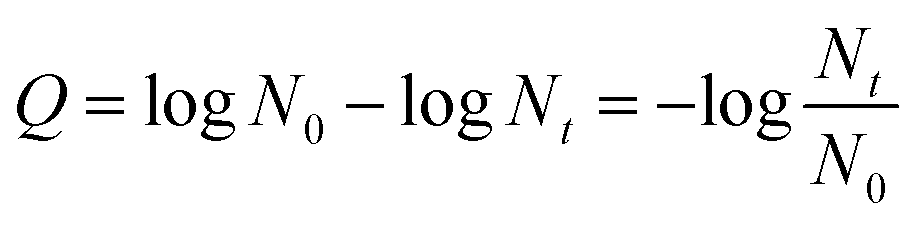DOI:
10.1039/C7RA07770J
(Paper)
RSC Adv., 2017,
7, 52172-52179
Electrospinning Cu–TiO2 nanofibers used for photocatalytic disinfection of bacteriophage f2: preparation, optimization and characterization
Received
14th July 2017
, Accepted 2nd November 2017
First published on 10th November 2017
Abstract
The presence of pathogenic viruses in drinking water threatens public health severely. However, there is little information about how to use photocatalysts to disinfect viruses. In this report, one-dimensional Cu–TiO2 nanofibers were fabricated using the electrospinning method and used for the removal of bacteriophage f2. The results showed that the optimum doping ratio and calcination temperature of the Cu–TiO2 nanofibers was n(Cu)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(Ti) = 1:8 and 450 °C, respectively. In addition, bacteriophage f2 with an initial concentration of 105 PFU mL−1 was completely inactivated with a dosage of 50 mg L−1 of Cu–TiO2 nanofibers under visible light irradiation for 4 h. Furthermore, the results from characterization of the nanofibers by various techniques, including scanning electron microscopy (SEM), energy dispersive spectrometry (EDS), X-ray diffractometry (XRD), X-ray photoelectron spectroscopy (XPS) and UV-vis spectrophotometry demonstrated that TiO2 existed in the anatase phase and Cu2+ substituted Ti4+ in the TiO2 lattice. The introduction of Cu into TiO2 effectively extended the spectral response of TiO2 to visible light. On the basis of this evidence, the mechanism of virus inactivation by Cu–TiO2 nanofibers was proposed.
:n(Ti) = 1:8 and 450 °C, respectively. In addition, bacteriophage f2 with an initial concentration of 105 PFU mL−1 was completely inactivated with a dosage of 50 mg L−1 of Cu–TiO2 nanofibers under visible light irradiation for 4 h. Furthermore, the results from characterization of the nanofibers by various techniques, including scanning electron microscopy (SEM), energy dispersive spectrometry (EDS), X-ray diffractometry (XRD), X-ray photoelectron spectroscopy (XPS) and UV-vis spectrophotometry demonstrated that TiO2 existed in the anatase phase and Cu2+ substituted Ti4+ in the TiO2 lattice. The introduction of Cu into TiO2 effectively extended the spectral response of TiO2 to visible light. On the basis of this evidence, the mechanism of virus inactivation by Cu–TiO2 nanofibers was proposed.
Introduction
It is of great significance for human beings to have access to potable water. Ensuring water quality, especially microbiological quality, has been regarded as the top priority when supplying drinking water.1 Pathogenic microorganisms in water, including bacteria, viruses, parasites, and so on, have received much attention for the microbial contamination they cause. Viruses are smaller in size and have stronger pathogenicity, posing a serious threat to public health after coming into contact with humans.2 In the US, viruses led to a few outbreaks of waterborne diseases and they were more difficult to analyze than bacterial pathogens.3 This calls for effective technologies to remove viruses from water.
Traditional disinfection methods can inactivate viruses to some extent, while there are also a series of problems. Chlorination is a dominant disinfection method, but it would produce disinfection by-products (DBPs), which are harmful to humans.4–6 UV disinfection can avoid DBPs, but it requires comparatively more energy and the photoreactivation phenomenon occurs frequently.7,8 Ozone is able to inactivate microorganisms in a short time owing to its strong oxidizing property, but it is unstable and has low solubility in water.9,10 Membranes can control pathogenic bacteria and protozoa effectively; however, the outcome turns to be reversed for viruses because their sizes are far smaller than those of membrane pores.11,12 In addition, the high cost and fouling of membrane are also limiting factors for its extensive application.
TiO2 photocatalysis is a new advanced oxidation technology that rises rapidly in recent years. TiO2 produces photoinduced electrons and holes after being excited by high-energy photons. The electrons and holes further react with the oxygen and hydroxyl group in water, then produce various reactive oxygen species (ROS) including ˙OH, ˙O2−, H2O2, 1O2. These ROS have strong oxidizability and are capable of degrading organic pollutants or inactivating microorganisms.13–15 Over the last decades, TiO2 photocatalysis has been widely studied and proved to be an effective, reliable disinfection method.16 However, TiO2 possesses large band gap, it can only be activated by UV and the utilization of the visible light is limited.17 In addition, the photo-generated electrons/holes tend to recombine during the photocatalytic process, thus reducing the overall quantum efficiency.18 For these problems, it has been well documented that metal doping is a promising method to extend the spectral response of TiO2 to the visible light as well as decrease the electron–hole recombination rate.19 Compared to other metal elements that are used as dopants, copper is relatively low-cost and widely used in daily life and industrial production, attracting many researchers' attention. Cu–TiO2 nanoparticles have been proved to show considerable photocatalytic performance in several literatures.20,21 Nevertheless, the high aggregation tendency of nanoparticles significantly reduces the photocatalytic efficiency. One-dimensional (1D) nanofibers would be a better option due to their low aggregation tendency, high axial ratio, good mechanical properties and large surface areas, thus promoting the photocatalytic activity.22 Electrospinning has been identified as the most advantageous method for nanofibers preparation owing to its simple operation, low cost and high efficiency.23
Most researchers prepared electrospinning Cu–TiO2 nanofibers to produce hydrogen or degrade organic pollutants,24–26 while very few pay attention to disinfection. Yousef et al. fabricated CuO/TiO2 nanofibers via one-step electrospinning, and achieved high removal efficiency of pathogenic Klebsiella pneumoniae under visible light.27 Despite that, no relevant research has been done for the disinfection of virus. There are differences between bacteria and viruses in structural and geometric properties.28 Viruses are relatively smaller in size and have a strong resistance to traditional disinfection technologies, so the results of bacterial disinfection cannot be translated to viral disinfection, and it is significant to evaluate the antiviral performance of electrospinning Cu–TiO2 nanofibers.
In this study, Cu–TiO2 nanofibers were fabricated by electrospinning method. Several parameters including doping ratio, calcination temperature and fiber diameter were optimized. The optimized Cu–TiO2 nanofibers were then characterized by scanning electron microscope (SEM), energy dispersive spectrometer (EDS), X-ray diffractometer (XRD), X-ray photoelectron spectroscopy (XPS) and UV-vis spectrophotometer. On basis of these characterizations, the disinfection mechanism of Cu–TiO2 nanofibers was discussed in detail.
Experimental
Chemicals
Tetrabutyl titanate (TBOT), polyvinylpyrrolidone (PVP), cupric nitrate, anhydrous alcohol, acetic acid, hydrochloric acid, sodium hydroxide, nutrient agar medium, agar and nutrient broth. All chemicals were analytically pure and purchased from Sinopharm Chemical Reagent Beijing. The ultrapure water was obtained from a Milli Q system (Millipore, US).
