DOI:
10.1039/C7RA08835C
(Paper)
RSC Adv., 2017,
7, 52180-52186
Design, synthesis and biological evaluation of 2,3-dihydroimidazo[1,2-c]quinazoline derivatives as novel phosphatidylinositol 3-kinase and histone deacetylase dual inhibitors†
Received
10th August 2017
, Accepted 5th November 2017
First published on 10th November 2017
Abstract
Histone deacetylase (HDAC) inhibitors are known to induce multiple epigenetic modifications affecting signaling networks and act synergistically with phosphatidylinositol 3-kinase (PI3K) inhibitors for the treatment of cancer. Herein we present a novel design approach for cancer drug development by incorporating HDAC inhibitory functionality into a PI3K inhibitor pharmacophore to construct dual-acting inhibitors. The designed compounds were synthesized and showed inhibitory activities against PI3K and HDAC. The representative dual PI3K/HDAC inhibitors, compounds 12a–j, showed potent antiproliferative activities against K562 and Hut78 in cellular assays. This work may lay the foundation for developing novel dual PI3K/HDAC inhibitors as potential anticancer therapeutics.
Introduction
The phosphoinositide 3-kinase (PI3K) family of lipid kinases generate 3′-phosphoinositides that activate a variety of cellular targets important for cell proliferation, survival, differentiation, and migration.1,2 Collectively, PI3Ks act as intermediate signaling molecules and activate the serine–threonine kinase AKT and other downstream effectors.3 Based on their primary structure and mechanism of action, PI3Ks are divided into three major classes: class I, II, and III.4–6 Class I PI3K is composed of a catalytic subunit, p110, and a regulatory subunit, p85 or p101. The catalytic subunit consists of four isoforms: p110α, β, γ, δ.7 In many human cancers, the PIK3CA gene that encodes p110α is also frequently mutated. Thus class I PI3Ks, particularly p110α, are potential therapeutic targets for cancers.8,9 Over the past decade, researchers have made significant progress in developing pan-PI3K or isoform-specific PI3K inhibitors, such as pictilisib, GSK1059615, PI-103 and copanlisib, which have shown promising activity against various types of cancers and most are currently under clinical investigation (Fig. 1).10 Unfortunately, numerous studies reported that the efficacy of PI3K inhibitors is limited by concurrent activation of other survival- and growth-related pathways.11,12
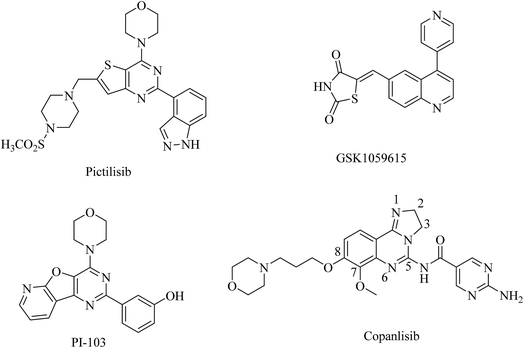 |
| | Fig. 1 Representative structures of PI3K inhibitors. | |
In addition to genetic mutations, epigenetic changes, such as dysregulation of histone deacetylases (HDAC), contribute to cancer cell initiation and growth, by altering the cell phenotype and gene expression and by disturbing homeostasis.13 Thus, HDAC inhibitors are emerging drugs for cancer therapeutics.14 Extensive efforts have resulted in the identification of many potent HDAC inhibitors, including vorinostat (SAHA), belinostat (PXD-101), and panobinostat (LBH-589), which have been approved by the FDA, respectively, to treat cutaneous T-cell lymphoma (CTCL), peripheral T-cell lymphoma (PTCL), and multiple myeloma (MM) (Fig. 2). Several small molecule HDAC inhibitors have entered clinical trials for the treatment of a variety of haematological and solid tumors.15
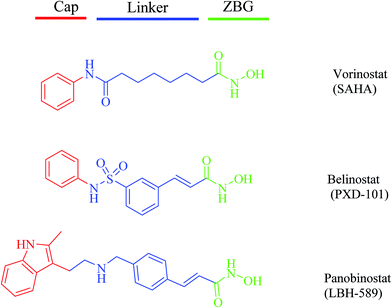 |
| | Fig. 2 Representative structures of HDAC inhibitors. | |
Recently, it has been reported that the combination of an HDAC inhibitor and a kinase inhibitor, such as epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor or PI3K inhibitor, can overcome kinase inhibitor resistance and induce apoptosis in human solid cancers in a synergistic manner.16–18 Therefore, development of inhibitors targeting both HDAC and PI3K would be advantageous as anticancer drug candidates. Indeed, a dual inhibitor of HDAC and PI3K, CUDC-907, is currently under evaluation in phase I clinical trials in patients with lymphoma or multiple myeloma, and in patients with advanced solid tumors.19,20 Another HDAC inhibitor, FK228, has been also demonstrated to directly inhibit PI3K activity and potently induce apoptosis through HDAC/PI3K dual inhibition (Fig. 3).21 The evidence has suggested that simultaneous PI3K and HDAC inhibition could be a promising approach in cancer therapy.
