
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlpha7-nicotinic acetylcholine receptors involve the imidacloprid-induced inhibition of IgE-mediated rat and human mast cell activation
Linbo Shiab,
Huaping Xuc,
Yujie Wuab,
Xin Liad,
Li Zouab,
Jinyan Gaod and
Hongbing Chen *ab
*ab
aState Key Laboratory of Food Science and Technology, Nanchang University, Nanchang, 330047, China. E-mail: chenhongbing@ncu.edu.cn; Fax: +86-791-8333708; Tel: +86-791-88334552
bSino-German Joint Research Institute, Nanchang University, 235 Nanjing Donglu, Nanchang, 330047, China
cDepartment of Rehabilitation, The First Affiliated Hospital of Nanchang University, Nanchang, 330006, China
dSchool of Food Science and Technology, Nanchang University, Nanchang, 330047, China
First published on 8th November 2017
Abstract
Although our recent study indicated that imidacloprid, a widely used neonicotinoid insecticide, inhibited IgE-mediated rat mast cell RBL-2H3 activation, little information is available on the relationship between imidacloprid and IgE-mediated human mast cell activation, and the inhibition mechanism still remains unclear. In the present work, the IgE-sensitized RBL-2H3 cells and human basophilic cell KU812 were incubated with imidacloprid or methyllycaconitine (MLA, the antagonist of α7-nAChRs) prior to the treatment of imidacloprid, followed by challenging the cells with dinitrophenyl–human serum albumin and β-lactoglobulin, respectively. The allergic mediator release, Ca2+ influx in cells, cPLA2 activity, the phosphorylation contents of PLC-γ and NF-κB in FcεRI signaling pathway were tested. The results indicated that imidacloprid could suppress the production of allergic mediators, Ca2+ mobilization, cPLA2 activity and the expression of the phosphorylated antibodies of PLC-γ and NF-κB in the RBL-2H3 and KU812 cells. Moreover, an IgE-dependent passive cutaneous anaphylaxis model was used to determine whether α7-nAChRs involved the suppressive effects of imidacloprid in vivo. It was shown that MLA alleviated the imidacloprid-induced inhibition on the absorbance value of vascular extravasation in mice. It is the first time it has been demonstrated that α7-nAChRs involve the inhibitory effects of imidacloprid on the IgE-mediated activation of mast cells.
1. Introduction
Mature mast cells express the high-affinity IgE receptor (FcεRI) and play an important role in immunoglobulin E (IgE)-mediated allergic disease. The binding of allergen to IgE-FcεRI causes the release of diverse allergic mediators, cytokines, and chemokines.1 IgE-induced crosslinking of FcεRI activates the spleen tyrosine kinase (Syk) and then induces subsequent tyrosine phosphorylation of cellular proteins, such as phospholipase C-γ (PLC-γ). Finally, phosphorylated PLC-γ increases calcium (Ca2+) mobilization and mast cell degranulation. These signaling cascades also activate mitogen-activated protein kinases (MAPKs), such as extracellular-signal-regulated kinases 1/2 (ERK1/2), c-Jun N-terminal kinases (JNKs), and p38. Each of these kinases can promote the activation of specific sets of transcription factors including nuclear factor kappa B (NF-κB), inducing cytokine and chemokine generation.2–4Imidacloprid, 1-[(6-chloro-3-pyridinyl)-methyl]-N-nitro-2-imidazolidinimine, a neonicotinoid insecticide which acted as a nicotinic acetylcholine receptor (nChR) agonist, was commonly used to protect crop worldwide in the last decade.5 Human exposure to imidacloprid is relatively possible as its extensive use which leads to environmental persistence and residues in food, raising the possibility that imidacloprid may negatively affect human health.6–8 Recent advances showed that imidacloprid can suppress adaptive and inflammatory immune responses of rats.9,10 Moreover, our previous study showed that low levels of imidacloprid can inhibit rat mast cell RBL-2H3 (RBL-2H3 cells) degranulation, suggesting a possible negative association of imidacloprid with allergic disease.11 However, the underlying mechanism for it still remains unclear, although we speculated α7-nAChRs may be involved in imidacloprid-induced changes in RBL-2H3 cell function as α7 is the most found nAChR subunit on immune cells and α7-nAChRs are involved in the pathology of IgE-mediated allergy.12–15 Furthermore, the results from RBL-2H3 cell couldn't reflect the real relationship between imidacloprid and IgE-mediated allergy as RBL-2H3 cell is an animal origin cell line. Accordingly, it is interesting to further detect the possible effects of imdacloprid on IgE-mediated human mast cell activation and the underlying mechanism.
In the present study, the IgE-sensitized RBL-2H3 cells and IgE-sensitized human basophilic cell line KU812 were incubated with imidacloprid or methyllycaconitine (MLA, the antagonist of α7-nAChRs) prior to the treatment of imidacloprid. Then, the cells were challenged with antigens for appropriate periods. The allergic mediator release, Ca2+ influx in cells, and the phosphorylation contents of PLC-γ and NF-κB in FcεRI signaling pathway were determined. Moreover, an IgE-dependent passive cutaneous anaphylaxis (PCA) model of mice was used to test whether α7-nAChRs were involved in mediating the suppressive effects of imidacloprid in vivo, and the vascular extravasation of mice ear was evaluated. Nevertheless, this work aimed to detect the possible effects of imidacloprid on IgE-activated human mast cell degranulation and further explore the mechanisms by which imidacloprid affect mast cell degranulation.
2. Materials and methods
2.1 Reagents
The RBL-2H3 cell and the human basophilic cell line KU812 were purchased from the Type Cell Culture Collection of the Chinese Academy of Science (Wuhan, China). The imidacloprid, bovine β-Lg and 4-nitrophenyl-N-acetyl-β-D-glucosamide were got from Sigma-Aldrich Corporation (St. Louis, MO, USA). MLA was obtained from Merck Millipore (Billerica, MA). The primary antibodies used were as follows: antibodies against phosphorylated forms of NF-κB, PLC-γ and antibody against total NF-κB (Cell Signaling Technology [Danvers, Mass]). Antibodies against total PLC-γ was obtained from Abcam (Cambridge, Mass). The Iscove's Modified Dulbecco's Medium (IMDM) cell culture medium and other reagents were purchased from Thermo Fisher Scientific (Shanghai, China).2.2 Human sera preparation
Human sera were prepared as previously described.16 The sera were pooled from 10 milk allergy patients (Table 1). Bovine specific β-Lg IgE and total IgE levels were determined (specific IgE level > 1 IU per mL, total IgE level >100 IU per mL) with an ImmunoCAP 100E (Phadia AB, Uppsala, Sweden). Sera from non-allergic individuals (n = 4) were also pooled and used as negative serum.| Patient no. | Sex | Age (years) | Clinical symptoms | Total IgE Level (IU per mL) | Specific IgE Level (IU per mL) |
|---|---|---|---|---|---|
| a NK, Not Known. | |||||
| 1 | Female | 4 | Urticaria | >100 | 7.8 |
| 2 | Male | 10 | NKa | >100 | 10.5 |
| 3 | Male | 5 | Chronic rhinitis | >100 | 2.3 |
| 4 | Male | 15 | NKa | >100 | 6.5 |
| 5 | Female | 3 | Asthma, bronchitis | >100 | 3.4 |
| 6 | Female | 2 | Bronchial asthma | >100 | 6.9 |
| 7 | Male | 3 | NKa | >100 | 12.4 |
| 8 | Male | 4 | Urticaria | >100 | 3.8 |
| 9 | Female | 20 | Bronchial asthma | >100 | 8.4 |
| 10 | Female | 1 | Chronic rhinitis | >100 | 5.3 |
2.3 Culture, treatment and activation of cells
RBL-2H3 cells and KU812 cells were cultured in IMDM supplemented containing 20% FBS (Gibco, USA), 100 U per mL penicillin, 100 μg mL−1 streptomycin. RBL-2H3 cells were sensitized with 500 ng mL−1 anti-DNP IgE (Sigma-Aldrich Corporation, USA), and KU812 cells were sensitized with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 dilution of patient serum overnight at 37 °C. After sensitization, the cells were incubated with MLA (α7-nAChR antagonist) for 4 h, prior to being treated with various concentrations of imidacloprid for 4 h. Then, the cells were challenged respectively by 50 μg mL−1 dinitrophenyl–human serum albumin (DNP–HAS) (Biosearch Technologies, USA) and 50 μg mL−1 β-Lg in the presence or absence of MLA for different durations in various assays. The control group was only sensitized with IgE and stimulated with antigens for appropriate periods, whereas the naïve group was treated with nothing but the vehicles. Moreover, ketotifen fumarate salt in DMSO was used for the positive control.