Photocatalyst synthesis
3 g PVP was added into 45 mL anhydrous alcohol, and then the solution was stirred for several hours till it became uniform and transparent, which was marked as solution A. Certain amount of TBOT and cupric nitrate was successively added into the mixed solution of 20 mL anhydrous alcohol and 16 mL acetic acid, and then the solution was stirred for a certain time, which was marked as solution B. Finally, solution B was transferred into solution A slowly with stirring for about 1 h. Thus the precursor solution of electrospinning was successfully prepared.
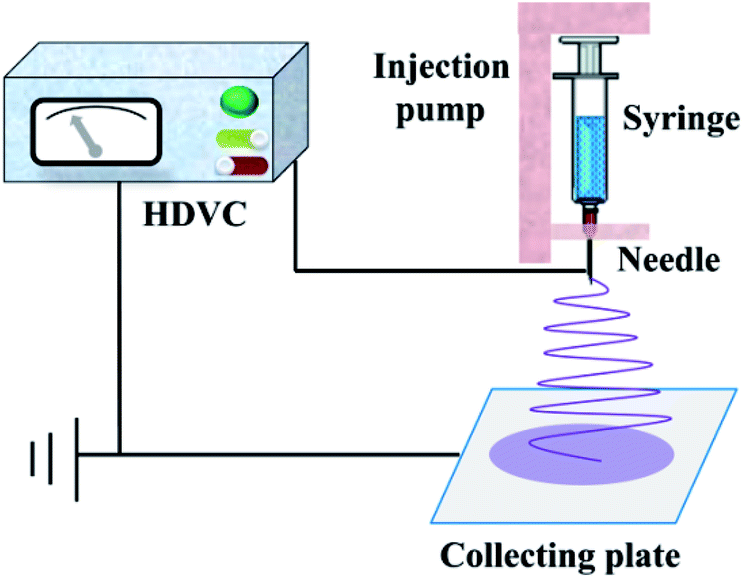
The diagrammatic sketch of the electrospinning device was shown in Fig. 1. First, the synthetized precursor solution was put into the syringe and then transferred to the injection pump. Second, the flow rate of injection pump and the operation voltage of high voltage DC power supply were adaptively adjusted, till the released liquid from the needle was sequentially stable and the jet trickle could be solidified into fibrous material before collected on the plate under the force of static electricity. After a period of time, the nanofiber mats were torn off from the collecting plate carefully and then calcined in a pipe electric furnace (SK-G10125K, ZhongHuan Electric Furnace Co. Ltd., China). The heating rate of the furnace was 2 °C min−1, and the thermal retardation time was 2 h at the stated temperature.
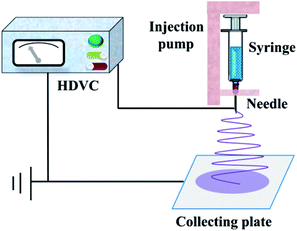 |
| | Fig. 1 Schematic diagram of the electrospinning device. | |
Catalyst characterization
A JSM-6301F field emission scanning electron microscopy (SEM, JEOL Ltd., Japan) was applied to investigating the surface morphology of the as-obtained photocatalysts. The photocatalyst samples were coated with aurum before SEM analysis to enhance electrical conductivity. An energy dispersive X-ray spectroscopy (EDX) was applied to study the elemental composition of the photocatalysts. Crystalline structure of the photocatalysts was investigated by a X-ray diffractometer (XRD, Rigaku Co., Japan) with Cu Kα (λ = 1.5418 Å) radiation at an operating voltage of 40 kV, an emission current of 40 mA. The surface composition was detected via X-ray photoelectron spectroscopy analysis (XPS, Thermo Fisher Co., America) with the scan step of 0.05 eV per step. The optical properties of the photocatalysts were obtained using a UV-visible spectrophotometer (Varian Co., America).
Virus preparation and assay
Bacteriophage f2 and its host bacteria (E. coli 285) were bought from institute of Hygiene and Environmental Medicine, Academy of Military Medical Sciences, China. The duplication and purification process of bacteriophage f2 was followed by the method described in a previous research.29 The concentration of bacteriophage f2 was determined by the double agar plate method. Petri dishes were put in the constant temperature incubator for 4 h at the temperature of 37 °C. Only the number of transparent plaques ranging from 30 to 300 on the plates was acceptable.
Disinfection experiment
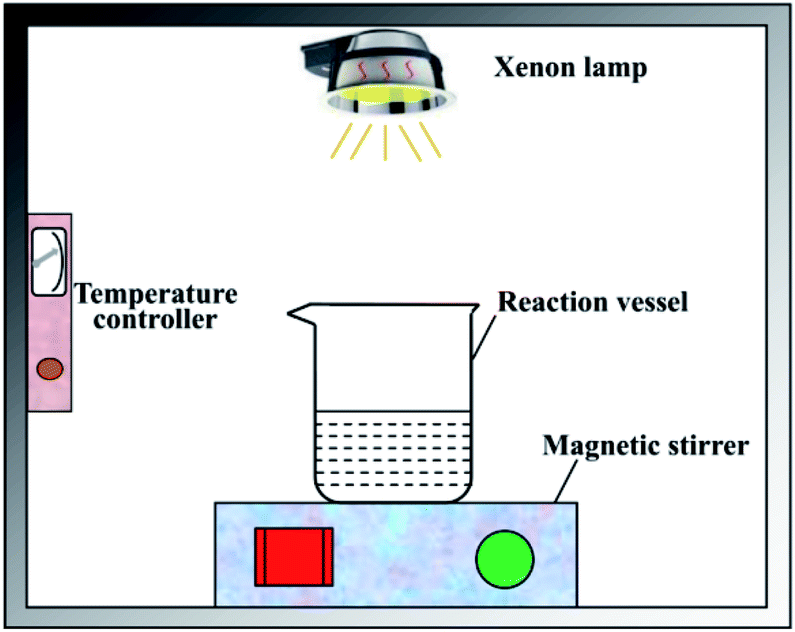
The disinfection experiment was carried out in a photocatalytic reactor which was shown in Fig. 2. The reaction vessel was a 500 mL beaker. First, 200 mL deionized water was added into the beaker after sterilization treatment. The pH value was adjusted to 7 by hydrochloric acid and sodium hydroxide solution. Second, the photocatalyst with a dose of 50 mg L−1 and concentrated liquid of bacteriophage f2 was put into the beaker successively, and the initial concentration of virus was 105 PFU mL−1. Then the beaker was placed on a magnetic stirrer in order to keep the photocatalyst well separated in the reaction solution during the experiment. The visible light source was provided by a CEL-HXUV300 Xenon lamp (Beijing ZhongJiaoJinYuan Tech Co. Ltd., China) with a 400 nm cutoff filter, the visible light intensity was set as 100 mW cm2. During the experiment, the whole system was kept closed in case of being interrupted by ambient light sources. The temperature of the reaction system was adjusted to 25 °C by a self-designed temperature controller, which consisted of a DLSB-2L/30 cryogenic coolant circulation pump (Gongyi YuHua Instrument Co. Ltd.) and a JRQM heater (Hunan HuaSi Instrument Co. Ltd.). Samples were taken from the beaker at certain time intervals. All the samples were treated for virus assay.