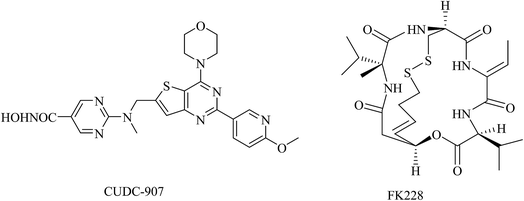 |
| | Fig. 3 Representative structures of PI3K/HDAC dual inhibitors. | |
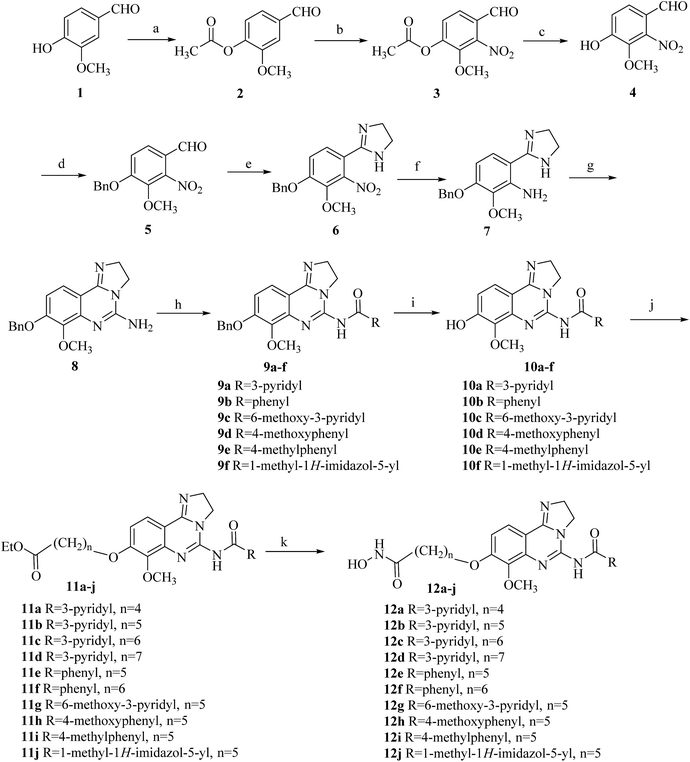
In this report, we designed and synthesized a novel series of dual-acting PI3K and HDAC inhibitors by incorporating HDAC inhibitory functionality into a PI3K inhibitor pharmacophore.22 The rationale for the dual-inhibitor design originated from our deep insights into previously reported X-ray crystal structure of copanlisib bound to PI3K (Fig. 4A) and vorinostat bound to a HDAC homolog (Fig. 4B).23,24 The imidazoline N1 nitrogen of copanlisib forms a critical hydrogen bond to V882 in the adenine pocket so as to possess strong hinge interactions. The C5 aminopyrimidine group fills the affinity pocket, forming hydrogen bonds with D836 and D841 through the amino group, and with K833 through a pyrimidine nitrogen. In addition, D964 appears to contribute a π-stacking interaction. Finally, the morpholine group of copanlisib lies over W812, and points to solvent. The long alkyl chain of vorinostat is located in a long narrow tubular channel. Hydroxamate forms three hydrogen bonds with Y297, H131 and H132, respectively. The capping phenyl group binds to the hydrophobic part on the protein surface. It is well established that the pharmacophore of HDAC inhibitors (Fig. 2) consists of a capping group, an appropriate linker and a zinc-binding group (ZBG). Generally, ZBG plays a significant role in the binding efficiency between HDAC inhibitors and enzyme. Hydroxamate is one of the most potent zinc ion chelating group among all types of ZBGs. Given the known binding modes of the PI3K inhibitor copanlisib and HDAC inhibitor vorinostat, it was envisaged that amalgamation of these pharmacophores would be a viable strategy to single molecules retaining both activities. We postulated that introduction of the linker and ZBG of vorinostat to the position C8 of copanlisib would be compatible with HDAC inhibition since that the channel pointing toward solvent of PI3K and the tubular channel of HDAC share considerable similarities (Fig. 4). The linker and ZBG of vorinostat may direct into the solvent to make it well occupied which may increase the affinity to PI3K. In the meantime, 2,3-dihydroimidazo[1,2-c]-quinazoline moiety of copanlisib may bind to the HDAC surface as the surface recognition capping group. The fused molecules were expected to retain the essential interactions with both proteins to exert desired biological functions.
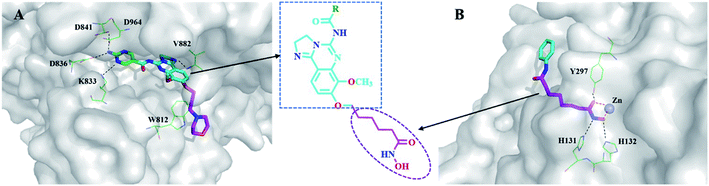 |
| | Fig. 4 Design of dual inhibitors of PI3K/HDAC. (A) X-ray crystal structures of copanlisib bound to PI3K (PDB entry: 5G2N), (B) vorinostat docked to an HDAC1 homolog (PDB entry: 1C3S). The proteins are shown as white surface or cartoon. Copanlisib and vorinostat are shown in sticks. The hydrogen bonds were denoted by black dash lines. Figures were prepared using PyMOL. | |
Results and discussion
Chemistry
The synthetic route of 12a–12j were outlined in Scheme 1.23,25 Vanillin was protected with acetate group, then was nitrated with fuming nitric acid to readily afford aldehyde 3. The acetate group of compound 3 was removed to provide phenol 4. Conversion into benzyl ether 5 followed ethanediamine/iodine oxidation gave the ring of 4,5-dihydro-1H-imidazole 6. Reduction provided aniline 7, and treatment with cyanogen bromide to give tricyclic amine 8, which was acylated using a variety of different acid chlorides to provide amide 9a–9f. Treatment of amide 9a–9f with trifluoroacetic acid afforded deprotected phenol 10a–10f, which could then be alkylated at C8 giving access to the aryl ethers 11a–11j. Finally, the ethyl ester groups of compounds 11a–11j were treated with freshly prepared hydroxylamine in methanol to produce target compounds 12a–12j.
 |
| | Scheme 1 Reagent and conditions: (a) (CH3CO)2O, Et3N, DMAP, rt, 3 h; (b) fuming HNO3, <5 °C, 2 h; (c) KOH, H2O, 100 °C, 15 min; (d) BnBr, K2CO3, DMF, rt, 4 h; (e) NH2CH2CH2NH2, I2, K2CO3, (CH3)3COH, 85 °C, 4 h; (f) Fe, AcOH, H2O, rt, 24 h; (g) BrCN, TEA, CH2Cl2, 0 °C to rt, 4 h; (h) RCOCl, Et3N, CH2Cl2, 0 °C to rt; (i) TFA, 60 °C, 5 h; (j) Br(CH2)nCO2Et, Cs2CO3, DMF, 80 °C, 2 h; (k) NH2OH·HCl, KOH, MeOH, rt. | |
Biological assays
In vitro HDAC and PI3K inhibition. To study the biological activity, compounds 12a–12j were evaluated against HDAC1 enzyme assay with vorinostat as a positive control and PI3Kα activity assay with copanlisib as a positive control using the fluorimetric activity assay.22,26 As shown in Table 1, most compounds exhibited moderate to good inhibitory activity against both PI3Kα and HDAC1. The length of the hydroxamic acid side chains of this series have marginal effects on HDAC1 inhibition (12b > 12c > 12d > 12a) and the optimal carbon chain length is five or six carbon atoms, such as 12b (IC50 = 107 nM) and 12c (IC50 = 86 nM), 12e (IC50 = 50 nM) and 12f (IC50 = 91 nM). Diversity in the length of side chains are tolerable to PI3K inhibition due to the long channel pointing to solvent of protein. These results illustrated that the linker between the hydroxamic acid side chain and the 2,3-dihydroimidazo[1,2-c]quinazoline skeleton has relatively significant effect on HDAC1 inhibition, but has little effect on PI3Kα protency.
Table 1 IC50 values of compounds 12a–12j against HDAC1 and PI3Kαa
Variation of the aromatic C5 amides moiety had significant effect on p110α potency (Table 1). Nicotinamide and substituted nicotinamide moiety exhibited almost the similar inhibitory activities against PI3K to copanlisib. Benzyl or substituted benzyl amides, such as 12e (IC50 = 2.89 nM) and 12f (IC50 = 3.33 nM), were slightly less active than compounds 12b (IC50 = 1.35 nM) and 12c (IC50 = 1.72 nM) with pyridine moiety. The same trends were observed in compounds 12g and 12h. This decreased activity may be due to the inability of these amides to act as hydrogen-bond acceptors. In addition, the five-membered heterocyclic amide 12j were significantly less active than six-membered aryl amides, such as 12b and 12e. This result may suggest that six-membered aromatic cycles are preferred to fit in the affinity pocket.
In order to further ascertain the pan-inhibition of HDAC enzymes and class I PI3K kinases, three representative compounds 12b, 12c and 12e with better HDAC1 and PI3Kα inhibition were selected and evaluated against recombinant human HDAC1, HDAC6, HDAC8 enzymes and PI3Kα, β, γ, δ, using vorinostat and copanlisib as the positive control compound. As shown on Table 2, it is noteworthy that this set of structures show promising pan-inhibition for HDACs and PI3Ks.
Table 2 Inhibition of HDAC and PI3K isoforms by compound 12b, 12c and 12ea
| Compd. |
IC50b (nM) |
| HDAC1 |
HDAC6 |
HDAC8 |
PI3Kα |
PI3Kβ |
PI3Kγ |
PI3Kδ |
| ND: not determined. Values were the average of three experiments, SD < 10%. |
| 12b |
107 |
97 |
327 |
1.35 |
10.2 |
19.3 |
1.97 |
| 12c |
86 |
162 |
412 |
1.72 |
9.6 |
15.8 |
2.02 |
| 12e |
50 |
62 |
153 |
2.98 |
14.2 |
29.5 |
4.52 |
| Vorinostat |
52 |
43 |
187 |
ND |
ND |
ND |
ND |
| Copanlisib |
ND |
ND |
ND |
1.42 |
9.12 |
16.3 |
1.72 |
In vitro cell growth inhibition. To test the anticancer activities of the synthesized compounds, we evaluated antiproliferative activities of representative compounds 12b, 12c, 12e, 12h and 12i against human colon cancer cell line (HCT116), human leukemia cell line (K562), and cutaneous T lymphocyte (Hut78) by applying the MTT colorimetric assay. The IC50 values were summarized on Table 3. According to the inhibition data, all tested compounds showed obvious anti-proliferative activities. Consistent with this observation of HDAC and PI3K inhibition, we found that 12b, 12c and 12e potently inhibits the growth of those cancer cells derived from both hematologic and solid tumors. Especially, compound 12e exhibited the better anti-proliferative activity in K562 and Hut78 cell lines than vorinostat and copanlisib.