:5 dilution of patient serum overnight at 37 °C. After sensitization, the cells were incubated with MLA (α7-nAChR antagonist) for 4 h, prior to being treated with various concentrations of imidacloprid for 4 h. Then, the cells were challenged respectively by 50 μg mL−1 dinitrophenyl–human serum albumin (DNP–HAS) (Biosearch Technologies, USA) and 50 μg mL−1 β-Lg in the presence or absence of MLA for different durations in various assays. The control group was only sensitized with IgE and stimulated with antigens for appropriate periods, whereas the naïve group was treated with nothing but the vehicles. Moreover, ketotifen fumarate salt in DMSO was used for the positive control.
2.4 Assessment of degranulation
To detect the levels of released histamine and β-hex, IgE-sensitized cells were challenged with 50 μg mL−1 antigens for 30 min at 37 °C. A histamine ELISA kit (Cayman Chemical) was used to measure the released histamine in accordance with the manufacturer's instructions. The β-hex activity in supernatants was measured by adding p-nitrophenyl N-acetyl-b-D-glucosaminide in 0.1 M citrate buffer (pH 4.5) to separate 96-well microtiter plates for 60 min at 37 °C. The stop solution (0.1 mol L−1 Na2CO3/NaHCO3) was added to each well to stop the reaction. The absorbance was measured at 405 nm wavelength. The results of histamine or β-hex released into cell culture were expressed as the percentage of the amount of histamine or β-hex in control group.2.5 Assays of LTC4 and TNF-α production
After treatments with various reagents, the cells were challenged with 50 μg mL−1 antigens for 4 h. The supernatant was collected after centrifugation. Then, the LTC4 and TNF-α production were assayed using an ELISA kit from Cayman Chemical (Ann Arbor, MI) and Abcam, Inc. (Cambridge, MA, USA), respectively, in accordance with the manufacturers' protocol.2.6 Western blot analysis of PLC-γ and NF-κB
Total cellular and nuclear proteins were prepared as previously described.17 The bicinchoninic acid method was then applied to determine protein concentrations. The proteins were resolved on Criterion Gels (Bio-Rad, Hercules, CA) and subsequently transferred onto nitrocellulose membranes. The proteins were incubated for 60 min at room temperature in 5% bovine serum albumin (Sigma-Aldrich) and then probed with appropriate antibodies overnight at 4 °C. After washing with TBS-T for three times, the membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary Abs at room temperature for 1 h. Afterwards, the immune reaction were developed with Pro-light HRP Chemiluminescent Kit (TIANGEN biotech, Beijing, China), and the images were recorded with Bio-Rad Gel Doc/Chemi Doc Imaging System (Bio-Rad, Hercules, USA).2.7 cPLA2 activity assay
The cPLA2 activity from cell extracts was assayed using a kit from Cayman Chemical (Ann Arbor, MI), in accordance with manufacturer's instructions.2.8 Intracellular calcium level assay
The intracellular calcium test was performed by the fluorescence assay as described previously.11 The cells were dyed with 5 μM Fluo-3/AM at 37 °C, after various treatments. Then, 30 min later, the cells were challenged with 50 μg mL−1 antigens. An inverted fluorescent microscope (Nikon Eclipse Ti-U, Nikon Instruments, Kanagawa, Japan) was used to observe the fluorescence images immediately. To further evaluate the possible effects of MLA on imidacloprid-induced change on the intracellular calcium levels, fluorescent intensity was measured using a Varioskan Flash microplate reader (Thermo Fisher Scientific, Waltham, MA, USA) at 488 nm excitation wavelength and 526 nm emission wavelength.2.9 IgE-mediated PCA in BALB/c mice
One ear of BALB/c mouse was intradermally injected 500 ng anti-DNP-specific IgE. Then, 24 h later, the ear was given MLA in 200 μL saline for 4 h, followed by another 4 hour treatment with 10−4 M imidacloprid in 200 μL DMSO. The mice were then challenged with an intravenous injection of 50 μg DNP–HSA in 100 μL saline containing 4% Evans blue. Then, after antigen challenge for 1 h, the mice were killed and the ears were removed to measure the amount of dye extravagated. Evans blue was extracted with formamide overnight at 63 °C, and the absorbance was measured at 630 nm a Varioskan Flash microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).2.10 Live subject statement
All animal-use protocols were approved by the Animal Care Committee of Nanchang University under the guidelines of the Care and Use of Laboratory Animals (NIH Publication no. 86-23, Revised 1996). Informed consent was obtained in all cases, and the availability of all serum samples was approved by the internal ethical committee of the hospital (Nanchang, China).2.11 Statistical analyses