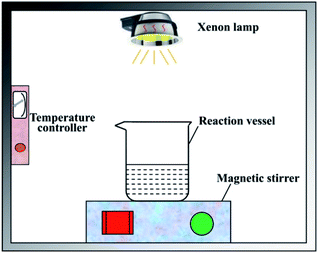 |
| | Fig. 2 Schematic diagram of the photocatalytic disinfection reactor. | |
The photocatalytic disinfection efficiency of bacteriophage f2 was calculated with the equation:
| |
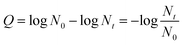 | (1) |
where
Q represents the virus removal efficiency,
t represents the reaction time,
N0 and
Nt is the virus concentration at the initial time and time
t, respectively.
Results and discussion
Optimization of Cu–TiO2 nanofibers
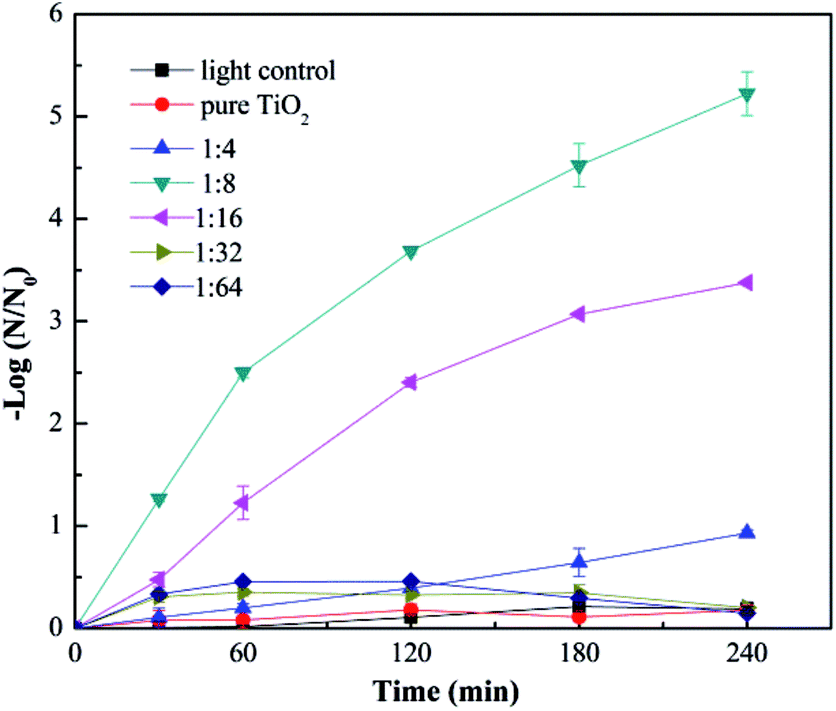
Fig. 3 shows the virus removal efficiency of TiO2 nanofibers doped with different contents of Cu. No significant virus removal was observed under visible light without any nanofibers, indicating that visible light didn't affect the activity of viruses. After adding pure TiO2 nanofibers into the reaction system, there was still no obvious virus removal, suggesting that pure TiO2 couldn't be activated by visible light. Then Cu-doped TiO2 nanofibers with different mole ratio of Cu and Ti were investigated. Doping Cu into TiO2 nanofibers significantly enhanced the virus removal efficiency. And the removal efficiency achieved the optimum when the mole ratio of Cu and Ti was 1:8. All viruses were inactivated after 4 h in this situation. This phenomenon confirmed that Cu modification successfully extended the spectral response of TiO2 into the visible region. However, when the mole ratio of Cu and Ti increased to 1:4, the virus removal efficiency dropped dramatically. This trend was consistent with many other researchers.30–32 Excessive content of Cu tended to cover the active sites on the TiO2 surface as well as acting as the recombination centers for photo-generated electrons and holes, thus reducing the disinfection efficiency.33,34
 |
| | Fig. 3 Photocatalytic disinfection of bacteriophage f2 by Cu–TiO2 with different mole ratios of Cu and Ti. Other conditions for nanofibers preparation: calcination temperature = 450 °C, TBOT dosage in precursor solution = 2 g. | |
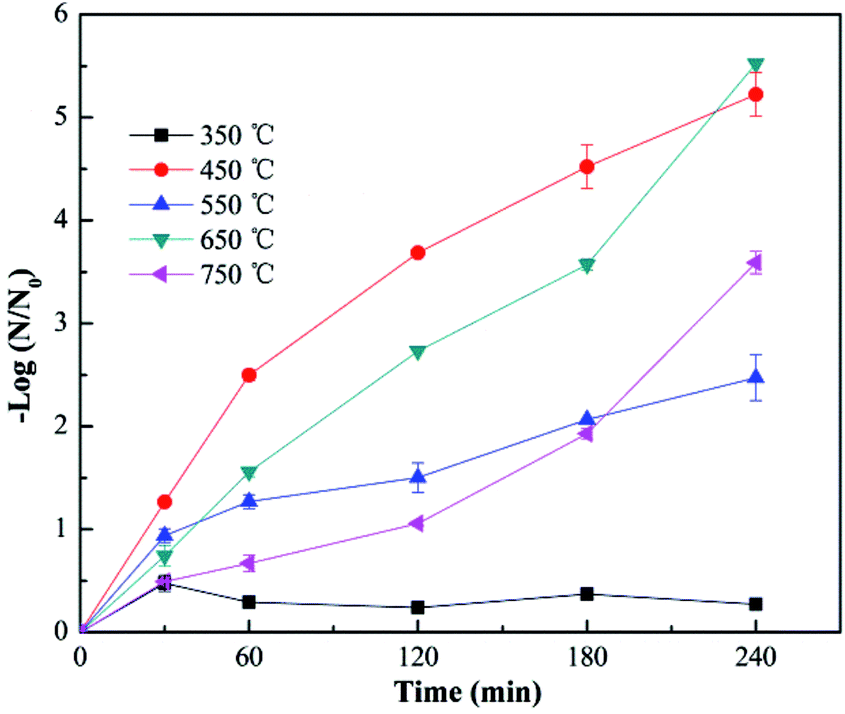
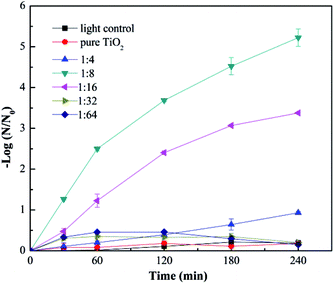
Fig. 4 shows the virus removal efficiency of Cu–TiO2 nanofibers treated at different calcination temperatures. No virus removal was achieved when the calcination temperature was 350 °C. As the temperature increased to 450 °C, all viruses were completely removed within 4 h. However, the removal efficiency decreased to some extent when the calcination temperature was over 450 °C. Calcination temperature could affect the crystal structure of TiO2, thus influencing the photocatalytic activity.35,36 Low removal efficiency at 350 °C might be due to the incomplete growth of crystals. As the temperature increased, TiO2 is gradually transformed into anatase, and then rutile,35 which was proved by the XRD test in Fig. 8. Normally, the anatase TiO2 exhibits relative higher photocatalytic activity than rutile TiO2, this could explain the optimum removal efficiency at 450 °C and lower removal efficiency at higher calcination temperatures. In addition, the nanocrystals tended to aggregate at over-high temperatures,35 resulting in a negative effect on virus removal efficiency, which was also a reasonable explanation for the as-described phenomenon.