Table 3 Antiproliferative activities of compounds 12b, 12c, 12e, 12h and 12i
| Compd. |
IC50a (μM) |
| HCT116 |
K562 |
Hut78 |
| Values were the average of two experiments, SD < 10%. |
| 12b |
0.52 |
0.32 |
0.17 |
| 12c |
0.41 |
0.48 |
0.21 |
| 12e |
0.33 |
0.095 |
0.062 |
| 12h |
0.89 |
0.77 |
0.51 |
| 12i |
0.64 |
0.53 |
0.15 |
| Vorinostat |
1.17 |
0.21 |
0.092 |
| Copanlisib |
0.26 |
0.73 |
0.12 |
Molecular docking studies
For further understanding of the interaction between these inhibitors and proteins (HDAC1 and PI3K) and guiding the structure–activity relationships, a molecular docking was performed using the C-DOCKER program within Discovery Studio 2.5 software package. We docked the representative compound 12c and 12e, the most potent compound of this series, in the active site of PI3K (PDB code: 5G2N) (Fig. 5) and HDAC homolog (PDB code: 1C3R) (Fig. 6), revealing excellent shape complementarity between ligand and the binding pocket. As shown in Fig. 5A and B, compound 12c and 12e have the binding mode similar to that of copanlisib as we expected. 2,3-Dihydroimidazo[1,2-c]quinazoline moiety occupied the ATP binding pocket. And the imidazoline N1 nitrogen forms a critical hydrogen bond to Val882 in the protein hinge region. The morpholine group lies over Trp812, apparently adding a hydrophilic shield to the hydrophobic heterocycle. The C5 aryl amide group appears to contribute a π-stacking interaction on Asp964. The C5 pyrimidine group of compound 12c can form a hydrogen bond with Lys833 through a pyrimidine nitrogen, but the compound 12e loses the hydrogen. This could be the reason that the compound 12e has a slightly lower activity against PI3K than compound 12b and 12c. In addition, the long alkyl chain at C8 of compound 12c and 12e just fits into the channel pointing toward solvent, and hydroxamate forms three or four hydrogen bonds with the residues located at the solvent. The docking mode of 12c and 12e in complex with HDAC reveals that this compound binds in the HDAC pocket in a similar manner and forms the same interactions as vorinostat (Fig. 6A and B). The hydroxamic acid group anchors 12c and 12e into the active site by chelating the essential catalytic zinc ion. Hydroxamate forms three hydrogen bonds with Y297, H131 and H132, respectively. The long alkyl chain of 12c and 12e exits the reaction centre along the long narrow tubular channel. 2,3-Dihydroimidazo[1,2-c]quinazoline moiety was placed in an open environment of the protein surface and projects outward into the solvent, corresponding to the fact that the SARs of the HDACs inhibitors do not strongly depend on variations in this region.
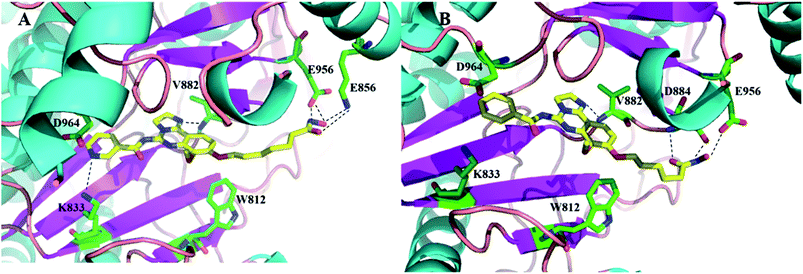 |
| | Fig. 5 The docking model of compounds 12c (A) and 12e (B) bound to PI3K (PDB entry 5G2N). 12c and 12e are represented by tube and colored by atom type (C, yellow; N, blue; O, red). The hydrogen bonds are denoted by black dash lines. The protein is shown as ribbons. The key amino acids forming the pocket are represented by tube with carbon atoms colored in green, oxygen atoms in red and nitrogen atoms in blue. Figures were prepared using PyMOL. | |
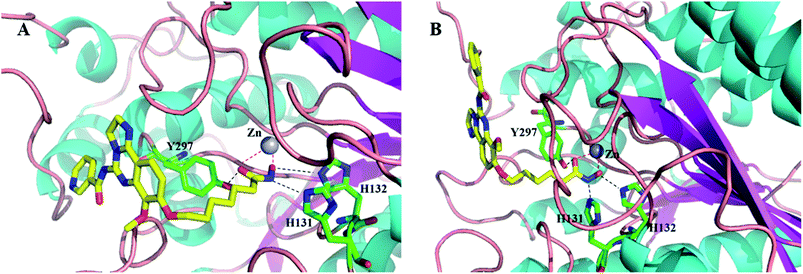 |
| | Fig. 6 The docking model of compounds 12c (A) and 12e (B) bound to HDAC1 (PDB entry 1C3S). 12c and 12e are represented by tube and colored by atom type (C, yellow; N, blue; O, red). The hydrogen bonds are denoted by black dash lines and chelation is shown by red dash lines. The protein is shown as ribbons. The key amino acids forming the pocket are represented by tube with carbon atoms colored in green, oxygen atoms in red and nitrogen atoms in blue and zinc in grey. Figures were prepared using PyMOL. | |
Conclusions
In summary, using structure-based design approach, we have successfully designed and synthesized a novel series of 2,3-dihydroimidazo [1,2-c]quinazoline with a hydroxamate group essential for chelation with the zinc ion in the active site of HDAC as novel PI3K/HDAC dual inhibitors. As expected, most of compounds exhibited distinct high to moderate inhibitory activities against both HDAC1 and PI3Kα. Moreover, the selectivity assay of compounds 12b, 12c and 12e shows that they are excellent pan inhibitors for HDACs and PI3Ks. In addition, compound 12e showed the most potent inhibitory activity against K562 and Hut78. By combining two distinct pharmacophores into one molecule, we have demonstrated the example of PI3K/HDAC dual inhibitors as a promising approach to search for efficient anticancer multi-target agents.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 81673301) and Fundamental Research Funds for the Central Universities (Grant No. 3010050069).
Notes and references
- L. C. Cantley, Science, 2002, 296, 1655–1657 CrossRef CAS PubMed.
- M. Osaki, M. Oshimura and H. Ito, Apoptosis, 2004, 9, 667–676 CrossRef CAS PubMed.
- P. Liu, H. Cheng, T. M. Roberts and J. J. Zhao, Nat. Rev. Drug Discovery, 2009, 8, 627–644 CrossRef CAS PubMed.
- M. Hayakawa, K. Kawaguchi, H. Kaizawa, T. Koizumi, T. Ohishi, M. Yamano, M. Okada, M. Ohta, S. Tsukamoto, F. I. Raynaud, P. Parker, P. Workman and M. D. Waterfield, Bioorg. Med. Chem., 2007, 15, 5837–5844 CrossRef CAS PubMed.
- M. A. Lawlor and D. R. Alessi, J. Cell Sci., 2001, 114, 2903–2910 CAS.
- V. Kumar and M. I. Siddiqi, RSC Adv., 2016, 6, 112455–112467 RSC.
- B. T. Hennessy, D. L. Smith, P. T. Ram, Y. Lu and G. B. Mills, Nat. Rev. Drug Discovery, 2005, 4, 988–1004 CrossRef CAS PubMed.
- Y. Samuels, Z. Wang, A. Bardelli, N. Silliman, J. Ptak, S. Szabo, H. Yan, A. Gazdar, S. M. Powell, G. J. Riggins, J. K. Willson, S. Markowitz, K. W. Kinzler, B. Vogelstein and V. E. Velculescu, Science, 2004, 304, 554 CrossRef CAS PubMed.
- M. Martini, E. Ciraolo, F. Gulluni and E. Hirsch, Front. Oncol., 2013, 3, 108 Search PubMed.
- C. Garcia-Echeverria and W. R. Sellers, Oncogene, 2008, 27, 5511–5526 CrossRef CAS PubMed.
- F. I. Raynaud, S. Eccles, P. A. Clarke, A. Hayes, B. Nutley, S. Alix, A. Henley, F. Di-Stefano, Z. Ahmad, S. Guillard, L. M. Bjerke, L. Kelland, M. Valenti, L. Patterson, S. Gowan, A. de Haven Brandon, M. Hayakawa, H. Kaizawa, T. Koizumi, T. Ohishi, S. Patel, N. Saghir, P. Parker, M. Waterfield and P. Workman, Cancer Res., 2007, 67, 5840–5850 CrossRef CAS PubMed.
- M. L. Sos, S. Fischer, R. Ullrich, M. Peifer, J. M. Heuckmann, M. Koker, S. Heynck, I. Stuckrath, J. Weiss, F. Fischer, K. Michel, A. Goel, L. Regales, K. A. Politi, S. Perera, M. Getlik, L. C. Heukamp, S. Ansen, T. Zander, R. Beroukhim, H. Kashkar, K. M. Shokat, W. R. Sellers, D. Rauh, C. Orr, K. P. Hoeflich, L. Friedman, K. K. Wong, W. Pao and R. K. Thomas, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18351–18356 CrossRef CAS PubMed.
- A. C. West and R. W. Johnstone, J. Clin. Invest., 2014, 124, 30–39 CrossRef CAS PubMed.
- A. A. Lane and B. A. Chabner, J. Clin. Oncol., 2009, 27, 5459–5468 CrossRef CAS PubMed.
- J. Tan, S. Cang, Y. Ma, R. L. Petrillo and D. Liu, J. Hematol. Oncol., 2010, 3, 5 CrossRef PubMed.
- T. Nakagawa, S. Takeuchi, T. Yamada, H. Ebi, T. Sano, S. Nanjo, D. Ishikawa, M. Sato, Y. Hasegawa, Y. Sekido and S. Yano, Cancer Res., 2013, 73, 2428–2434 CrossRef CAS PubMed.
- T. Yoshioka, S. Yogosawa, T. Yamada, J. Kitawaki and T. Sakai, Gynecol. Oncol., 2013, 129, 425–432 CrossRef CAS PubMed.
- W. P. Zhang, X. W. Zheng, T. Meng, H. S. You, Y. L. Dong, J. F. Xing and S. Y. Chen, RSC Adv., 2016, 6, 48072–48082 RSC.
- C. P. Wu, Y. J. Hsieh, S. H. Hsiao, C. Y. Su, Y. Q. Li, Y. H. Huang, C. W. Huang, C. H. Hsieh, J. S. Yu and Y. S. Wu, Mol. Pharm., 2016, 13, 784–794 CrossRef CAS PubMed.
- S. Kotian, L. Zhang, M. Boufraqech, K. Gaskins, S. K. Gara, M. M. Quezado, N. Nilubol and E. Kebebew, Clin. Cancer Res., 2017, 23, 5044–5054 CrossRef CAS PubMed.
- K. Saijo, J. Imamura, K. Narita, A. Oda, H. Shimodaira, T. Katoh and C. Ishioka, Cancer Sci., 2015, 106, 208–215 CrossRef CAS PubMed.
- X. Chen, S. Zhao, Y. C. Wu, Y. D. Chen, T. Lu and Y. Zhu, RSC Adv., 2016, 6, 103178–103184 RSC.
- W. J. Scott, M. F. Hentemann, R. B. Rowley, C. O. Bull, S. Jenkins, A. M. Bullion, J. Johnson, A. Redman, A. H. Robbins, W. Esler, R. P. Fracasso, T. Garrison, M. Hamilton, M. Michels, J. E. Wood, D. P. Wilkie, H. Xiao, J. Levy, E. Stasik, N. Liu, M. Schaefer, M. Brands and J. Lefranc, ChemMedChem, 2016, 11, 1517–1530 CrossRef CAS PubMed.
- M. S. Finnin, J. R. Donigian, A. Cohen, V. M. Richon, R. A. Rifkind, P. A. Marks, R. Breslow and N. P. Pavletich, Nature, 1999, 401, 188–193 CrossRef CAS PubMed.
- Y. Zhu, X. Chen, Z. Wu, Y. Zheng, Y. Chen, W. Tang and T. Lu, Arch. Pharmacal Res., 2012, 35, 1723–1732 CrossRef CAS PubMed.
- R. R. Yadav, S. K. Guru, P. Joshi, G. Mahajan, M. J. Mintoo, V. Kumar, S. S. Bharate, D. M. Mondhe, R. A. Vishwakarma, S. Bhushan and S. B. Bharate, Eur. J. Med. Chem., 2016, 122, 731–743 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedure, spectral data of all novel compounds. See DOI: 10.1039/c7ra08835c |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a

