The results obtained were expressed as the mean ± standard deviation (SD). All experiments were performed at least 3 times. The unpaired two-sided t tests were used to make a statistical comparison between groups. Results with p < 0.05 were considered statistically significant.3. Results
3.1 Effects of imidacloprid on the viability of RBL-2H3 cells and KU812 cells
The effects of imidacloprid on cell viability was assessed by the CCK-8 assay to ensure that the decreased level of mast cell granules was not due to cell death. As shown in Fig. 1, 10−3–10−11 M imidacloprid did not significantly affect the RBL-2H3 cell viability. Since our previous work already indicated that 10−3–10−11 M imidacloprid could inhibit RBL-2H3 cell degranulation,11 the dosage of 10−3, 10−5, 10−7 and 10−9 M imidacloprid were selected for the further studies about the inhibition mechanism on RBL-2H3 cell degranulation. Moreover, 10−4–10−12 M imidacloprid was used for the subsequent studies about KU812 cells as the CCK-8 assays indicated that imidacloprid of less than 10−3 M did not affect the cells viability (Fig. 1).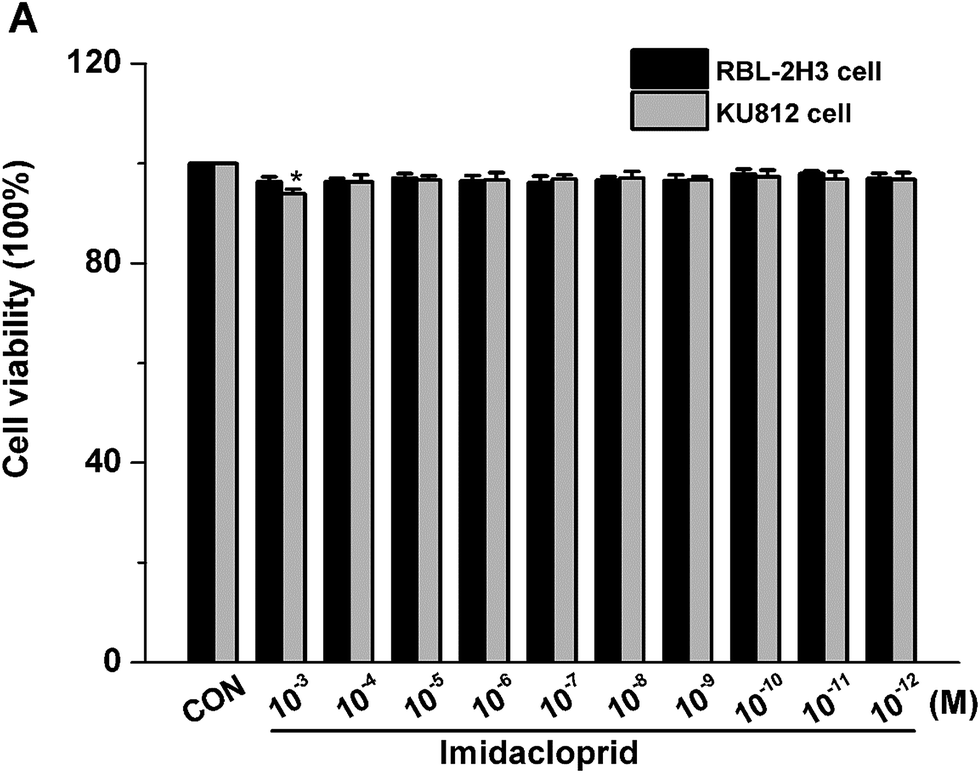 | ||
| Fig. 1 Effects of imidacloprid on the cell viability of RBL-2H3. Cells were treated with imidacloprid for 24 hours, and cytotoxicity was measured by cell counting kit-8 assay. *p < 0.05, compared with control (CON). | ||
3.2 Effects of different reagents on IgE-mediated mast cell degranulation
The release of several allergic mediators such as β-hex and TNF-α from mast cell were tested to determine which concentration of MLA can mostly resist the imidacloprid effect, and to make clear that whether there is any synergistic effect of MLA and imidacloprid. As shown in Fig. 2, 10−3 M imidacloprid inhibited significantly the release of β-hex and TNF-α from RBL-2H3 cells/KU812 cells as 10−3 M ketotifen fumarate salt of the positive control did. However, MLA resisted the inhibition of imidacloprid on IgE-mediated mast cell activation significantly. For example, the release of β-hex from RBL-2H3 cells increased from 56.73% ± 0.60 to 95.1% ± 0.70, 56.73% ± 0.60 to 95.26% ± 1.11, 56.73% ± 0.60 to 95.30% ± 0.60, respectively, when the concentration of MLA was 10−3 M, 10−2 M and 10−1 M, whereas MLA of 10−5 M and 10−4 M just increased the β-hex release from 56.73% ± 0.60 to 69.36% ± 1.11, 56.73% ± 0.60 to 82.13% ± 1.11, respectively (Fig. 2A). Moreover, MLA with different concentrations in the present study did not show any specific effects on the tested immunological characteristics of mast cells. Reasonably, 10−3 M MLA was used for the subsequent studies to further explore the mechanisms by which imidacloprid affect mast cell degranulation.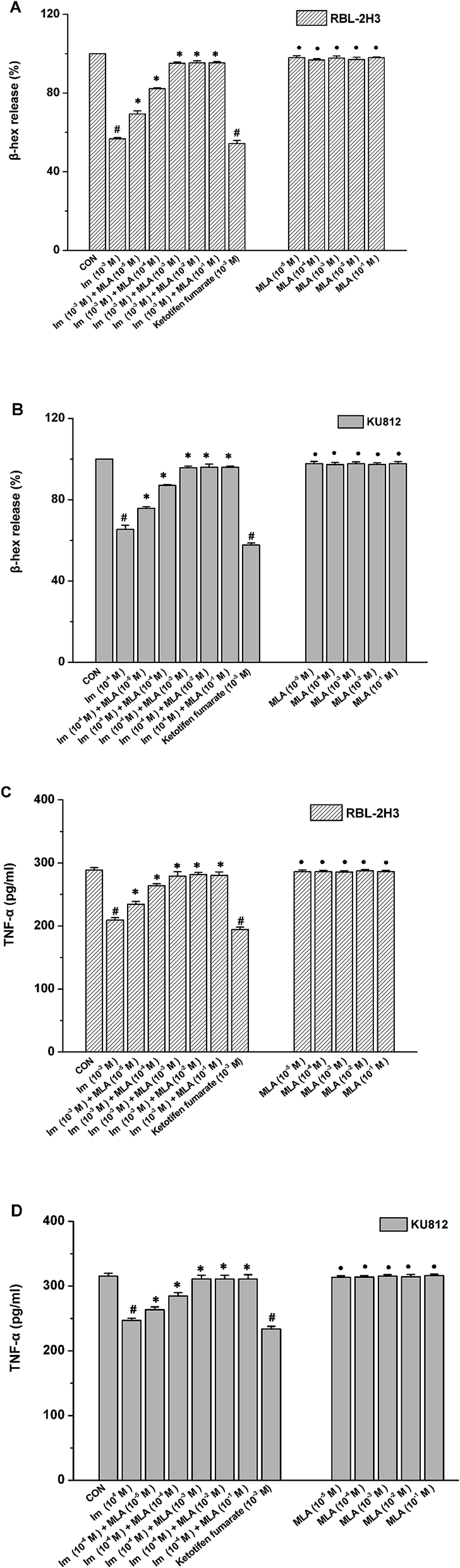 | ||
| Fig. 2 Effects of different reagents on mast cell degranulation. RBL-2H3 cells and KU812 cells were sensitized respectively with anti-DNP IgE or patient serum overnight at 37 °C. After sensitization, the cells were incubated with MLA (α7-nAChR antagonist) for 4 h, prior to being treated with various concentrations of imidacloprid for 4 h. Then, the cells were challenged with 50 μg mL−1 DNP–HAS or 50 μg mL−1 β-Lg, respectively. The imidacloprid-treated group and the MLA-treated group were treated only with imidacloprid and MLA, respectively. Then, the levels of β-hex and the production of TNF-α in supernatants were measured. (A) The levels of β-hex released from RBL-2H3 cells. (B) The levels of β-hex released from KU812 cells. (C) The production of TNF-α in RBL-2H3 cells. (D) The production of TNF-α in KU812 cells. #p < 0.05 (compared with the control group). *p < 0.05 (compared with the imidacloprid-treated group), •p > 0.05 (compared with the control group). | ||
3.3 Imidacloprid inhibited the degranulation of mast cells via α7-nAChRs
The release of β-hex and histamine from mast cell was tested to determine whether imidacloprid could affect mast cell degranulation via α7-nAChRs. It was shown in Fig. 3 that MLA could suppress the imidacloprid's effects on the release of histamine and β-hex from RBL-2H3 cells (Fig. 3B and C). MLA's effects on imidacloprid-induced inhibition could also be evidenced by more amounts of non-degranulated cells in the group treated with both imidacloprid and MLA when compared with that in the group treated with imidacloprid only (Fig. 3D). Similarly, MLA could also suppress the imidacloprid's inhibitory effects on the release of histamine and β-hex from IgE-activated KU812 cells (Fig. 3E and F). For example, the pretreatment of 10−3 M MLA can suppress the imidacloprid's effects by increasing the release of histamine and β-hex from RBL-2H3 cells by 6% (p < 0.05) and 4% (p < 0.05), respectively, and 10−3 M MLA also significantly increased the release of histamine and β-hex from KU812 cells by 7% (p < 0.05) and 5% (p < 0.05), respectively, when compared with the imidacloprid-only-treated group. Nevertheless, both of the release of histamine and β-hex in MLA-pretreated groups were lower than that of the control group. For an example, the release of histamine from RBL-2H3 and KU812 in 10−3 M-MLA-pretreatment group was 89.00% ± 0.50 and 92.73% ± 0.40, respectively, which were significant lower than 100% of the control group (p < 0.05).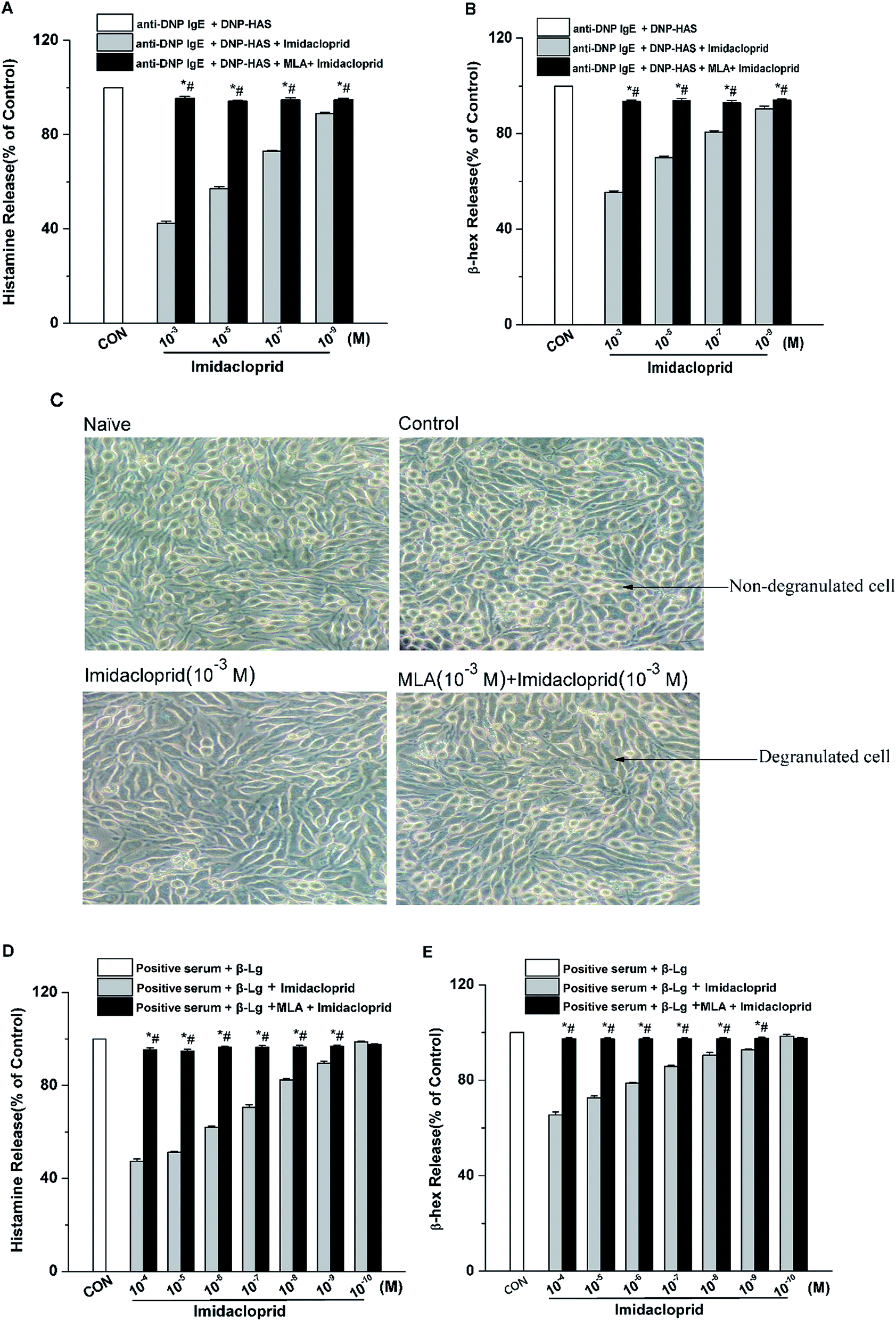 | ||