 |
| | Fig. 4 Photocatalytic disinfection of bacteriophage f2 by Cu–TiO2 treated at different calcination temperatures. Other conditions for nanofibers preparation: n(Cu):n(Ti) = 1:8, TBOT dosage in precursor solution = 2 g. | |
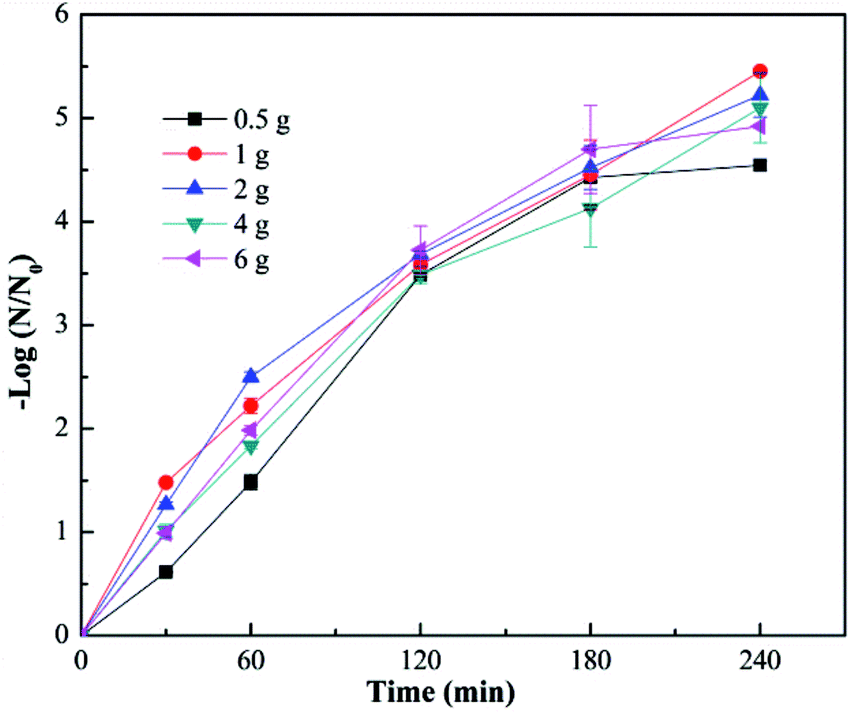
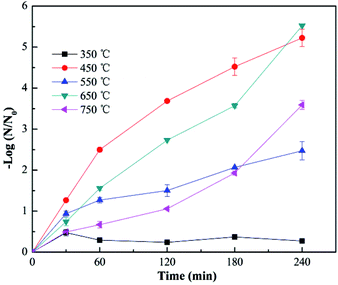
The effect of fiber diameter on virus removal efficiency was also investigated. The fiber diameter was adjusted by adjusting the addition amount of TBOT in the precursor solution, and the cupric nitrate dosage was adjusted correspondingly to maintain the mole ratios of Cu and Ti at 1:8. As shown in Fig. 5, the calcined Cu–TiO2 nanofibers were characterized by SEM, and the diameter of the nanofibers was measured. The fiber diameter increased from 55 nm to 146 nm with the increase of TBOT dosage from 0.5 g to 4 g, then decreased to 81 nm as the TBOT dosage increased to 6 g. Generally, the fiber diameter would increase with the increase of TBOT dosage. However, to maintain the mole ratios of Cu and Ti at 1:8, the increase of TBOT dosage correspondingly increased the concentration of Cu2+ in the precursor solution, and appropriate addition of metal ions could increase the surface charge density of charged jet and speed up the movement of charges under the external electric field, which was conducive to the refinement of the jet, thus the fiber diameter became smaller.37 The prepared Cu–TiO2 nanofibers with different diameters were applied to the disinfection experiment. As shown in Fig. 6, fiber diameter exhibited no significant effect on virus removal efficiency, almost all the viruses were completely inactivated in 4 h. Li et al. prepared g-C3N4, N–TiO2, Bi2WO6 and Ag@AgCl to inactivate MS2 under visible light,38 although the experimental operating conditions were not exactly the same, it could be concluded that the as-prepared Cu–TiO2 nanofibers showed better antiviral performance than N–TiO2, Bi2WO6, and showed comparable performance with g-C3N4 and Ag@AgCl.
 |
| | Fig. 5 SEM images of Cu–TiO2 nanofibers with different TBOT dosage in precursor solution. (A) 0.5 g, (B) 1 g, (C) 2 g, (D) 4 g, (E) 6 g. (F) The determined fiber diameter of Cu–TiO2 nanofibers with different TBOT dosage in precursor solution. Other conditions for nanofibers preparation: n(Cu):n(Ti) = 1:8, calcination temperature = 450 °C. | |
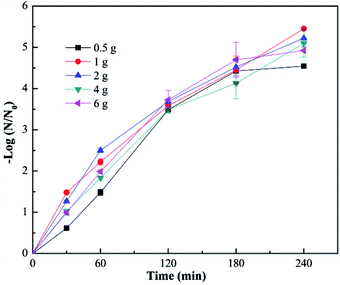 |
| | Fig. 6 Photocatalytic disinfection of bacteriophage f2 by Cu–TiO2 with different TBOT dosage in precursor solution. Other conditions for nanofibers preparation: n(Cu):n(Ti) = 1:8, calcination temperature = 450 °C. | |