| Fig. 3 Imidacloprid suppressed degranulation in mast cells via α7-nAChRs. RBL-2H3 cells and KU812 cells were sensitized respectively with anti-DNP IgE or patient serum overnight at 37 °C. Then, the IgE-sensitized cells were treated with MLA for 4 h, followed by incubation with imidacloprid. Afterwards, 4 h later, the cells were challenged with 50 μg mL−1 DNP–HAS or 50 μg mL−1 β-Lg, respectively for 30 min. Then, the levels of histamine and β-hex in supernatants were measured, and cell images were recorded using an inverted microscope. (A) The levels of histamine released from RBL-2H3 cells. (B) The levels of β-hex released from RBL-2H3 cells. (C) RBL-2H3 cell images were recorded using an inverted microscope. (D) The levels of histamine released from KU812 cells. (E) The levels of β-hex released from KU812 cells. Experiments were conducted in triplicates, *p < 0.05 (compared with the imidacloprid-treated group), #p < 0.05 (compared with the control group). | ||
3.4 Imidacloprid inhibited leukotriene C4 (LTC4) and TNF-α production via α7-nAChRs
Aggregation of FcεRI on mast cells also induces the de novo synthesis and secretion of LTs, such as LTC4, as well as pro-inflammatory cytokines, such as TNF-α and IL-6, which were suggested to initiate and maintain the late-phase allergic reaction.18 Accordingly, the effects of imidacloprid on the secretion of LTC4 and TNF-α from mast cells was investigated, and also the possible effect mechanism was detected. The enzyme immunoassays demonstrated that imidacloprid treatment significantly suppressed the production of LTC4 and TNF-α in both of RBL-2H3 cells and KU812 cells (Fig. 4). By contrast, MLA pretreatment attenuated the imidacloprid-induced inhibition on the production of LTC4 and TNF-α from the cells (Fig. 4). Fig. 4A and C shown that 10−3 M of MLA significantly increased the secretion of LTC4 from RBL-2H3 and KU812 to 94.67% ± 0.65 and 97.20% ± 0.26 respectively, when compared with the imidacloprid-only-treated group. Moreover, 10−3 M MLA also significantly attenuated the inhibitory effects of 10−9 M imidacloprid by promoting the production of TNF-α from RBL-2H3 and KU812 cells by 14.50 pg mL−1 (p < 0.05) and 12.3 (p < 0.05), respectively. Additionally, the production of LTC4 and TNF-α in MLA-pretreated groups were lower than that of the control group.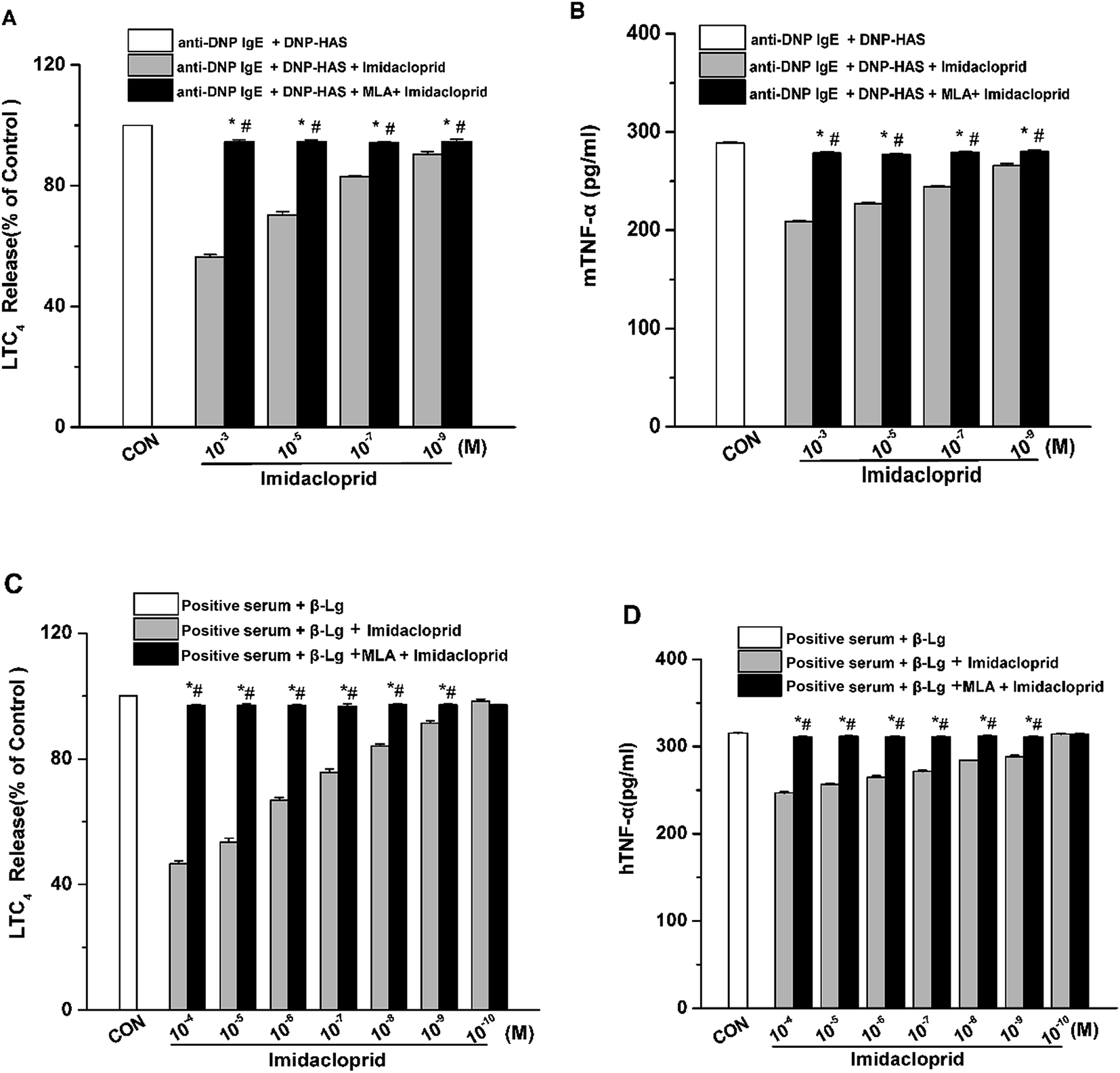 | ||
| Fig. 4 Imidacloprid inhibited the production of LTC4 and TNF-α in mast cells via α7-nAChRs. RBL-2H3 cells and KU812 cells were sensitized respectively with anti-DNP IgE or patient serum overnight at 37 °C. Then, the IgE-sensitized cells were treated with MLA for 4 h, followed by incubation with imidacloprid. Afterwards, 4 h later, the cells were challenged with 50 μg mL−1 DNP–HAS or 50 μg mL−1 β-Lg, respectively for 4 h. The enzyme immunoassays of the production of LTC4 and TNF-α were executed. (A) The production of LTC4 in RBL-2H3 cells. (B) The production of TNF-α in RBL-2H3 cells. (C) The production of LTC4 in KU812 cells. (D) The production of TNF-α in KU812 cells. Experiments were conducted in triplicate, *p < 0.05 (compared with the imidacloprid-treated group), #p < 0.05 (compared with the control group). | ||
3.5 α7-nAChRs mediated the imidacloprid-induced repression of Ca2+ mobilization
Since the increase of Ca2+ influx is vital for mast cell degranulation,19 10−3, 10−5, 10−7 and 10−9 M of imidacloprid were selected again for the further work about any possible modulating effects of MLA on the imidacloprid-induced change of Ca2+ mobilization in RBL-2H3 cells. As shown in Fig. 5A, 10−3, 10−5, 10−7 and 10−9 M of imidacloprid could inhibit the amplitude of the intracellular calcium level (ΔF − F0) in RBL-2H3 cells. However, the MLA pretreatment could significantly increase the ΔF − F0 from 0.65 ± 0.01 (10−3 M imidacloprid) to 0.96 ± 0, 0.75 ± 0 (10−5 M imidacloprid) to 0.96 ± 0, 0.83 ± 0.01 (10−7 M imidacloprid) to 0.96 ± 0, 0.92 ± 0 (10−9 M imidacloprid) to 0.96 ± 0, respectively. Moreover, the effects of MLA on the imidacloprid-induced change of Ca2+ mobilization in KU812 cells were also investigated by the same methods. As shown in Fig. 5B, the pretreatment of 10−3 M of MLA resulted in a significant increase of ΔF − F0, from 0.51 ± 0 (10−4 M imidacloprid) to 0.99 ± 0.02, 0.60 ± 0.01 (10−5 M imidacloprid) to 1.00 ± 0.02, 0.71 ± 0 (10−6 M imidacloprid) to 0.99 ± 0.03, 0.80 ± 0.01 (10−7 M imidacloprid) to 1 ± 0.02, 0.86 ± 0 (10−8 M imidacloprid) to 1 ± 0.02, 0.96 ± 0 (10−9 M imidacloprid) to 0.99 ± 0.02, respectively. MLA's effects on imidacloprid-induced inhibition could also be evidenced by much more rise in intracellular calcium level in MLA-treated KU812 cells (Fig. 5C and D). In addition, the changes of Ca2+ mobilization in MLA-pretreated group were lower than those for the control group. For an example, the value of ΔF − F0 for 10−3 M MLA group in KU812 cells was 0.99 ± 0.02, which was significantly different when compared with 1.07 ± 0 for the control (p < 0.05).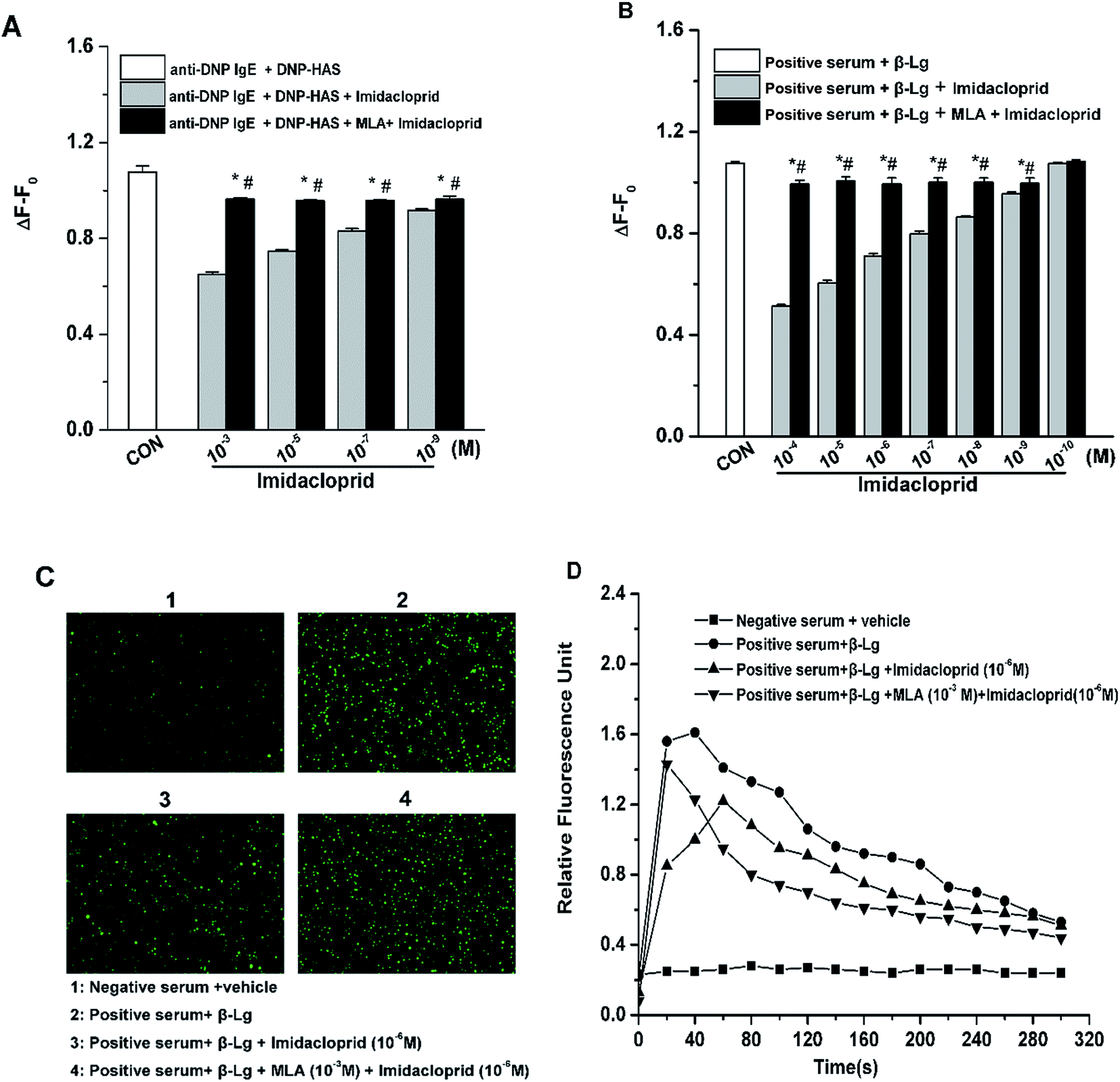 | ||
| Fig. 5 α7-nAChRs mediated the imidacloprid-induced repression of Ca2+ mobilization. RBL-2H3 cells and KU812 cells were sensitized respectively with anti-DNP IgE or patient serum overnight. Then, the IgE-sensitized cells were treated with MLA for 4 h, followed by incubation with imidacloprid. Afterwards, 4 h later, the Fluo 3-AM fluorescent dye was incubated with the cells for 30 min, followed by the challenge with 50 μg mL−1 DNP–HAS or 50 μg mL−1 β-Lg, respectively. The fluorescence intensity was measured with a Varioskan Flash microplate reader and fluorescence image was recorded using an inverted microscope. (A) The amplitude of the intracellular calcium level (ΔF − F0) in RBL-2H3 cells. (B) The amplitude of the intracellular calcium level (ΔF − F0) in KU812 cells. (C) The fluorescence images of KU812 cells were recorded using an inverted microscope. (D) Ca2+ response to β-Lg IgE/serum activation in KU812 cells. *p < 0.05 (compared with the imidacloprid-treated group), #p < 0.05 (compared with the control group). | ||
3.6 α7-nAChRs mediated the imidacloprid-induced change of the FcεRI signaling pathway
IgE-induced crosslinking of FcεRI activates PLC-γ to regulate mast cell degranulation at the initial stage of FcεRI signaling pathway, and the activation of NF-κB upregulates cytokine gene and chemokine gene expression to produce pro-inflammatory cytokines in mast cells at the late period of the pathway.20 Accordingly, western blot analysis was applied to test whether imidacloprid influenced the FcεRI-mediated activation of PLC-γ and NF-κB in mast cells. It was shown in Fig. 6 that 10−9 M imidacloprid suppressed phosphorylation but did not diminish the total content of PLC-γ and NF-κB in mast cells (Fig. 6A, results for RBL-2H3 cells not shown). To further confirm whether the inhibitory effects of imidacloprid on mast cells were mediated by the α7-nAChRs, the cells were pre-incubated with the MLA before imidacloprid treatment. It was demonstrated that the MLA pretreatment resulted in a 16% and 10% increase in p-NF-κB and p-PLC-γ (Fig. 6B), respectively, which were significantly different when compared with the groups treated with imidacloprid only (p-PLC-γ relative intensity not shown). Additionally, the relative intensity of phosphorylated antibodies in the MLA-pretreated groups (47.7% ± 0.7 for p-NF-κB and 55.8% ± 0.5 for p-PLC-γ) was significantly lower than that for the control groups (51.6.7% ± 0.9 and 60.9% ± 0.4 for p-NF-κB and p-PLC-γ, respectively, p < 0.05).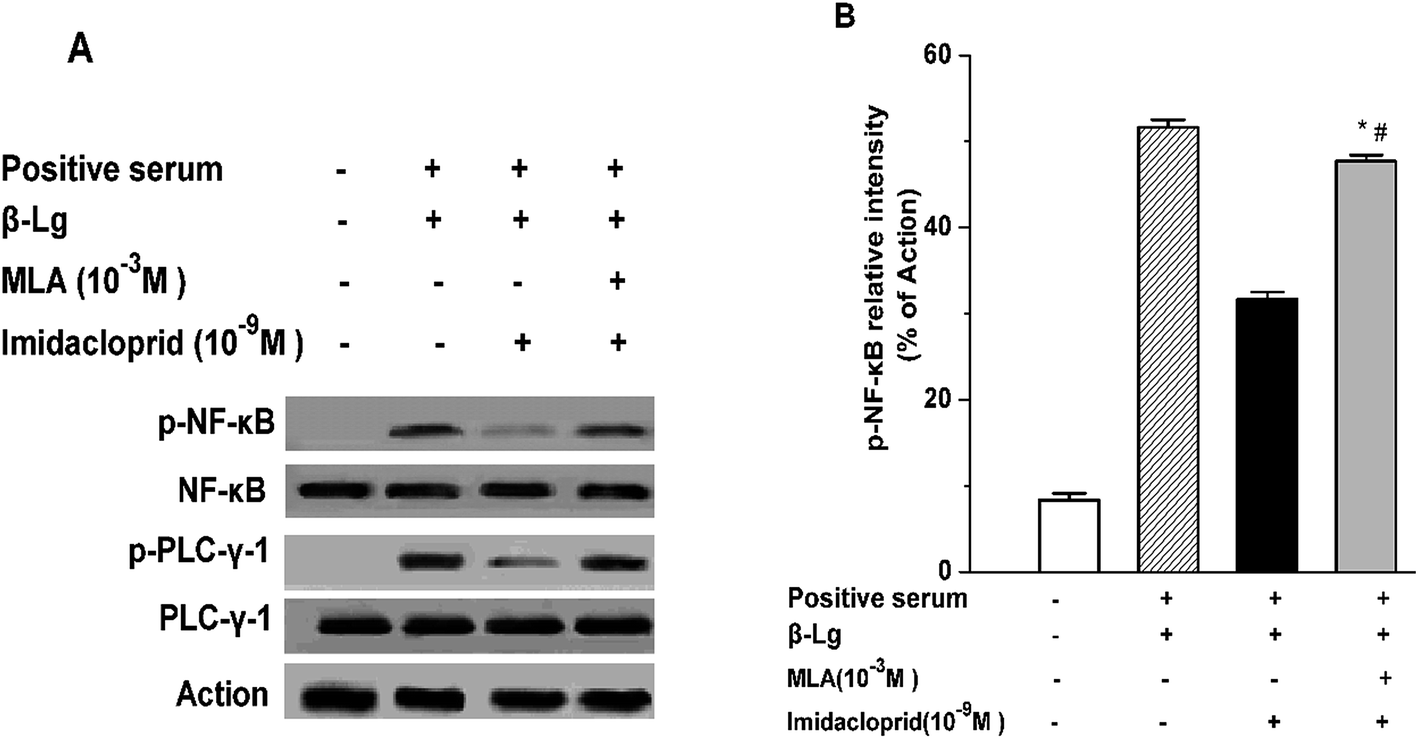 | ||
| Fig. 6 α7-nAChRs mediated the imidacloprid-induced inhibition of the FcεRI signaling pathway. Mast cells were sensitized with anti-DNP IgE or patient serum overnight at 37 °C. Then, the IgE-sensitized cells were treated with MLA for 4 h, followed by incubation with imidacloprid. Afterwards, 4 h later, the cells were challenged with 50 μg mL−1 DNP–HAS or 50 μg mL−1 β-Lg for 20 min. Whole-cell extracts were fractionated on sodium dodecyl sulfate-polyacrylamide gel electrophores, followed by being transferred to a nitrocellulose membrane, and probed with appropriate antibodies. β-Actin was used as loading control. The image was recorded (A) and the band intensity was digitized (B). *p < 0.05 (compared with the imidacloprid-treated group), #p < 0.05 (compared with the control group). | ||
3.7 Imidacloprid inhibited the IgE-induced cPLA2 activation via α7-nAChRs
Cytosolic phospholipase A2 (cPLA2) is rapidly activated by increased intracellular Ca2+ concentration and phosphorylated by MAPKs, and this action accounts for IgE-activated arachidonic acid production.21 In the present study, it was investigated that whether imidacloprid affected cPLA2 activity via α7-nAChRs. Fig. 7 shown that the 10−9 M imidacloprid decreased the cPLA2 activity to 85.80% ± 0.26 (for RBL-2H3 cells) and 90.20% ± 0.68 (for KU812 cells), which were significantly lower than that of the control groups (p < 0.05). Moreover, the addition of MLA significantly increased the cPLA2 activity (93.4% ± 0.96 for RBL-2H3 cells and 95.06% ± 0.87 for KU812 cells), which were obviously higher than that of the imidacloprid-only-treated groups. Additionally, the cPLA2 activity of the cells in the MLA-pretreated groups was significantly lower than that for the control groups (p < 0.05).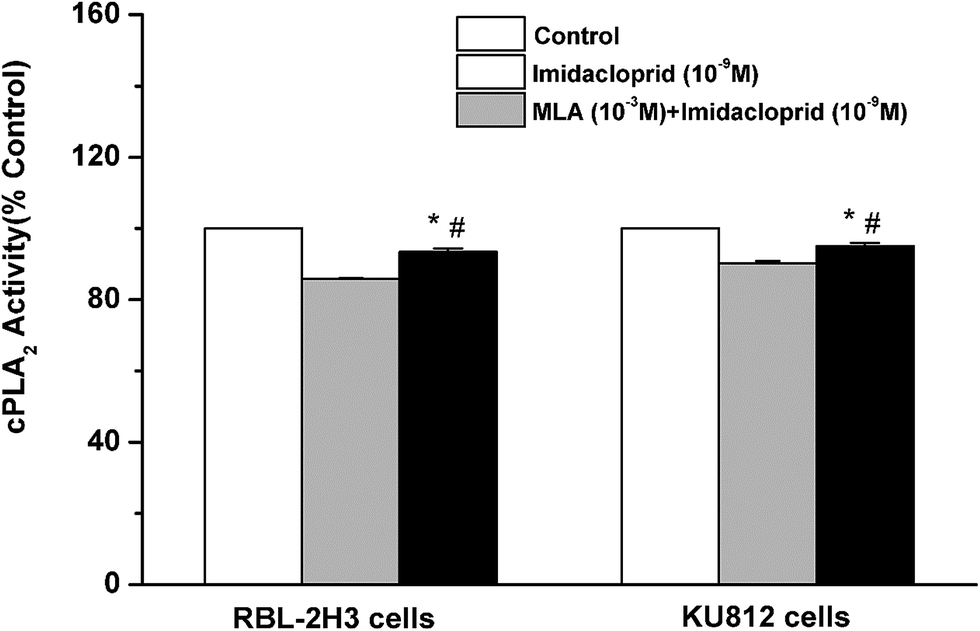 | ||