Characterization of Cu–TiO2 nanofibers
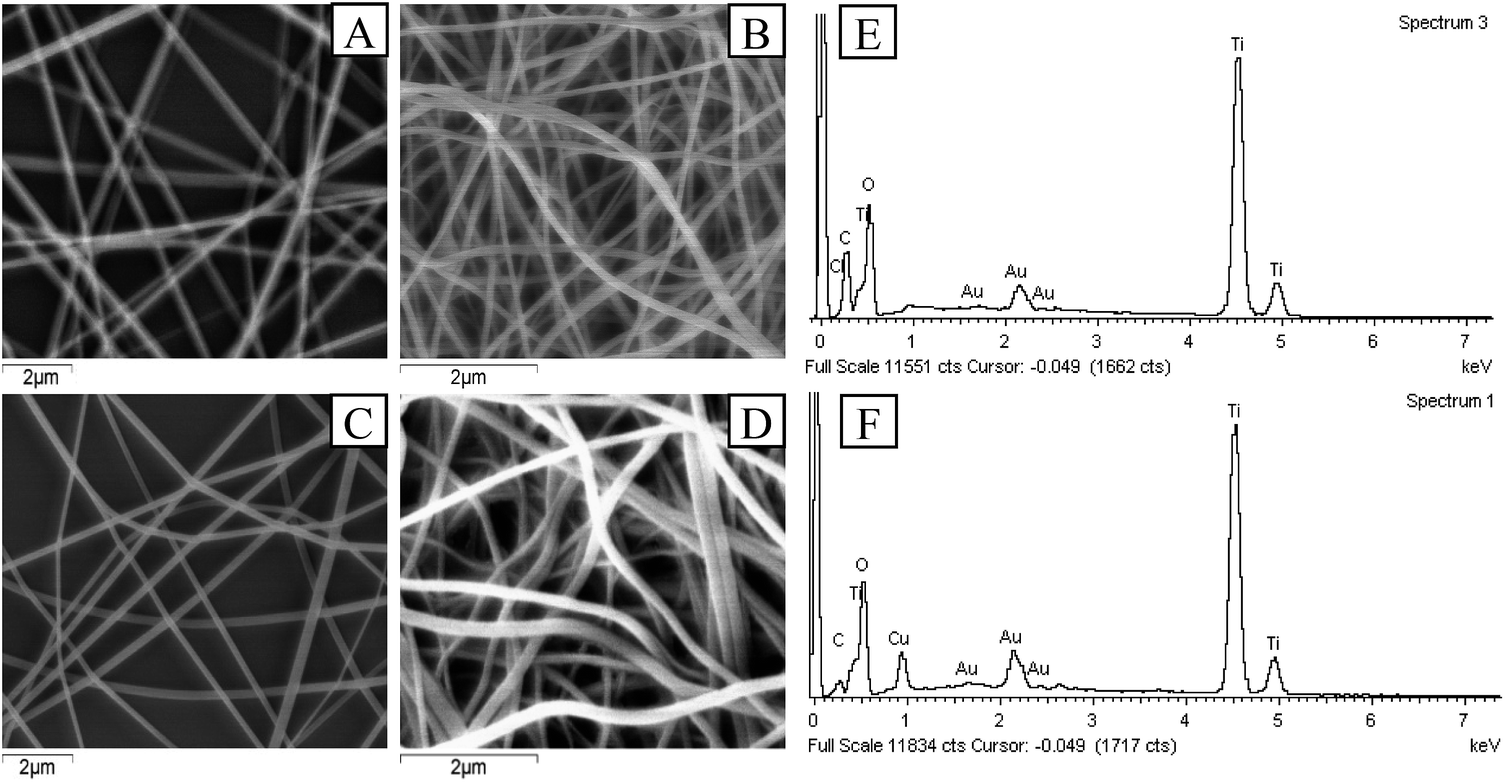
The Cu–TiO2 composite nanofibers were synthesized under the optimum condition (n(Cu):n(Ti) = 1:8, calcination temperature = 450 °C, TBOT dosage in precursor solution = 2 g) for further characterization. In the first place, SEM was applied to investigating the morphology of the nanofibers. As shown in Fig. 7, after calcination, both of the diameters of TiO2 and Cu–TiO2 decreased from over 200 nm to about 140 nm, and all nanofibers maintained a good one dimensional structure. The thermal decomposition of PVP was responsible for the decrease of fiber diameters.27 The melting point of PVP was about 150 °C, which was far below the calcination temperature (450 °C). In addition, it was observed that the surfaces of TiO2 and Cu–TiO2 nanofibers became rough after calcination, which might be caused by the crystallization of TiO2.39 The rough surfaces were reported to be conductive to improve photocatalytic activity.26 It could also be observed that there were no significant differences in the morphology and diameter between TiO2 and Cu–TiO2 nanofibers. EDS spectrums of TiO2 and Cu–TiO2 nanofibers were shown in Fig. 7(e) and (f). The presence of Ti, O, Au and C elements were observed in TiO2 nanofibers, and the mole ratio of O and Ti was 2.3:1, which was a possible evidence for the formation of TiO2. Au element was derived from the application of gold film in the pre-treatment process of the samples. C element was possibly due to the residue of PVP after calcination. In the EDS spectrum of Cu–TiO2 nanofibers, Cu element was observed at 0.93 keV in addition to Ti, O, Au and C elements, and the mole ratio of Cu and Ti was around 1:7, which was close to the designed ratio (n(Cu):n(Ti) = 1:8). It could be illustrated that Cu element was successfully introduced into TiO2 nanofibers.
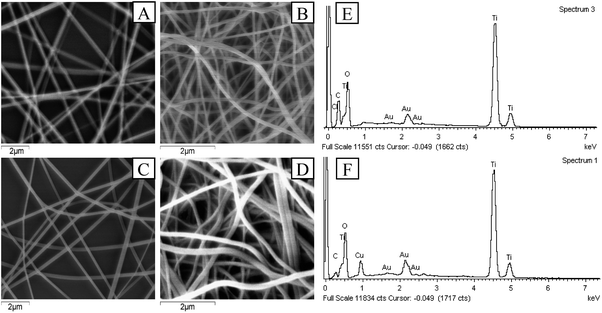 |
| | Fig. 7 SEM images of (A) TiO2 before calcination, (B) TiO2 after calcination, (C) Cu–TiO2 before calcination, (D) Cu–TiO2 after calcination; EDS spectrums of (E) TiO2 nanofibers and (F) Cu–TiO2 nanofibers. | |
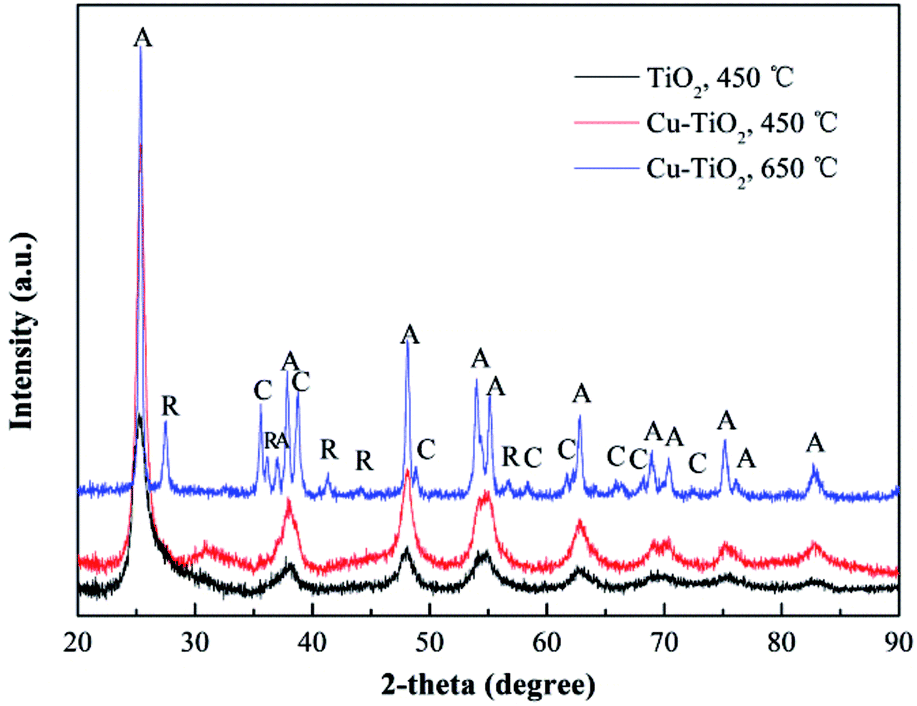
XRD spectrums were used to study the phase structures of TiO2 and Cu–TiO2 nanofibers. As shown in Fig. 8, for both of the nanofibers at the calcination temperature of 450 °C, the diffraction peaks at 2θ of 25.3°, 37.9°, 47.9°, 54.0°, 62.6°, 68.8°, 70.2°, 75.0°, 83.0° were correspond to the (101), (103), (200), (105), (213), (116), (220), (107), (303) crystal planes of the anatase TiO2 phase. The rutile TiO2 phase was not presented. Compared to pure TiO2, the diffraction peaks of Cu–TiO2 at 450 °C were enhanced, indicating that doping Cu into TiO2 promoted the formation of anatase TiO2. The diffraction peaks of Cu or Cu oxides in Cu–TiO2 were not observed at 450 °C, which was consistent with the research conducted by Lin et al.40 The possible reason may be that Cu is highly dispersed within TiO2 crystal lattice or that the content of Cu or Cu oxides is below the detection limit of XRD. However, the diffraction peaks of CuO were observed at 650 °C, ruling out the latter possibility. Therefore, it could be inferred that Cu was successfully doped into the crystal lattice of TiO2 at 450 °C.41 In addition, a slight shift in the peaks of Cu–TiO2 calcined at 450 °C compared to pure TiO2 was observed, which was also an evidence of the incorporation of Cu ions into the TiO2 lattice.41
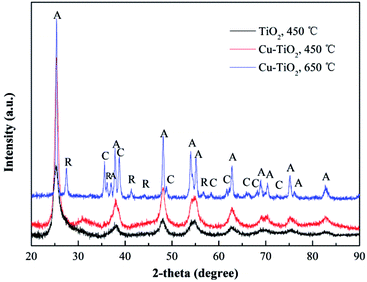 |
| | Fig. 8 XRD patterns of TiO2 and Cu–TiO2 nanofibers. n(Cu):n(Ti) = 1:8. A: anatase, R: rutile, C: CuO. | |
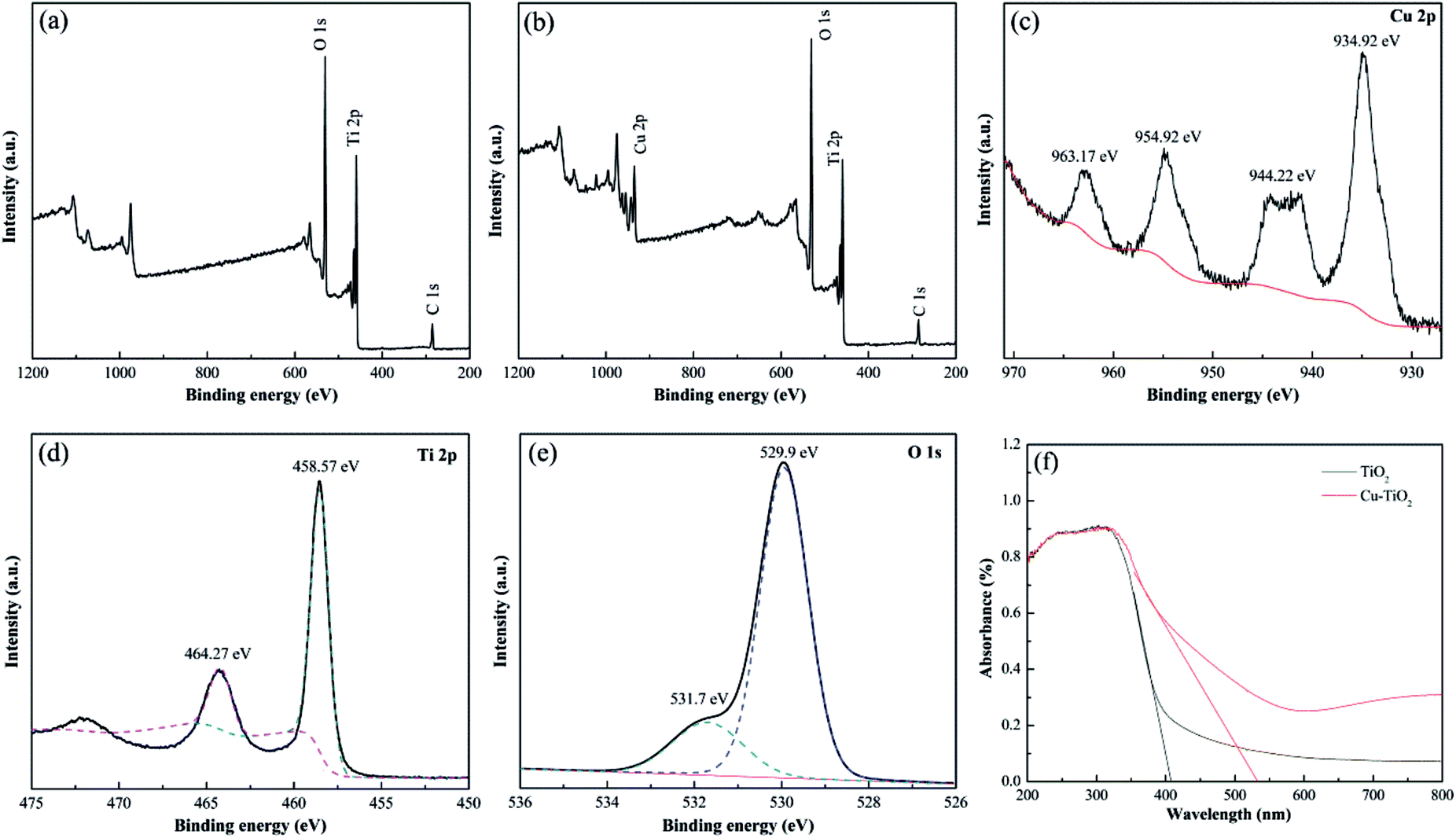
The chemical states of the elements in the calcinated nanofibers were determined by XPS analysis. C 1s peak at binding energy of 284.6 eV was applied as the reference for calibration. The XPS survey spectrum shown in Fig. 9(b) confirmed the introduction of Cu element in Cu–TiO2 nanofibers compared to pure TiO2 nanofibers (Fig. 9(a)), which was in accordance with the results of EDS. High resolution XPS spectrums for Cu 2p, Ti 2p and O 1s of Cu–TiO2 nanofibers were shown in Fig. 9(c)–(e). The peaks at binding energy of 934.92 eV and 954.92 eV were respectively correspond to Cu 2p3/2 and Cu 2p1/2 (Fig. 9(c)), indicating that Cu existed as Cu2+ in Cu–TiO2 nanofibers.31 The shakeup satellite peaks at 944.22 eV and 963.17 eV ruled out the existence of Cu+, further demonstrating that the only oxidation state of Cu is +2. The peaks around binding energy of 458.57 eV and 464.27 eV represented Ti 2p3/2 and Ti 2p1/2 respectively (Fig. 9(d)), which was correspond to the tetragonal crystal structure of Ti4+. Meanwhile, the asymmetry of O 1s spectrum illustrated that at least two kinds of binding state of oxygen were existed in Cu–TiO2 nanofibers (Fig. 9(e)). The peak at 529.97 eV was associated with the lattice oxygen in Cu–TiO2 nanofibers.42 The peak at higher binding energy of 531.7 eV was indicative of surface contamination by hydroxides from the atmosphere.43 According to the results of XRD and XPS, it could be inferred that Cu2+ penetrated into the TiO2 lattice and substituted the Ti4+, forming new Cu–O–Ti bonds in Cu–TiO2 nanofibers.