| Fig. 7 Imidacloprid inhibited the IgE-induced cPLA2 activation via α7-nAChRs. RBL-2H3 cells and KU812 cells were sensitized with anti-DNP IgE or patient serum overnight at 37 °C. Then, the IgE-sensitized cells were treated with 10−3 M MLA for 4 h, followed by incubation with 10−9 M imidacloprid. Afterwards, 4 h later, the cells were challenged with 50 μg mL−1 DNP–HAS or 50 μg mL−1 β-Lg for 30 min for the enzyme immunoassay of cPLA2 activity. *p < 0.05 (compared with the imidacloprid-treated group), #p < 0.05 (compared with the control group). | ||
3.8 α7-nAChR mediated the repression of imidacloprid on IgE-induced PCA
Our previous studies already indicated that 10−3–10−5 M imidacloprid could inhibit IgE-dependent PCA in the ear of BALB/c mice.11 In this study, 10−3–10−5 M of imidacloprid were used again to further evaluate whether α7-nAChRs were required to mediate the suppressive effects of imidacloprid on PCA. As demonstrated in Fig. 8A, the group pretreated with MLA showed higher absorbance values of Evans blue dye than that values of the imidacloprid-only-treated groups. The ear in the group pretreated with 10−3 M MLA showed more severe dye extravasation with a higher absorbance value of Evans blue dye (OD = 1.30 ± 0.01) than that of the imidacloprid-only-treated groups with a lower absorbance value of 1.01 ± 0.02, which was significantly different when compared with the control (Fig. 8B). Moreover, the ears pretreated with MLA owned lower absorbance values of Evans blue dye than that of the control group (Fig. 8).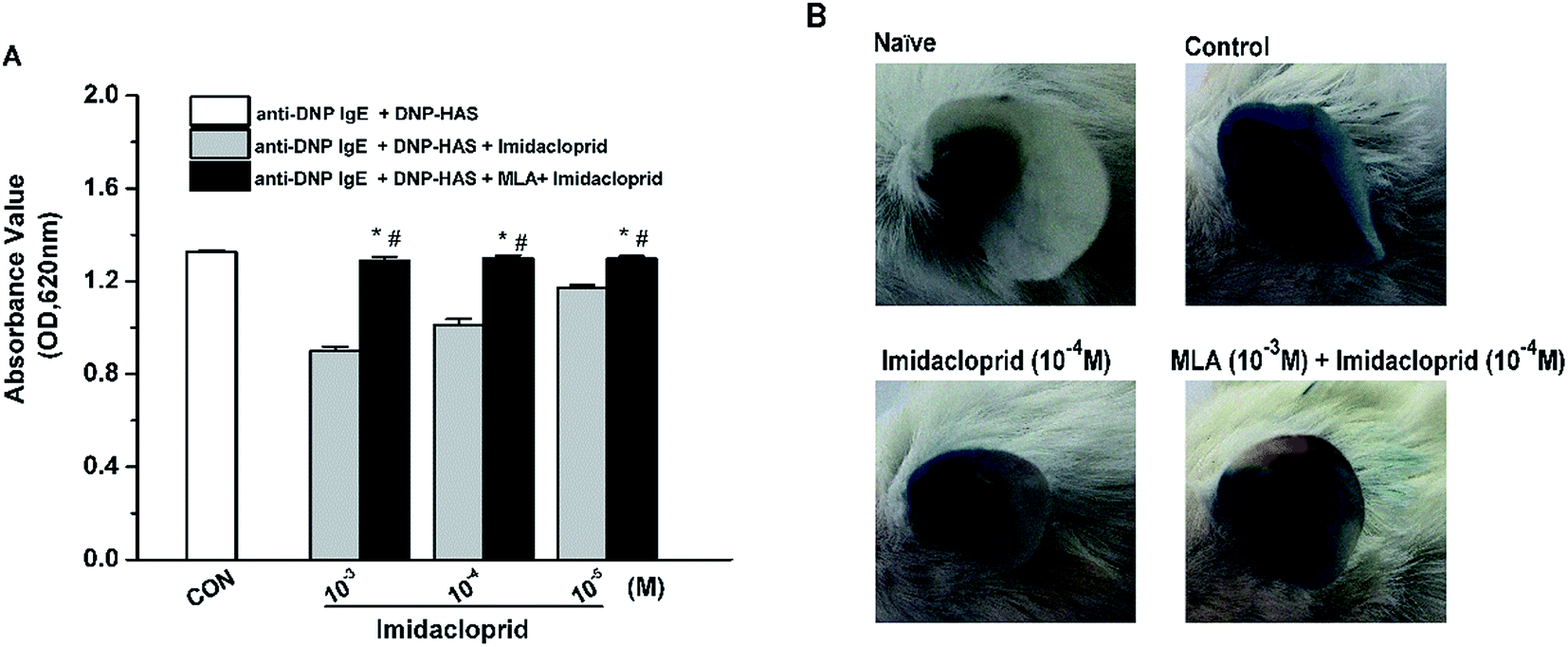 | ||
| Fig. 8 MLA alleviated imidacloprid repression on anti-DNP IgE-induced PCA. One ear of BALB/c mouse was intradermally injected 500 ng anti-DNP-specific IgE. Then, 24 h later, the ear was given 10−3 M MLA in 200 μL saline for 4 h, followed by another 4 hour treatment with 10−4 M imidacloprid in 200 μL DMSO. The mice were then challenged with an intravenous injection of 50 μg DNP–HSA in 100 μL saline containing 4% Evans blue. The naïve group was treated with the vehicle only. Then, after antigen challenge for 1 h, the amount of Evans blue dye was measured (A) and the ear Evans blue extravasation was photographed (B). Data represent the mean ± SD. *p < 0.05 (compared with the imidacloprid-treated group), #p < 0.05 (compared with the control group). | ||
4. Discussion
The nAChRs are ligand-gated ion channels consisting of five subunits that form a central, cation-permeable channel with a gated opening in response to the binding of the neurotransmitter acetylcholine (ACh), mediating synaptic transmission.22,23 The α7 nAChR subtype in the brain has been implicated to play a key role in conditions such as schizophrenia, Alzheimer's disease and epilepsy.24 However, nicotinic receptors were also presented in the cells from the immune system.25,26 The presence of these receptors in non-neuronal cells suggested the distinct functions beyond neurotransmission.27 Some studies reported that the α7-nAChRs on mast cells may be involved in the pathology of allergy.12–15,28,29 Consistent with these studies, our present study also demonstrated that α7-nAChRs also were involved in IgE-mediated mast cell activation (Fig. 8).Imidacloprid acted as a nicotinic acetylcholine receptor (nAChR) agonist, and it was most commonly used to protect crop worldwide. Our previous study showed indicated that imidacloprid could inhibit IgE-mediated rat mast cell RBL-2H3 degranulation. The possible effects of imidacloprid on IgE-mediated human mast cell degranulation and the imidacloprid's effect mechanism were investigated in the present work. Firstly the data in Fig. 3 and 4 demonstrated that imidacloprid treatment inhibited the IgE-mediated secretion of histamine, β-hex, LTC4 and TNF-α from RBL-2H3 and KU812 cells. However, MLA pretreatment attenuated the imidacloprid-induced inhibition on the production of the above mediators. All these results suggested that α7-nAChRs play an important role in imidacloprid-induced changes of the IgE-mediated degranulation of mast cells. Besides, 10−3–10−12 M imidacloprid could inhibit the IgE-mediated RBL-2H3 activation, yet 10−4–10−12 M imidacloprid could do that on KU812 cells. It is possible that the variability of imidacloprid concentration is due to the functional differences in the two cell types.
To further define the intracellular mechanisms of imidacloprid action, the activation status of signaling molecules in the FcεRI signaling pathway was examined in this study. Mast cell activation was initiated by IgE-induced crosslinking of FcεRI and then the degranulation was totally dependent on the increased influx of Ca2+, while PLC-γ activation was critical for Ca2+ mobilization and mast cell degranulation. As shown in Fig. 5 and 6, imidacloprid suppressed the activation of PLC-γ and Ca2+ mobilization in mast cells, while the antagonist of α7-nAChRs could block the imidacloprid-induced repression of PLC-γ and Ca2+ mobilization in mast cells. Reasonably, imidacloprid could block mast cell activation at the initial stage of the FcεRI signaling pathway via α7-nAChRs. Moreover, Fig. 6 indicated that α7-nAChR antagonist also alleviated the inhibitory effects of imidacloprid on NF-κB activation which occurred after the activation of MAPK and regulated the expression of pro-inflammatory cytokines, such as TNF-α.20 MLA also attenuated the inhibitory effects of imidacloprid on cPLA2 activity, accounting for the decreased arachidonic acid production by mast cells treated with imidacloprid. All the above-mentioned works suggested that imidacloprid blocked the mast cell activation from the initial stage to the late stage of the FcεRI pathway through the α7-nAChRs.
Finally, an IgE-dependent passive cutaneous anaphylaxis (PCA) model of mice was used to determine whether α7-nAChRs were involved to mediate the suppressive effects of imidacloprid in vivo. This investigation evidenced that intradermal pretreatment of MLA to mice ear suppressed the inhibition of imidacloprid on the permeability of IgE-mediated PCA because the MLA-pretreated group had more severe vascular extravasation with higher absorbance values of Evans blue dye than that of imidacloprid-only-treated group (Fig. 8). Logically, α7-nAChRs were also involved in mediating the repressive effects of imidacloprid in IgE-activated vascular extravasation in mice.
The prevalence of allergic disease has increased dramatically in the last few decades, and numerous efforts were made to develop more potent anti-allergic therapeutic approaches. Up to date, some emphasis was put on targeting α7-nAChRs, which is widely believed to play an important role in allergic diseases. MLA, one of the most potent and specific α7-nAChR antagonist, alleviated imidacloprid-induced inhibition on IgE-mediated mast cell activation in vitro and in vivo, indicating a protective effect of agonist treatment of α7-nAChR on IgE-mediated mast cell activation. In addition, the cholinergic anti-inflammatory pathway via α7-nAChR is becoming increasingly elucidated to be important in maintaining immune homeostasis in diverse inflammatory disease, such as ischemia-reperfusion injury, pancreatitis, peritonitis, DSS-induced colitis, postoperative ileus, inflammatory bowel disease and other gastrointestinal pathologies.30 It was reported that cholinergic activation via nAChRs, including α7-nAChR, serves as an anti-inflammatory pathway in Th2-related diseases, such as allergic diseases.15,31 The therapeutic potential of the cholinergic agonist, such as nicotine, in inflammatory diseases was recently demonstrated, even though it formerly was reported to have adverse side effects, such as nausea, dizziness, headache and addiction because of non-selectivity of cholinergic agonist for subtypes of nAChRs.32
Interestingly, in our present study, the MLA-pretreated groups showed a lower production of allergic mediators than that of the control group, and the ears pretreated with MLA had slighter vascular extravasation with lower absorbance values of Evans blue dye than that of the control group. These observations implied that the imidacloprid-induced effects on mast-cell function could not be blocked wholly by treatment with α7-nAChR antagonist. It is possible other AChR subunits may synergize with α7-nAChRs to mediate the imidacloprid-induced inhibition on mast-cell degranulation. Actually, α7, α9, and α10 subunits of nAChR on RBL cells and α3, α5, and α10 subunits of nAChR on human skin mast cells have potentially interaction and form hybrid nAChR receptors to affect mast-cell function.12–14,33 The specific antagonists of other subunits of nAChRs should be applied for further study about the exact mechanism of imidacloprid effect. Moreover, there is no single conserved mode of binding of neonicotinoids and related nicotinic ligands to their target receptor, as it is a variety of binding pockets depending on the combination of receptor subunits, the receptor subtype, its functional state, the structural flexibility of both the binding pockets and the ligands, etc.34 Consequently, it is better to get a descend imidacloprid effect curve with the ascend concentrations of antagonists of nAChRs, in order to provide new insights to understand exactly the mode of binding of nAChR antagonists to their cellular targets.