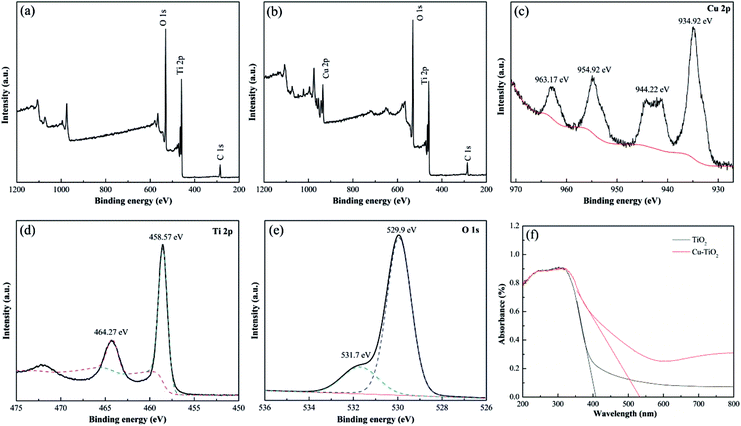 |
| | Fig. 9 XPS survey spectrums for (a) TiO2 nanofibers and (b) Cu–TiO2 nanofibers; high resolution XPS spectrums of Cu–TiO2 nanofibers for (c) Cu 2p, (d) Ti 2p, (e) O 1s; (f) UV-vis absorption spectrums of TiO2 and Cu–TiO2 nanofibers. | |
The UV-vis absorption spectrums of TiO2 and Cu–TiO2 nanofibers were displayed in Fig. 9(f). For pure TiO2 nanofibers, characteristic peak attributed to adsorption of UV irradiation were obvious from 200 to 400 nm. However, there was no absorption peak at 400–800 nm, indicating that pure TiO2 cannot absorb visible light irradiation. For Cu–TiO2 nanofibers, the absorption intensity of UV irradiation was increased compared to pure TiO2 nanofibers, indicating that doping Cu into TiO2 improved the utilization ratio of UV irradiation. In addition, the characteristic peak of Cu–TiO2 was obviously extended to the visible light region, resulting in the red shift of absorption spectrum. It was calculated by Kubelkae–Munk function that the band gaps of pure TiO2 and Cu–TiO2 nanofibers were 3.01 eV and 2.30 eV respectively. This illustrated that Cu–TiO2 nanofibers were promising photocatalysts under visible light.
Mechanism of virus inactivation by Cu–TiO2 nanofibers
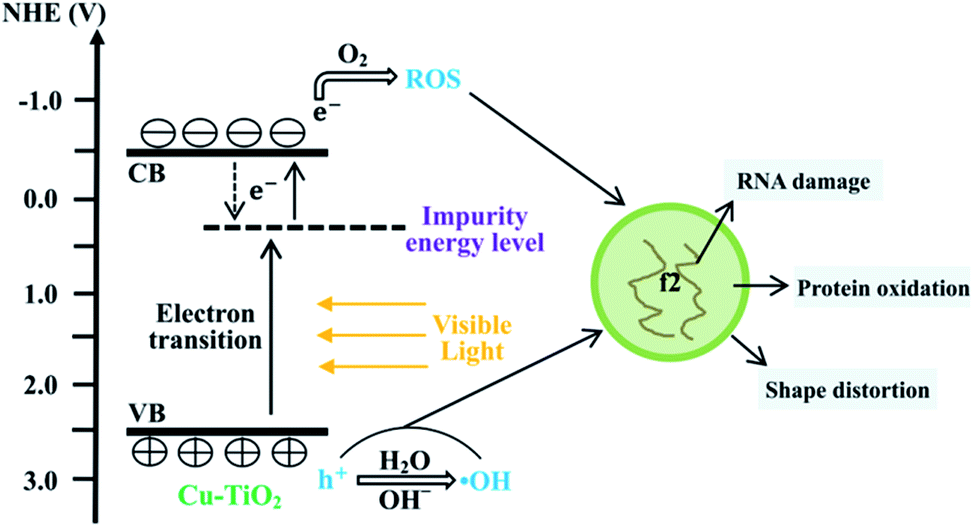
The first step of the virus inactivation process is the adsorption of bacteriophage f2 onto the surfaces of Cu–TiO2 nanofibers. The SEM images (Fig. 7) showed that the Cu–TiO2 nanofibers maintained a good one dimensional structure and their surfaces became rough after calcination, which was conductive to the adsorption process, ensuring desirable disinfection efficiency consequently. Pure TiO2 nanofibers were not able to inactivate bacteriophage f2 under visible light (Fig. 3), since its bind gap was up to 3.01 eV as we calculated (Fig. 9(f)), which was too high to be excited by visible light. Optimized Cu–TiO2 nanofibers achieved 100% inactivation of bacteriophage f2 in 4 h under visible light, since the introduction of Cu2+ successfully narrowed the bind gap of Cu–TiO2 nanofibers to 2.30 eV (Fig. 9(f)), which could be activated by visible light. According to the XRD and XPS results, the doped Cu2+ substituted Ti4+ in the crystal lattice of TiO2. This resulted in the formation of an impurity energy level below the conduction band of TiO2.14 As shown in Fig. 10, under visible light irradiation, the electrons in TiO2 were excited from its valence band (VB) to the impurity energy level first and then transferred to the conduction band (CB). Part of the electrons in CB would transfer back to the impurity energy level due to potential difference while the others took part in the reaction with surface adsorbed O2 to produce ˙O2− (eqn (3)). Subsequently, ˙O2− underwent facile disproportionation to produce ˙OH, H2O2 and 1O2 (eqn (4)–(6)).44 The separated holes in the VB of TiO2 were positive enough to oxidize H2O/OH− into ˙OH (eqn (7) and (8)) as well as attack and oxidize the viruses directly.45 Generally, holes and ROS generated from electrons and holes contributed to the inactivation of bacteriophage f2 collectively (eqn (9)). Furthermore, the oxidative holes and ROS tended to distort the viral particle shapes and damage the surface proteins primarily, and then destroy the major types of RNA genes, resulting in definite viral death.38| | |
TiO2 + hν (λ > 400 nm) → TiO2 + e− + h+
| (2) |
| | |
˙O2− + H2O → 1O2 + OH−
| (4) |
| | |
1O2 + 1O2 → H2O2 + O2
| (5) |
| | |
H2O2 + e− → ˙OH + OH−
| (6) |
| | |
ROS (˙O2−, 1O2, H2O2, ˙OH) + h+ + f2 → organic debris of virus
| (9) |
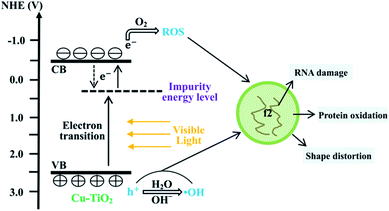 |
| | Fig. 10 Sketch map of proposed mechanism for viral inactivation by Cu–TiO2 nanofibers under visible light irradiation. | |
Conclusions
Cu–TiO2 nanofibers were prepared with electrospinning method and exhibited excellent antiviral performance under visible light. The optimum doping ratio and calcination temperature were n(Cu):n(Ti) = 1:8 and 450 °C respectively. Cu–TiO2 nanofibers with various diameters (55–146 nm) didn't show obvious differences in virus removal efficiency. Characterization of Cu–TiO2 nanofibers demonstrated that Cu2+ substituted Ti4+ in the TiO2 lattice and formed an impurity energy level below the conduction band of TiO2, which enabled the nanofibers to realize visible light response. This study provides a promising technology for viral disinfection in drinking water.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The study was supported by Beijing Natural Science Foundation (8164054) and the Special Funds of the Construction of World-class Universities (Disciplines) and Guidance of Characteristic Developments for the Central Universities (Renmin University of China, 2017), which are greatly acknowledged.
Notes and references
- M. Khraisheh, L. Wu, A. H. Ala and M. A. Al-Ghouti, J. Ind. Eng. Chem., 2015, 28, 369–376 CrossRef CAS.
- S. Pedley and K. Pond, Emerging issues in water and infectious disease, World Health Organization, 2003 Search PubMed.
- J. L. Liang, E. J. Dziuban, G. F. Craun, V. Hill, M. R. Moore, R. J. Gelting, R. L. Calderon, M. J. Beach and S. L. Roy, MMWR Surveillance Summaries, 2006, 55, 31–65 Search PubMed.
- S. E. Hrudey, Water Res., 2009, 43, 2057–2092 CrossRef CAS PubMed.
- R. Sadiq and M. J. Rodriguez, Sci. Total Environ., 2004, 321, 21–46 CrossRef CAS PubMed.
- J. Sohn, G. Amy, J. Cho, Y. Lee and Y. Yoon, Water Res., 2004, 38, 2461–2478 CrossRef CAS PubMed.
- R. A. Rodriguez, S. Bounty, S. Beck, C. Chan, C. McGuire and K. G. Linden, Water Res., 2014, 55, 143–149 CrossRef CAS PubMed.
- M. Guo, J. Huang, H. Hu, W. Liu and J. Yang, Water Res., 2012, 46, 4031–4036 CrossRef CAS PubMed.
- G. Shin and M. D. Sobsey, Appl. Environ. Microbiol., 2003, 69, 3975–3978 CrossRef CAS PubMed.
- V. Lazarova, P. Liechti, P. Savoye and R. Hausler, J. Water Reuse Desalin., 2013, 3, 337–345 CrossRef CAS.
- J. G. Jacangelo, S. S. Adham and J. Laîné, J. - Am. Water Works Assoc., 1995, 87, 107–121 CAS.
- S. S. Madaeni, A. G. Fane and G. S. Grohmann, J. Membr. Sci., 1995, 102, 65–75 CrossRef CAS.
- W. Wang, G. Huang, C. Y. Jimmy and P. K. Wong, J. Environ. Sci., 2015, 34, 232–247 CrossRef PubMed.
- S. Malato, P. Fernández-Ibáñez, M. I. Maldonado, J. Blanco and W. Gernjak, Catal. Today, 2009, 147, 1–59 CrossRef CAS.
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- C. McCullagh, J. M. Robertson, D. W. Bahnemann and P. K. Robertson, Res. Chem. Intermed., 2007, 33, 359–375 CrossRef CAS.
- S. G. Kumar and L. G. Devi, J. Phys. Chem. A, 2011, 115, 13211–13241 CrossRef CAS PubMed.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. Dunlop, J. W. Hamilton, J. A. Byrne and K. O'Shea, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- C. Karunakaran, G. Abiramasundari, P. Gomathisankar, G. Manikandan and V. Anandi, J. Colloid Interface Sci., 2010, 352, 68–74 CrossRef CAS PubMed.
- B. Wu, R. Huang, M. Sahu, X. Feng, P. Biswas and Y. J. Tang, Sci. Total Environ., 2010, 408, 1755–1758 CrossRef CAS PubMed.
- S. K. Choi, S. Kim, S. K. Lim and H. Park, J. Phys. Chem. C, 2010, 114, 16475–16480 CAS.
- D. Li, J. T. McCann, Y. Xia and M. Marquez, J. Am. Ceram. Soc., 2006, 89, 1861–1869 CrossRef CAS.
- A. Yousef, N. A. Barakat and H. Y. Kim, Appl. Catal., A, 2013, 467, 98–106 CrossRef CAS.
- S. S. Lee, H. Bai, Z. Liu and D. D. Sun, Water Res., 2013, 47, 4059–4073 CrossRef CAS PubMed.
- S. S. Lee, H. Bai, Z. Liu and D. D. Sun, Appl. Catal., B, 2013, 140, 68–81 CrossRef.
- A. Yousef, N. A. Barakat, T. Amna, S. S. Al-Deyab, M. S. Hassan, A. Abdel-hay and H. Y. Kim, Ceram. Int., 2012, 6, 4525–4532 CrossRef.
- F. Chen, X. Yang, H. K. Mak and D. W. Chan, Build Environ., 2010, 45, 1747–1754 CrossRef.
- R. Cheng, G. Li, C. Cheng, P. Liu, L. Shi, Z. Ma and X. Zheng, Chem. Eng. J., 2014, 252, 150–158 CrossRef CAS.
- S. Qin, F. Xin, Y. Liu, X. Yin and W. Ma, J. Colloid Interface Sci., 2011, 356, 257–261 CrossRef CAS PubMed.
- T. Pham and B. Lee, Appl. Surf. Sci., 2014, 296, 15–23 CrossRef CAS.
- W. Chen, V. Jovic, D. Sun-Waterhouse, H. Idriss and G. I. Waterhouse, Int. J. Hydrogen Energy, 2013, 38, 15036–15048 CrossRef CAS.
- J. Wu, C. Li, X. Zhao, Q. Wu, X. Qi, X. Chen, T. Hu and Y. Cao, Appl. Catal., B, 2015, 176, 559–569 CrossRef.
- P. Khemthong, P. Photai and N. Grisdanurak, Int. J. Hydrogen Energy, 2013, 38, 15992–16001 CrossRef CAS.
- J. Yu, H. Yu, B. Cheng and C. Trapalis, J. Mol. Catal. A: Chem., 2006, 249, 135–142 CrossRef CAS.
- D. J. Kim, S. H. Hahn, S. H. Oh and E. J. Kim, Mater. Lett., 2002, 57, 355–360 CrossRef.
- X. Zong, K. Kim, D. Fang, S. Ran, B. S. Hsiao and B. Chu, Polymer, 2002, 43, 4403–4412 CrossRef CAS.
- Y. Li, C. Zhang, D. Shuai, S. Naraginti, D. Wang and W. Zhang, Water Res., 2016, 106, 249–258 CrossRef CAS PubMed.
- J. Lee, S. Divya, P. Shanmugasundaram and P. I. Gouma, J. Nanoeng. Nanomanuf., 2014, 4, 140–145 CrossRef CAS.
- C. Lin and W. Yang, Chem. Eng. J., 2014, 237, 131–137 CrossRef CAS.
- X. Yang, W. Shu, H. Sun, X. Wang and J. Lian, Trans. Nonferrous Met. Soc. China, 2015, 25, 504–509 CrossRef CAS.
- L. Zhu, M. Hong and G. W. Ho, Nano Energy, 2015, 11, 28–37 CrossRef CAS.
- S. Xu, A. J. Du, J. Liu, J. Ng and D. D. Sun, Int. J. Hydrogen Energy, 2011, 36, 6560–6568 CrossRef CAS.
- C. A. Castro, P. Osorio, A. Sienkiewicz, C. Pulgarin, A. Centeno and S. A. Giraldo, J. Hazard. Mater., 2012, 211, 172–181 CrossRef PubMed.
- D. Xia, T. An, G. Li, W. Wang, H. Zhao and P. K. Wong, Water Res., 2016, 99, 149–161 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Jing Dongb
*a and
Jing Dongb