5. Conclusion
This study is the first time to demonstrate that α7-nAChRs can mediate the imidacloprid-induced inhibition of IgE-mediated mast cell activation. This work implies furtherly that imidacloprid likely contributes partly in considerably the lower incidence of allergy in rural areas of some countries with widespread imidacloprid use and increased inhabitant exposure. Moreover, this work presents well the mechanisms underlying the effects of imidacloprid, an AChR agonist, on IgE-meditated mast cell activation, suggesting that the nAChRs presented on mast cells including α7-nAChRs maybe a potential target to treat allergic diseases.Author contributions
H. C. and L. S. conceptualized, designed and wrote the manuscript. L. S., L. Z. and Y. W. performed experiments. J. G. and H. X. contributed to the animal test, and revised the manuscript carefully for important intellectual content. All authors read and approved the final manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The work was supported by the International Science & Technology Cooperation Program of China (No. 2013DFG31380), the Key Program of Natural Science Foundation of Jiangxi Province, China (No. 20133ACB20009), and the Research Program of State Key Laboratory of Food Science and Technology (No. SKLF-ZZA-201612).References
- S. Wernersson and G. Pejler, Nat. Rev. Immunol., 2014, 14, 478–494 CrossRef CAS PubMed.
- A. M. Gilfillan and C. Tkaczyk, Nat. Rev. Immunol., 2006, 6, 218–230 CrossRef CAS PubMed.
- C. C. Wu, S. C. Hsu, H. M. Shih and M. Z. Lai, Mol. Cell. Biol., 2003, 23, 6442–6444 CrossRef CAS PubMed.
- V. Parravicini, M. Gadina, M. Kovarova, S. Odom, C. Gonzalez-Espinosa, Y. Furumoto, S. Saitoh, L. E. Samelson, J. J. O'Shea and J. Rivera, Nat. Immunol., 2003, 3, 741–748 Search PubMed.
- K. Matsuda, M. Shimomura, M. Ihara, M. Akamatsu and D. B. Satelle, Biosci., Biotechnol., Biochem., 2005, 69, 1442–1452 CrossRef CAS PubMed.
- P. Mineau and C. Palmer, The impact of the nation's most widely used insecticides on birds, American Bird Conservancy, 2013, www.abcbirds.org/abcprograms/policy/toxins/Neonic_FINAL.pdf Search PubMed.
- F. M. Fishel, Pesticide toxicity profile: neonicotinoid pesticides, University of Florida, IFAS, 2005, http://edis.ifas.ufl.edu/pi117 Search PubMed.
- J. Kimura-Kuroda, Y. Komuta, Y. Kuroda, M. Hayashi and H. Kawano, PLoS One, 2012, 7, e32432 CAS.
- P. C. Badgujar, S. K. Jain, A. Singh, J. S. Punia, R. P. Gupta and G. A. Chandrate, Environ. Toxicol. Pharmacol., 2013, 35, 408–418 CrossRef CAS PubMed.
- M. M. Gatne, P. S. Bhoir and M. D. Deore, Toxicol. Int., 2006, 13, 89–92 CAS.
- L. B. Shi, L. Zou, J. Y. Gao, H. P. Xu, X. Y. Shi and H. B. Chen, Asia. Pac. Allergy, 2016, 6, 140–147 Search PubMed.
- F. Kindt, S. Wiegand, V. Niemeier, J. Kupfer, C. Löser, M. Nilles, H. Kurzen, W. Kummer, U. Gieler and R. V. Haberberger, Br. J. Dermatol., 2008, 159, 847–857 CrossRef CAS PubMed.
- R. T. Boyd, Crit. Rev. Toxicol., 1997, 27, 299–318 CrossRef CAS PubMed.
- N. C. Mishra, R. T. Boyd, S. P. Singh, S. Gundavarapu, R. J. Langley, S. Razani-Boroujerdi and M. L. Sopori, J. Immunol., 2010, 13, 591–595 Search PubMed.
- T. Yamamoto, T. Kodama, J. Lee, N. Utsunomiya, S. Hayashi, H. Sakamoto, H. Kuramoto and M. Kadowaki, PLoS One, 2014, 9, e85888 Search PubMed.
- X. Y. Meng, Y. X. Bai, J. Y. Gao and H. B. Chen, Food Chem., 2017, 219, 290–296 CrossRef CAS PubMed.
- B. Shi, J. Liang, X. H. Yang, Y. Wang, Y. N. Zhao, H. J. Wu, L. Y. Sun, Y. Zhang, Y. P. Chen, R. F. Li, Yu. Zhang, M. Hong and Y. F. Shang, Mol. Cell. Biol., 2007, 27, 5105–5119 CrossRef CAS PubMed.
- S. J. Galli, New concepts about the mast cell, N. Engl. J. Med., 1993, 328, 57–65 Search PubMed.
- M. El-Sibai and J. M. Backer, Eur. J. Immunol., 2007, 37, 261–270 CrossRef CAS PubMed.
- H. J. Jeong, H. N. Koo, H. J. Na, M. S. Hong, J. W. Eom, K. S. Kim, T. Y. Shin and Y. M. Kim, Cytokines, 2002, 18, 252–259 CrossRef CAS.
- N. Hirasawa, F. Santini and M. A. Beaven, J. Immunol., 1995, 154, 5391–5402 CAS.
- J. L. Galzi, F. Revah, A. Bessis and J. P. Changeux, Annu. Rev. Pharmacol. Toxicol., 1991, 31, 37–72 CrossRef CAS PubMed.
- A. Sobel, M. Weber and J. P. Changeux, Eur. J. Biochem., 1997, 80, 215–224 CrossRef.
- D. Paterson and A. Nordberg, Prog. Neurobiol., 2000, 61, 75–111 CrossRef CAS PubMed.
- K. Kawashima and T. Fujii, Life Sci., 2003, 74, 675–696 CrossRef CAS PubMed.
- W. Kummer, K. S. Lips and U. Pfeil, Cell Biol., 2008, 130, 219–234 CAS.
- J. D. Minna, J. Clin. Invest., 2003, 111, 31–33 CrossRef CAS PubMed.
- N. Kageyama-Yahara, Y. Suehiro, T. Yamamoto and M. Kadowaki, Biochem. Biophys. Res. Commun., 2008, 377, 321–325 CrossRef CAS PubMed.
- L. Mirotti, J. M. Castro, F. A. Costapinto and M. Russo, J. Allergy, 2010, 491928 CAS.
- R. Willemze, M. D. Luyer, W. A. Buurman and W. De Jonge, Nat. Rev. Gastroenterol. Hepatol., 2015, 12, 353–362 CrossRef CAS PubMed.
- C. Tilp, H. Bucher, H. Haas, M. J. Duechs, E. Wex and K. J. Erb, Clin. Exp. Allergy, 2016, 46, 957–972 CrossRef CAS PubMed.
- R. D. Pullan, J. Rhodes, S. Ganesh, V. Mani, J. S. Morris, G. T. Williams, R. G. Newcombe, M. A. H. Russell, C. Feyerabend, G. Thomas and U. Sawe, N. Engl. J. Med., 1994, 330, 811–815 CrossRef CAS PubMed.
- I. Wessler and C. J. Kirkpatrick, Br. J. Pharmacol., 2008, 154, 1558–1571 CrossRef CAS PubMed.
- H. Kayser, K. Lehmann, M. Gomes, W. Schleicher, K. Dotzauer, M. Moron and P. Maienfisch, Pest Manage. Sci., 2016, 72, 2166–2175 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |