
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene oxide/β-cyclodextrin composite as fiber coating for high efficiency headspace solid phase microextraction of organophosphate ester flame retardants in environmental water†
Jiali Denga,
Pengcheng Zhangb,
Tingting Jina,
Hongbin Zhoua and
Jing Cheng *a
*a
aKey Laboratory of Pesticide and Chemical Biology, Ministry of Education, Institute of Environmental Chemistry, College of Chemistry, Central China Normal University, Wuhan 430079, China. E-mail: chengjingok@mail.ccnu.edu.cn; Tel: +86-27-67867961
bDepartment of Biological Science and Technology, School of Environmental Studies, China University of Geosciences, Wuhan 430074, China
First published on 28th November 2017
Abstract
A novel solid-phase microextraction fiber was prepared using graphene oxide/β-cyclodextrin (GO/β-CD) composite as a coating material immobilized on a stainless steel wire using sol–gel technique. The coating has large surface area with uniform porous structure, stable performance at high temperature, and good coating preparation reproducibility. The prepared GO/β-CD sol–gel stainless steel fiber used as a headspace solid phase microextraction (HS-SPME) fiber was evaluated by the determination of organophosphate ester flame retardants (OPFRs) in water samples coupled to a gas chromatography/nitrogen phosphorus detector (GC/NPD). The studied OPFRs have a wide range of polarities and volatilities, and are not easily extracted by conventional commercial SPME fibers simultaneously. The prepared GO/β-CD SPME fiber showed good extraction performance for all the studied OPFRs. The developed HS-SPME-GC/NPD method provided low limits of detection ranging between 1.1 and 60.4 ng L−1. Good intra- and inter-day precision expressed as relative standard deviations for a single fiber were in the range of 2.2–9.6% and 2.7–9.7%, respectively, and fiber-to-fiber reproducibility was in the range of 6.3–10.9%. The calibration curves were linear in the concentration range from 10 to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ng L−1 (R > 0.9955). The developed method was applied for the analysis of tap, lake and river water samples and the relative recoveries were found to be in the range from 82.1 to 116.9%. Based on these features, the proposed fiber has great potential for widespread use as an effective and useful extraction tool.
000 ng L−1 (R > 0.9955). The developed method was applied for the analysis of tap, lake and river water samples and the relative recoveries were found to be in the range from 82.1 to 116.9%. Based on these features, the proposed fiber has great potential for widespread use as an effective and useful extraction tool.
1. Introduction
For several decades, a variety of flame retardant additives have been applied to polymers and resins found in commercial products, including electronics, furniture, and textiles, to serve to slow the combustion of treated materials to meet flammability standards. Organophosphate flame retardants (OPFRs) are used as replacements for the commercial polybrominated diphenyl ethers (PBDEs) that were phased out in 2004 owing to their persistence, bioaccumulation, and toxicity.1,2 Worldwide restrictions on the use of PBDEs in new products have resulted in the increasing use of OPFRs. The annual consumption of OPFRs in the EU increased from 84000 tons in 2004 to 91000 tons in 2006.3 OPFRs are just mixed and not chemically bonded with the materials, thus they can easily leach out of the material into the environment via volatilization, abrasion, and dissolution. As a result of these processes, OPFRs are widely distributed in the environment. Although OPFRs are generally considered to be less toxic and harmful than the brominated flame retardants (BFRs), recent studies showed that many kinds of OPFRs have obvious nerve toxicity, carcinogenicity, genetic toxicity, and can cause skin irritation and dermatitis,4–7 and especially halogenated OPFRs may affect neurodevelopment with similar or greater potency compared to organophosphorus pesticide and PBDEs organic pollutants.4 Furthermore, some of these chemicals (e.g. chlorinated alkyl phosphates) have a low degradation potential and thus may be persistent.8 The toxicity and growing usage of OPFRs raised concerns among authorities, and then the analysis of them in environmental media is therefore a hot spot of analytical research at present.9,10
In complex environmental media, the analyte is often found at trace level of concentration. As a result, the step of pretreatment is the most challenging and important in the whole analytical procedure. Until now, a number of pretreatment techniques have attempted to extract OPFRs from environmental samples, including liquid–liquid extraction (LLE),11 solid-phase extraction (SPE),12 membrane-assisted solvent extraction (MASE),13 dispersive liquid–liquid microextraction (DLLME),14 microporous membrane liquid–liquid extraction (MMLLE),15 hollow fiber liquid phase microextraction (HF-LPME)16 and solid phase microextraction (SPME).9,10 LLE and SPE have been the most often used extraction methods. However, these methods are tedious, labor-intensive and consume large amounts of organic solvents, and suffer from poor recovery of tri (2-chloroethyl) phosphate (TCEP) (31%)11 or tri(2-ethylhexyl) phosphate (TEHP) (50%).12 MASE employs a nonporous polypropylene membrane to improve the selectivity in the LLE of OPFRs from water samples, but the extraction efficiency in MASE for the most polar TCEP only 5%, and MASE is also quite time-consuming (3 h).13 DLLME was introduced since it drastically reduces organic solvents consumption which usually happened in the classical large-scale methods. However, DLLME exhibited unsatisfactory recovery for TEHP (about 40%).14 Although MMLLE is simple, inexpensive and has no carryover problems, it is unsuitable for the determination of TCEP and TEHP in water samples.15 Additionally, the extraction efficiencies of HF-LPME for both TEHP and TCEP were lower than other OPFRs and resulted in lower recoveries as well.16
Solid-phase microextraction (SPME) is based on the equilibrium of the target analytes between the sample and fiber coating. Thus, the stationary phase coated onto the fiber plays a key role for the extraction efficiency.17 There are some commercially available SPME fibers, such as polydimethylsiloxane (PDMS), polyacrylate (PA), divinylbenzene (DVB), carboxen (CAR), and carbowax (CW), and the home-made coating materials include polypyrrole,18 polyaniline,19 polymeric ionic liquid,20 anodized aluminum wire,21 ionic liquids,9 metal–organic framework,22 carbon materials based coatings23–25 and so on. The commercial polydimethylsiloxane (PDMS)/divinylbenzene (DVB) SPME fiber and home-made ionic liquid-based sol–gel SPME fiber have been reported to extract OPFRs in environmental media.9,10 However, the commercial PDMS/DVB fiber used in headspace-solid phase microextraction (HS-SPME) method, was still not suitable for extracting TEHP due to the low recovery (26.7%) and unacceptable deviation (64.8%),9 and a home-made ionic liquid-based sol–gel SPME fiber has been reported to extract OPFRs from environmental water samples and exhibited good extraction capability and precisions when it applied for the simultaneous extraction of different polarity OPFRs, even including TCEP and TEHP.10 However, ionic liquid is expensive and adsorbs water so easily in the air that it is not suitable for practical application.
Graphene oxide (GO), as a graphene precursor, exhibits similar adsorption ability to graphene which shows excellent extraction performance as coating materials of SPME fiber.26 Compared with graphene, GO abounds with many oxygen-containing chemical groups across the basal planes, such as epoxy groups, hydroxyl groups, and carboxyl groups, which indicates that it can be readily interacted with other materials.27 Furthermore, GO is much cheaper than graphene, suitable for large scale production. Therefore, GO is more practical than graphene as an adsorbent material. However, there are many hydroxy and epoxy groups on the GO sheets which make GO adsorb polar molecules easily through electrostatic interaction. Thus, introducing a new functional molecule to GO through covalent or noncovalent strategies to bring new or enhanced functions to GO is highly desirable and important.
Cyclodextrins (CDs) are cyclic oligosaccharides consisting of six, seven, or eight (α-1,4)-linked-D-glucopyranose units (α, β, or γ-CD, respectively), which are toroidal in shape with a hydrophobic inner cavity and a hydrophilic exterior.28,29 Such characteristics make them selectively bind many kinds of inorganic, organic, and biological molecules into their cavities, even if only partially, to form supramolecular complexes without structural changes through host–guest interactions (van der Waals force, hydrophobic interaction, electrostatic affinity, dipole–dipole interaction, and hydrogen bonding).29 β-CD is the most largely produced CD used in many fields, including pharmaceuticals, food, cosmetics, chemical products, and technologies.30 The applications include chemical separations,31,32 adsorbents,27,33 food processing,34 and pharmaceutical excipients.35 Once GO is functionalized with β-CD, it is likely to gain a new material simultaneously having the properties of both GO (high surface-to-weight ratio and large delocalized-electron system) and β-CD (high supramolecular recognition and enrichment capability) through combining their individual characteristics. However, as far as we know, there has been no report yet for the application of the β-CD-functionalized GO as a fiber coating material in SPME for the analysis of volatile or semivolatile OPFRs.
The new preparation methods of a SPME fiber, including direct use of uncoated fibers,36 epoxy–glue solid sorbent,37 physical deposition,38 electrochemical deposition,39 and sol–gel technique.9,18 Among the various coating preparation methods, sol–gel technique is popular to improve the performance of SPME coating. The synthetic conditions of sol–gel are usually mild, and the coating prepared by the sol–gel technique is often of high purity and homogeneity, high thermal and solvent stability, and high surface areas with porous structure.40 To overcome the fragile of fused silica fibers, the coating fiber was immobilized on stainless steel wire due to its rigidity, low cost, non-toxicity and durability. The integrated use of the sol–gel technique and the stainless steel wire for the preparation of the SPME fibers would take the advantages of the both and possibly enhance the durability of the prepared fiber.
In this work, β-CD decorated GO material was prepared under the conditions of ultrasound irradiation. The β-CD macromolecules were covalently grafted onto the surface of GO through 3-aminopropyltriethoxysilane for forming amido bond with GO and forming silicon oxygen bond with β-CD. A novel β-CD–GO-coated SPME fiber was prepared by immobilizing the β-CD–GO through a sol–gel technique onto a prefunctionalized stainless-steel wire. The extraction performance of the β-CD–GO-coated fiber was evaluated with volatile and semivolatile OPFRs as the model analytes because of their toxicity and widespread environmental occurrence. The main experimental parameters including the extraction time and temperature, desorption temperature and time, the ratio of the volume of water solution to headspace (HS), stirring rate, and ionic strength were investigated to achieve a good extraction efficiency. In addition, the fiber lifetime was also evaluated. Finally, a HS-SPME with the β-CD–GO coated fiber followed by GC with nitrogen phosphorus detector (NPD) for the determination of some OPFRs was developed. The method was applied for the determination of the OPFRs in tap, lake and river water samples.
2. Experimental
2.1 Reagents and materials
Graphite powder (50 meshes), potassium permanganate (KMnO4), sulfuric acid (H2SO4, 98%), hydrogen peroxide (H2O2), sodium chloride (NaCl), β-cyclodextrin (β-CD), ethanol, acetone, concentrated hydrochloric acid and other chemicals were all of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Methyltrimethoxysilane (MTMOS, 97%), poly (methylhydrosiloxane) (PMHS), and (3-mercaptopropyl) trimethoxysilane (MPTS, 95%) were purchased from J&K Scientific Ltd. (Beijing, China). Trifluoroacetic acid (TFA) and 3-aminopropyltriethoxysilane (3-APTES) were purchased from Aladdin Reagent Co., Ltd. (Shanghai, China). Tripropyl phosphate (TPrP), tri-n-butyl phosphate (TnBP), tri(2-chloroethyl) phosphate (TCEP) and tri(2-ethylhexyl) phosphate (TEHP) were purchased from Dr Ehrenstorfer Gmbh (Augsburg, Germany). Tri(2-chloroisopropyl) phosphate (TCPP), 2-ethylhexyl diphenyl phosphate (EHDPP) were obtained from Sigma-Aldrich (Louis, USA), and triphenyl phosphate (TPhP), tricresyl phosphate (TCrP) were purchased from Aucc stand (NewHaven, USA). The physicochemical properties of the nine OPFRs are listed in Table S1 (see ESI†).2.2 Apparatus
A magnetic stirrer (model: MC-MAG HS7) purchased from IKA instrument equipment Co., Ltd. (IKA, Guangzhou, China) was employed for stirring and heating samples during the extraction. Ultrasonic instrument KQ-100DE was purchased from Kunshan Ultrasonic Instrument Co., Ltd. (Jiangsu, China). The commercial fibers coated with 100 μm thickness of polydimethylsiloxane (PDMS) coating and 80 μm thickness of polyamide (PA) coating were used for comparative purposes.The analytes were separated and detected on a TRACE 1300 system (Thermo Fisher Scientific, MA, USA), equipped with split/splitless injector, a TR-1 capillary column (30 m × 0.32 mm i.d., 0.25 m film thickness) and nitrogen–phosphorous detector (NPD). Ultrapure nitrogen (>99.999%) was used as carrier gas at a constant flow of 2 mL min−1 and it also used as make-up gas at 15 mL min−1. Synthetic air and hydrogen (99.999%) were used as detector gases at flow rates of 60 and 2.3 mL min−1, respectively. The oven temperature was programmed as follows: initial temperature from 80 °C (held for 1.0 min), heating at 20 °C min−1 to 170 °C (held for 1.0 min), then at 4 °C min−1 to 270 °C (held for 1.0 min). The injector and the detector temperatures were set at 270 and 320 °C, respectively.
Surface structure of the prepared fiber was investigated by JSM-6700F scanning electron microscopy (SEM) (FESEM, JEOL, Japan). Fourier transform infrared spectroscopy was performed with a Thermo Nicolet Is50 FT-IR instrument (Boston, MA, USA). The thermal stability of the developed coatings was investigated by a STA 409 PC thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) instrument. The samples were heated from 30 °C to 500 °C under a nitrogen atmosphere at a heating rate of 10 °C min−1.
2.3 Water sample
River water was collected from the Changjiang River (Wuhan, China). Lake water was collected from East Lake (Wuhan, China). Tap water was collected from Central China Normal University (Wuhan, China). All water samples were stored in amber glass bottles at 4 °C in darkness before use.2.4 Preparation of GO/β-CD sol–gel stainless steel fiber and HS-SPME procedures
As shown in Fig. 1, the whole process of preparation of GO/β-CD sol–gel stainless steel fiber and its HS-SPME procedures were presented as follows: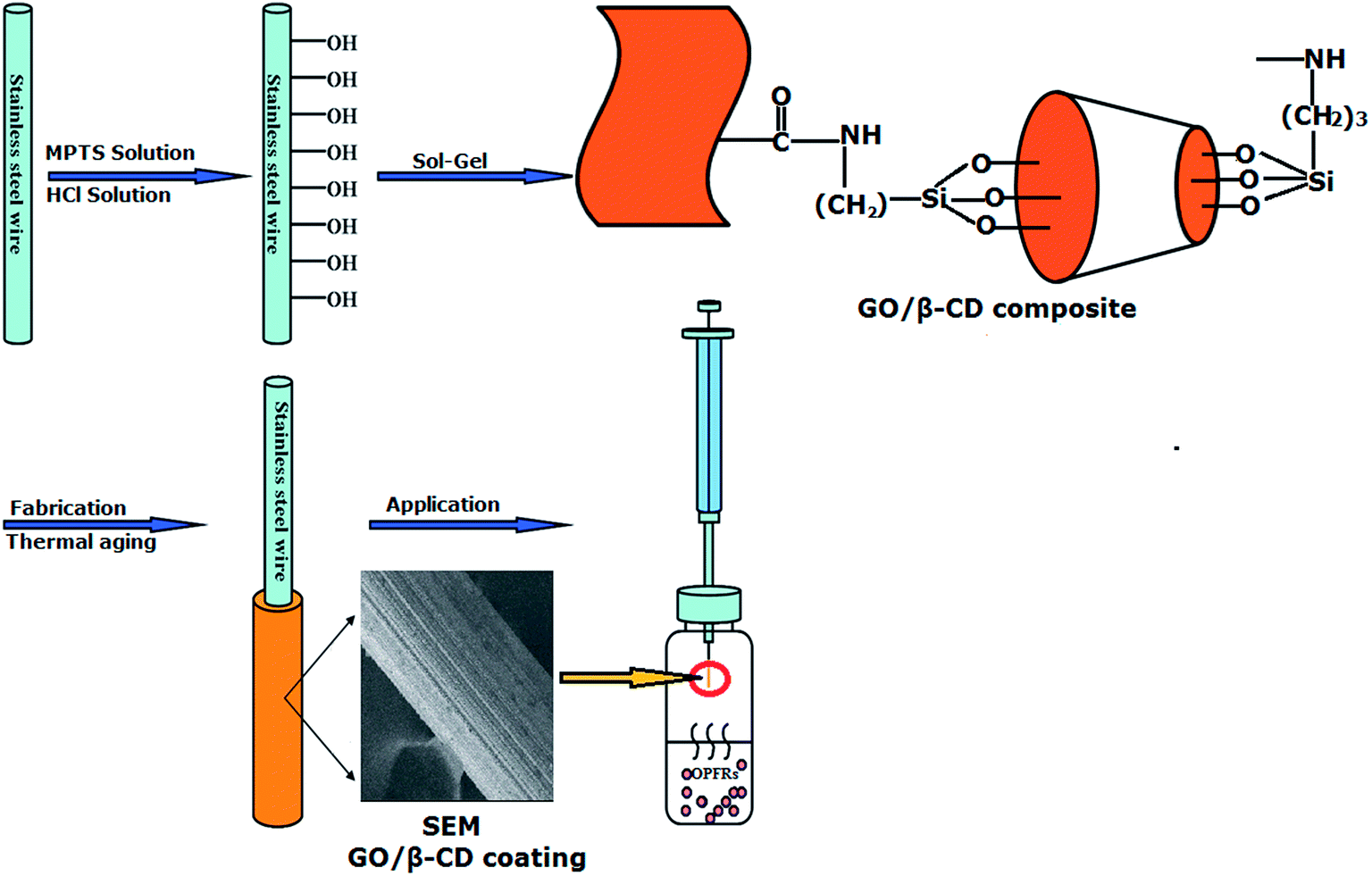 | ||
| Fig. 1 Schematic presentation of preparation of GO/β-CD sol–gel stainless steel fiber coating and its application for HS-SPME of OPFRs. | ||
Firstly, the stainless steel wire was modified. As shown in Fig. S1 (see ESI†), a stainless steel wire (about 2.5 cm of the wire end) vibrated ultrasonically in ethanol, acetone and again in ethanol for 10 min in each to remove any contaminants. The MPTS was used to modify the surface of the steel wire. For modification, 1.5 cm of the wire end was dipping into a solution of 10−3 mol L−1 MPTS in ethanol for 3 h. It is generally known that under ambient conditions, surfaces of common metals and alloys such as stainless steel are covered with oxide species. They can react with MPTS to form a monolayer.39 Afterwards, the wires were rinsed with ethanol and dried under nitrogen atmosphere. Finally, the modified stainless steel wire was immersed in hydrochloric acid (0.1 M in water) for 1 h to hydrolyze the methoxy groups.
Secondly, GO/β-CD composite was prepared. Graphene oxide (GO) was prepared by oxidizing natural graphite powders and then transformed into GO sheets.39 Then, the schematic synthesis process for GO/β-CD composite was shown in Fig. S2.† 30 mg GO was dissolved in 30 mL DMF by sonication and then 3 mL of APTES was added to the above GO/DMF mixture. The mixture was stirred at 70 °C for 30 min. GO would react with APTES by forming amido bond between the –COOH, epoxy groups from GO and –NH2 from APTES. After the reaction finished, 1 g β-CD was added to the obtained mixture, after stirring 5 h, the mixture was centrifuged at 10000 rpm for 8 min and then, the resultant precipitate was washed by DMF to remove the unreacted β-CD and finally dried under at 60 °C to yield GO/β-CD composite.
Thirdly, 3 mg of GO/β-CD composite was dissolved in 100 μL DMF in a 1.5 mL micro-centrifuge tube, and the tube was vibrated sonically for 2.5 h. Then 300 μL MTMOS and 50 μL PMHS, used as sol–gel precursor and deactivation reagent, respectively, were added to the above-mentioned micro-centrifuge tube and vibrated sonically for 20 min. 60 μL 95% TFA solution, used as acid catalyst, was added and mixed for 10 min. The GO/β-CD composite sol–gel solution was obtained. Finally, the modified stainless fire wire was vertically dipped into the GO/β-CD sol–gel solution 1.5 cm deep for 10 min and a gel coating was formed on the wire surface. Then, it was pulled out and dried at room temperature for 20 min. This coating process was repeated several times until the desired thickness of the coating (about 10 μm) was obtained. Then, the coated fiber was put in a drier for curing at room temperature for 24 h. Prior to use, the fiber was assembled into a 5 μL GC microsyringe and then conditioned in the GC injector at 100 °C for 0.5 h, each additional 50 °C for 0.5 h to 270 °C.
The sol–gel coated fiber was prepared with the same procedures in the absence of GO/β-CD composite. The GO sol–gel coating fiber and β-CD sol–gel coating fiber were prepared with the same procedures as the GO/β-CD coating fiber except that GO and β-CD were used instead of GO/β-CD composite, respectively.
Finally, HS-SPME procedures were performed in a 20 mL amber glass vial containing 14 mL aqueous solution which was tightly sealed with an aluminum cap. After adding 4.2 g NaCl, the 20 mL amber glass vial with the fiber exposed to the headspace above the sample was put in a 80 °C oil bath on the magnetic stirrer for 30 min at a constant stirring rate of 600 rpm. After extraction, the fiber was withdrawn into the needle of the GC micro-syringe and removed from the sample vial. Finally, the GC micro-syringe was inserted into the GC injector to desorb the analytes at 270 °C for 4 min and then for GC/NPD analysis.
3. Results and discussion
3.1 Characterization of the prepared fiber
The SEM images of a stainless steel fiber, the prepared β-CD sol–gel coating bonded on the stainless steel fiber, the prepared GO sol–gel coating bonded on the stainless steel fiber and the prepared GO/β-CD sol–gel coating bonded on the stainless steel fiber were illustrated in Fig. S3a–d,† respectively. Fig. S3† let us see clearly the morphological structure of β-CD sol–gel coating, GO sol–gel coating and GO/β-CD coating which were homogeneously bonded on the stainless steel fiber with fluted structures. GO sol–gel coating owns similar fluted morphological structure with GO/β-CD coating. Both GO sol–gel coating and GO/β-CD coating are much rougher than β-CD coating.As shown in Fig. 2a, the strong adsorption peak around 3450 cm−1 in the FT-IR spectra was the O–H stretching vibration from both GO and β-CD. The O–H stretching vibration adsorption peaks of both GO/β-CD composite and GO/β-CD sol–gel coating moved to lower wave number at around 3420 cm−1 due to the formation of hydrogen bond between GO and β-CD. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration adsorption peak at 1719 cm−1 was moved to lower wave number at 1680 cm−1 and 1670 cm−1 due to the formation of amido bond from the GO/β-CD composite and GO/β-CD sol–gel coating, respectively. The typical C–O/C–C stretching vibrations at 1030/1080 cm−1 and CH2 stretching vibrations at 2928/2988 cm−1 of β-CD were found in GO/β-CD composite and GO/β-CD sol–gel coating. All these results confirmed that the β-CD were covalently bonded to the surface of GO. As illustrated in Fig. 2b, the thermal stability of GO/β-CD sol–gel coating was evaluated by the TGA and DSC. There is no obvious weight loss (only 5% weight loss) from 70–350 °C which might be due to the loss of water. The result indicates that the coating has good thermal stability and it can endure the high temperature in the GC/NPD injector. The lifetime of the fiber is very crucial for practical application. The high temperature of the injection port of GC and the solvent in the matrix are the main factors which damaged the coating. As shown in Fig. S4,† the extraction performance of the prepared fiber without significant deterioration after being used more than 150 times in an adsorption/desorption cycle. The long lifetime of the prepared fiber may be due to its good thermal and mechanic stability.
O stretching vibration adsorption peak at 1719 cm−1 was moved to lower wave number at 1680 cm−1 and 1670 cm−1 due to the formation of amido bond from the GO/β-CD composite and GO/β-CD sol–gel coating, respectively. The typical C–O/C–C stretching vibrations at 1030/1080 cm−1 and CH2 stretching vibrations at 2928/2988 cm−1 of β-CD were found in GO/β-CD composite and GO/β-CD sol–gel coating. All these results confirmed that the β-CD were covalently bonded to the surface of GO. As illustrated in Fig. 2b, the thermal stability of GO/β-CD sol–gel coating was evaluated by the TGA and DSC. There is no obvious weight loss (only 5% weight loss) from 70–350 °C which might be due to the loss of water. The result indicates that the coating has good thermal stability and it can endure the high temperature in the GC/NPD injector. The lifetime of the fiber is very crucial for practical application. The high temperature of the injection port of GC and the solvent in the matrix are the main factors which damaged the coating. As shown in Fig. S4,† the extraction performance of the prepared fiber without significant deterioration after being used more than 150 times in an adsorption/desorption cycle. The long lifetime of the prepared fiber may be due to its good thermal and mechanic stability.
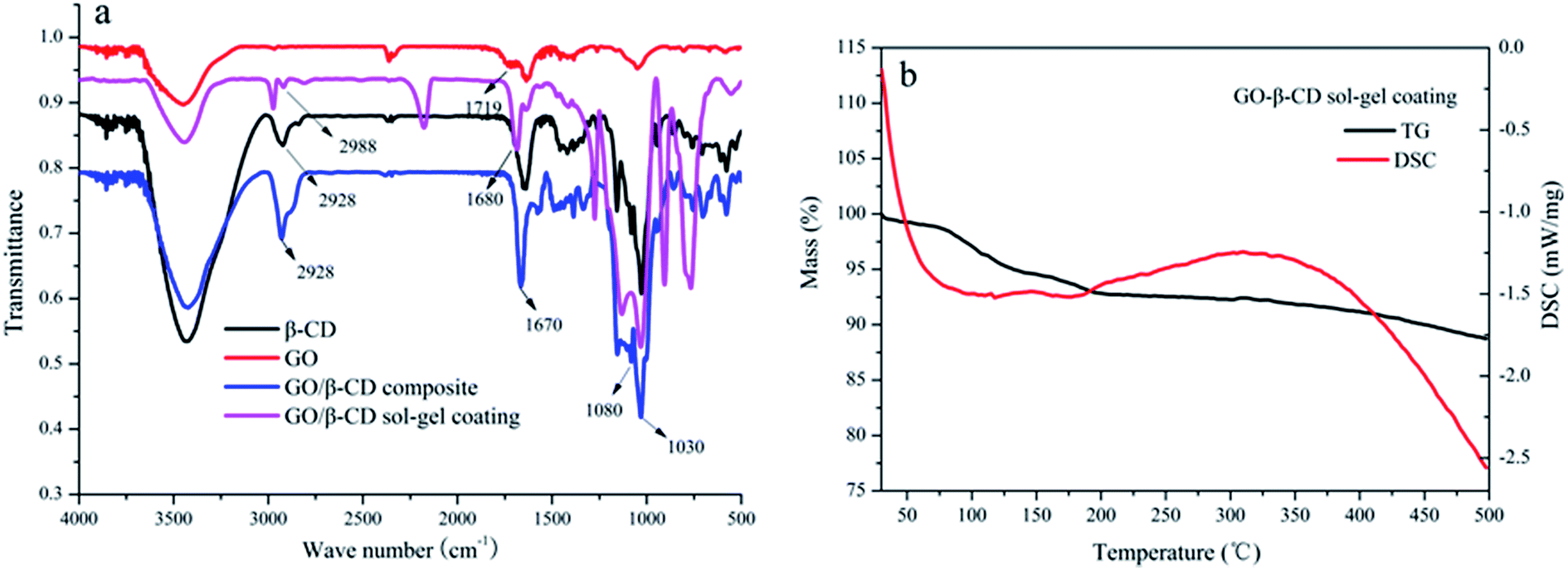 | ||
| Fig. 2 FT-IR spectra of β-CD, GO, GO/β-CD composite and GO/β-CD sol–gel coating (a). TGA and DSC curves of the GO/β-CD sol–gel coating (b). | ||
As shown in Fig. 3, the N2 adsorption desorption isotherms of sol–gel coating (a), GO sol–gel coating (b), β-CD sol–gel coating (c) and GO/β-CD sol–gel coating (d) all showed IV type curves, which correspond to the capillary condensation phenomenon of porous adsorbents. The hysteresis loops of Fig. 3a–c display H3 type curves, which meant the pore structure was very irregular. While the hysteresis loops of Fig. 3d showed a prominent adsorption platform which reflected uniform pore size distribution of GO/β-CD sol–gel coating. The pore structure properties are shown in Table 1. The specific BET surface area, mean pore diameter and the mesopore volume of GO/β-CD sol–gel coating were bigger than pure sol–gel coating, GO sol–gel coating and β-CD sol–gel coating, indicating that the extraction capacity of GO/β-CD sol–gel coating fiber would be the highest.
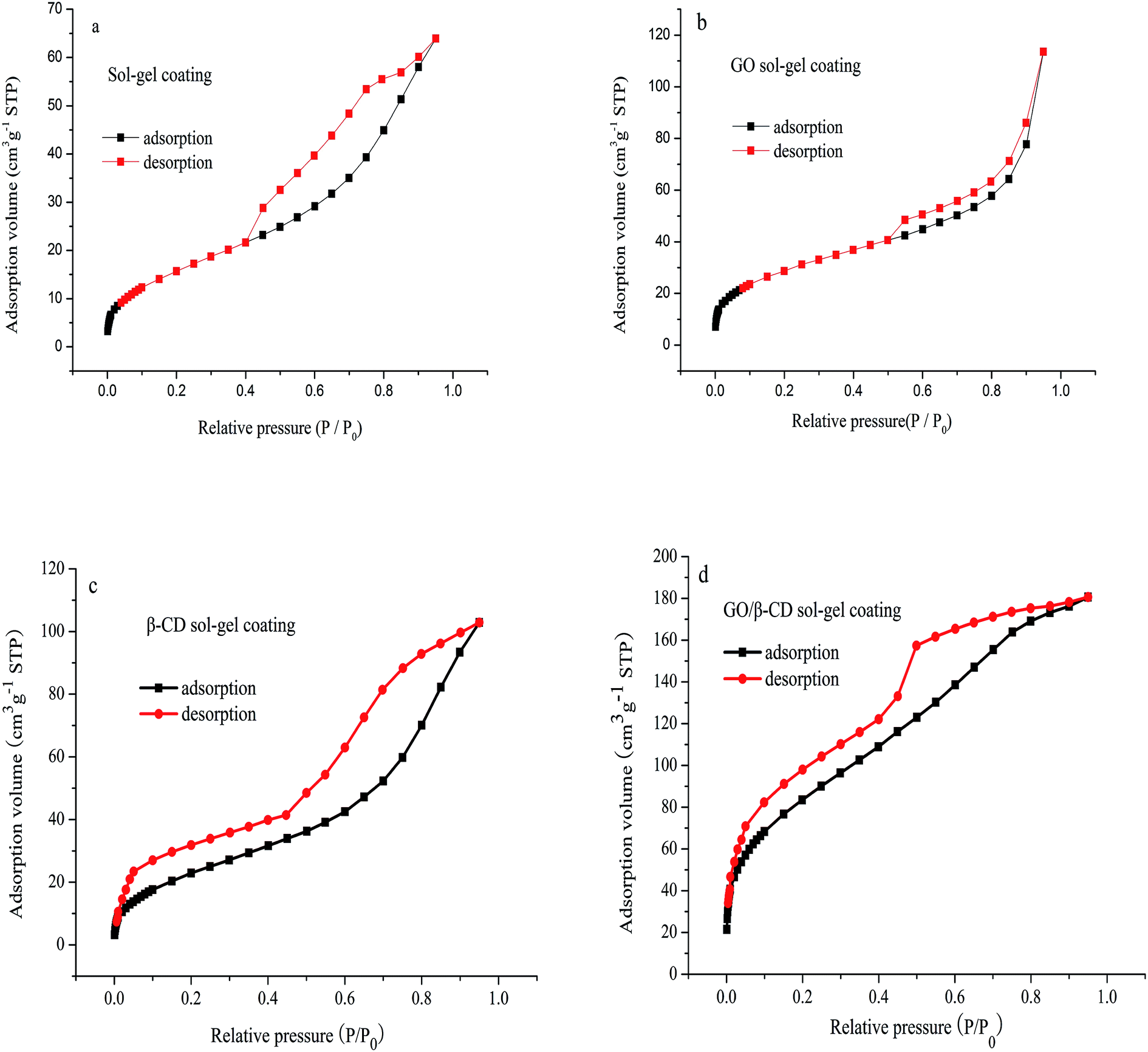 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms of the pure sol–gel coating (a); GO sol–gel coating (b); β-CD sol–gel coating (c) and GO/β-CD sol–gel coating (d). | ||
| Samples | Mesopore volume (cm3 g−1) | Mean pore diameter (Å) | SBET (m2 g−1) |
|---|---|---|---|
| a SBET: specific area calculated according to BET method. | |||
| Sol–gel coating | 0.165 | 3.179 | 62.127 |
| GO-sol–gel coating | 0.095 | 7.032 | 107.478 |
| β-CD-sol–gel coating | 0.153 | 3.179 | 73.248 |
| β-CD/GO-sol–gel coating | 0.280 | 8.079 | 229.375 |
3.2 Optimization of the HS-SPME conditions
HS-SPME mode can minimize the matrix interference from sample and prolong the fiber lifetime when it was used for the extraction of the semi-volatile and volatile compounds.40 To achieve the best extraction efficiency for the nine OPFRs, the following parameters were investigated and optimized: (a) extraction temperature; (b) extraction time; (c) volume of water sample; (d) stirring rate (e) ionic strength; (f) desorption time. Each experiment was performed in triplicate. Respective data and figures are given in Fig. S5.† The following experimental conditions were found to give best results: (a) 80 °C was chosen as the extraction temperature. (b) 30 min was selected as the preferred extraction time. (c) 14 mL water was used to perform in 20 mL sample vial. (d) Stirring rate of 600 rpm was used. (e) 30% (w/v) NaCl was chosen as the optimum content. (f) All desorption processes were performed at 270 °C for 4 min (Table 2).| Analytes | Linear range (ng L−1) | R | LOD (ng L−1) | LOQ (ng L−1) | RSD (%, n = 6)a | EFs | ||
|---|---|---|---|---|---|---|---|---|
| Repeatability (single fiber) | Reproducibility (fiber-to-fiber) | |||||||
| Intra-day | Inter-day | |||||||
| a Spiking level: 10 μg L−1 TnBP, TPrP, TPhP, EHDPP, TCrP, TEHP and 50 μg L−1 TCEP, TCPP, TDCP. | ||||||||
| TPrP | 50–5 × 104 | 0.9995 | 5.7 | 14.2 | 2.2 | 6.0 | 8.5 | 176.8 |
| TnBP | 10–5 × 104 | 0.9976 | 1.1 | 2.7 | 4.3 | 7.6 | 7.1 | 663.9 |
| TCEP | 1 × 103–1 × 105 | 0.9969 | 60.4 | 170.5 | 9.6 | 5.2 | 9.0 | 22.5 |
| TCPP | 100–5 × 104 | 0.9955 | 20.7 | 56.9 | 8.2 | 8.9 | 8.3 | 141.5 |
| TDCP | 100–5 × 104 | 0.9998 | 12.5 | 28.8 | 6.5 | 7.2 | 6.3 | 158.5 |
| TPhP | 50–5 × 104 | 0.9967 | 5.2 | 16.8 | 8.3 | 9.7 | 10.9 | 1307.6 |
| EHDPP | 50–5 × 104 | 0.9970 | 6.9 | 20.4 | 7.2 | 8.1 | 8.4 | 1079.4 |
| TEHP | 50–5 × 104 | 0.9974 | 7.2 | 19.9 | 8.4 | 9.4 | 7.1 | 432.9 |
| TCrP | 50–5 × 104 | 0.9986 | 17.7 | 48.5 | 6.8 | 3.3 | 8.1 | 1270.5 |
3.3 Method validation
Under the optimized conditions, the prepared GO/β-CD sol–gel coating fiber was used for determination of nine OPFRs in different environmental water coupled to GC/NPD. A series of experiments with regard to the linearity, limits of detection (LOD), limits of quantification (LOQs), repeatability and enrichment factors was performed to validate the HS-SPME-GC/NPD method. As shown in Table 3, the method using a GO/β-CD sol–gel coating stainless steel wire as the HS-SPME fiber showed a wide linear range with correlation coefficients (R) ranging from 0.9955 to 0.9998. The LODs and LOQs for the nine analytes ranged from 1.1 to 60.4 ng L−1 and 2.7 to 170.5 ng L−1 based on the ratio of signal-to-noise identical to 3 and 10, respectively. The intra- and inter-day precisions of the method, expressed as relative standard deviations (RSDs) were less than 9.7%. Enrichment factor (EF) was defined as the ratio of the sensitivity of an analyte after extraction to that before extraction (i.e., by direct injection of 1 μL of standard solution) using the chromatographic peak area for quantification, EFs for all analytes were in the range from 22.5 to 1307.5. The higher polarity and boiling point of the TCEP may result in the lower EF than the other OPFRs. The proposed method had high enrichment factors for most analytes because the fiber combined the advantages of β-CD with the hydrophobic interior cavity and hydrophilic peripheral face and GO with large surface and highly delocalized conjugate system of the π-electron and the π–π interaction. The fiber-to-fiber reproducibility was also determined using three different GO/β-CD coating stainless steel fibers fabricated in the same batch, and the RSDs ranged from 6.3 to 10.9%, which suggests that the fabrication of fiber is reproducible.| Method | RSD (%) | LOD (ng L−1) | Recovery (%) | EFs | Ref. |
|---|---|---|---|---|---|
| LLE/GC-MS | 12–33 | 1.9–6.1 | 31–107 | — | 11 |
| SPE/LC-MS | 4–13 | 0.9–24.5 | 50–128 | — | 12 |
| MASE/LC-MS/MS | 2–19 | 3–25 | 5–98 | 9–196 | 13 |
| DLLME/GC-NPD | 2–17 | 10–80 | 23–109 | 195–906 | 14 |
| MMLLE/GC-NPD | 4–16 | 20–120 | 4–51 | 34–1400 | 15 |
| HF-LPME/GC-MS | 2.1–10.4 | 2.6–120.0 | 24–132 | 132–294 | 16 |
| SPME (PDMS–DVB fiber)/GC-NPD | 5.3–64.8 | 4.5–7.6 | 26.7–119.2 | — | 9 |
| SPME (IL-based sol–gel fiber)/GC-FPD | 3.3–7.6 | 0.7–11.6 | 73.2–101.8 | 13.4–69.1 | 10 |
| SPME (β-CD–GO-based sol–gel fiber)/GC-NPD | 2.2–9.6 | 1.1–60.4 | 82.1–116.9 | 22.5–1307.6 | This study |
3.4 Comparison with the commercial fibers and some published methods
The extraction efficiency of GO/β-CD sol–gel coating fiber was compared with pure sol–gel coating fiber (10 μm), GO sol–gel coating (10 μm), β-CD sol–gel coating fiber (10 μm), commercial PDMS (100 μm) and PA fiber (80 μm) under their optimized conditions, respectively. As shown in Fig. 4, except for the OPFRs containing benzene ring, GO/β-CD sol–gel coating fiber showed the highest extraction efficiency among all the other fibers. GO sol–gel coating fiber displayed slightly higher extraction efficiency for three OPFRs containing benzene ring than GO/β-CD sol–gel coating fiber. That may be because the GO has higher delocalized conjugate system of the π-electron and π–π interaction than GO/β-CD, so it has higher affinity for aromatic compounds. The performance of GO/β-CD-coating materials for extraction in molecules with different benzene or non-polar groups will be further deeply investigated in our group.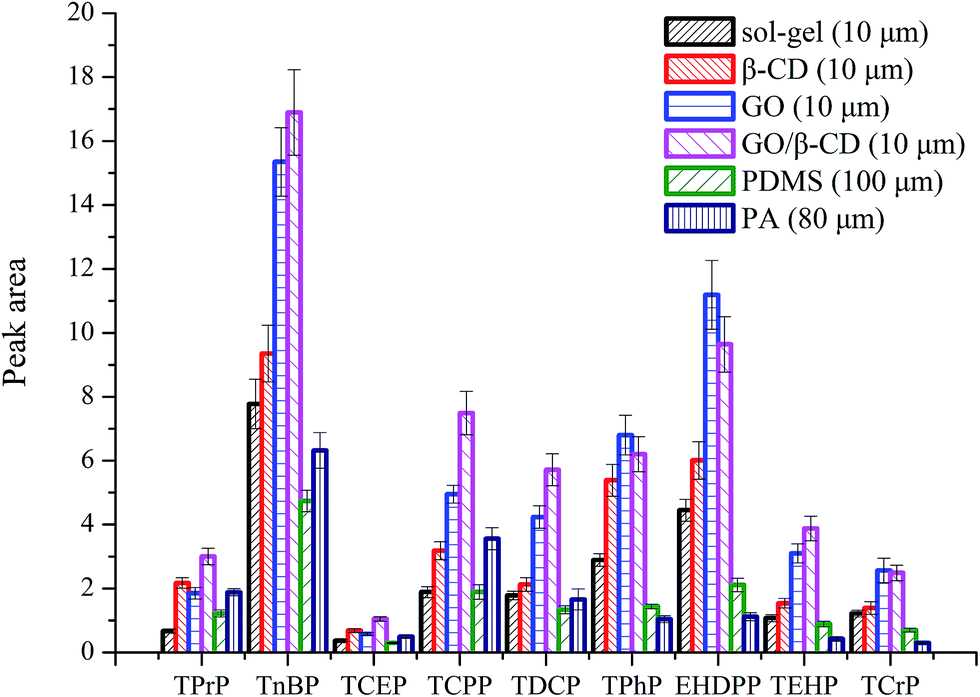 | ||
| Fig. 4 Comparison of the extraction efficiencies of OPFRs by the bare sol–gel fiber, β-CD fiber, GO fiber, GO/β-CD fiber, PDMS and PA fiber. Experimental conditions: concentration of OPFRs, 50 ng mL−1 TPrP, TnBP, TPhP, EHDPP, TEHP, TCrP, 250 ng mL−1 TCEP, TCPP, TDCP; extraction time, 30 min; extraction temperature, 80 °C; stirring rate, 600 rpm; the concentration of salt, 20% (w/v); desorption time, 4 min. | ||
To illustrate the advantages of the GO/β-CD-coating materials as a novel SPME coating fiber, the comparative study of our developed method with other reported sample preparation procedures9–16 is performed and the results are presented in Table 3. As shown in Table 3, the LODs obtained by the developed β/CD–GO-based sol–gel fiber SPME/GC-NPD method are comparable to those reported in other papers, and the recoveries are better than those published methods.
3.5 Application for analysis of environmental water
The feasibility of the proposed HS-SPME/GC method for determining OPFRs in lake water, river water, and tap water was investigated by analyzing 14 mL of the real water samples spiked with 10 μL of acetone. In Fig. 5 and Table 4, no target analytes were detected in the tap water. TnBP (97.7 ng L−1), TCPP (150.4 ng L−1), TPhP (162.1 ng L−1), TEHP (185.3 ng L−1) detected in the river water, TEHP (205.4 ng L−1) was detected in the lake water. TEHP was detected in both river and lake water due to its poor degradability, which was consistent with Letcher's study.35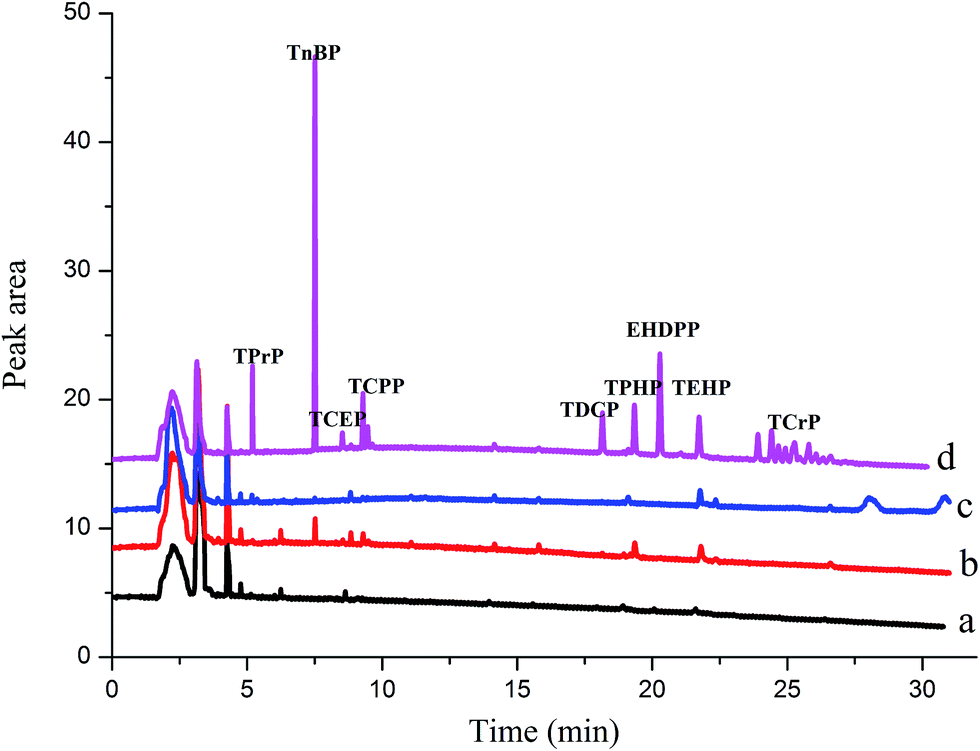 | ||
| Fig. 5 Chromatograms of blank tap water after extraction (a), blank river water after extraction (b), blank lake water after extraction (c) and river water spiked with OPFRs after extraction (d) (spiking level: 1 ng mL−1 for TPrP, TnBP, TPHP, EHDPP, TEHP, TCrP, 5 ng mL−1 TCEP, TCPP, TDCP). | ||
| Analytes | Spiked (ng L−1) | Recovery ± RSD (%) (n = 5) | ||
|---|---|---|---|---|
| Lake water | River water | Tap water | ||
| TPrP | 0 | nd | nd | nd |
| 100 | 97.0 ± 3.0 | 97.0 ± 3.0 | 96.7 ± 6.7 | |
| 1000 | 82.1 ± 4.3 | 83.0 ± 4.3 | 87.0 ± 7.7 | |
| TnBP | 0 | nd | 97.7 ± 7.8 | nd |
| 100 | 96.0 ± 5.2 | 83.5 ± 5.2 | 97.2 ± 3.9 | |
| 1000 | 86.7 ± 9.7 | 108.2 ± 9.7 | 92.9 ± 8.3 | |
| TCEP | 0 | nd | nd | nd |
| 500 | 113.2 ± 10.4 | 100.3 ± 10.4 | 99.9 ± 5.6 | |
| 5000 | 94.2 ± 8.2 | 106.8 ± 7.8 | 108.6 ± 4.2 | |
| TCPP | 0 | nd | 150.4 ± 6.5 | nd |
| 500 | 101.8 ± 3.6 | 97.6 ± 9.5 | 101.7 ± 3.3 | |
| 5000 | 106.2 ± 5.9 | 108.8 ± 4.5 | 83.2 ± 8.6 | |
| TDCP | 0 | nd | nd | nd |
| 500 | 101.0 ± 9.4 | 95.0 ± 6.2 | 83.2 ± 9.7 | |
| 5000 | 112.1 ± 9.4 | 94.5 ± 9.7 | 98.4 ± 8.6 | |
| TPhP | 0 | nd | 162.1 ± 8.8 | nd |
| 100 | 99.7 ± 7.7 | 98.2 ± 6.0 | 95.6 ± 3.4 | |
| 1000 | 85.4 ± 8.9 | 111.6 ± 8.8 | 94.7 ± 8.7 | |
| EHDPP | 0 | nd | nd | nd |
| 100 | 113.9 ± 3.2 | 108.5 ± 7.3 | 85.0 ± 10.1 | |
| 1000 | 86.8 ± 7.6 | 89.4 ± 3.7 | 98.1 ± 9.4 | |
| TEHP | 0 | 205.4 ± 6.9 | 185.3 ± 9.2 | nd |
| 100 | 109.8 ± 4.9 | 96.8 ± 6.5 | 90.6 ± 5.5 | |
| 1000 | 105.0 ± 8.8 | 89.4 ± 7.7 | 99.0 ± 4.3 | |
| TCrP | 0 | nd | nd | nd |
| 500 | 96.8 ± 9.3 | 94.1 ± 8.1 | 100.9 ± 7.8 | |
| 5000 | 84.7 ± 7.9 | 110.2 ± 3.1 | 86.5 ± 6.8 | |
Recovery testing was carried out by spiking a standard mixture prepared in acetone to 14 mL of real water matrices and the amount of acetone was kept at 1% (v/v). The relative recoveries and the RSDs are listed in Table 4. The results showed that the relative recoveries of OPFRs (n = 3) ranged from 82.1 to 116.9%, and the RSDs were between 3.0% and 10.4%. The results also prove that the proposed method is robust and can be applied to the determination of OPFRs in different water samples with satisfying accuracy and precision. In addition, the good recoveries and RSDs of TEHP also showed that the GO/β-CD sol–gel coating fiber may be more suitable for extracting OPFRs than PDMS/DVB fiber.
4. Conclusions
In this work, a simple, low-cost and sensitive HS-SPME-GC/NPD method using GO/β-CD sol–gel stainless steel coating fiber was proposed for the determination of nine OPFRs from water samples. Under the optimal conditions, the proposed method exhibits good precision, detection limit and linear response over a wide concentration range of OPFRs. The proposed method had been successfully applied for the determination of OPFRs in the real water samples with good recoveries and RSDs, including TEHP. Based on these highly efficient results, the proposed approach provides another new and alternative GO-based SPME fiber for detecting OPFRs in water samples, which goes forward a step on our previous study.36 Moreover, this is also the first report about the preparation and application of GO-based composite sol–gel fiber. The novel coating showed higher extraction capability for most of OPFRs than the commercial fibers and GO sol–gel fiber. It also has the advantages of high stability, long lifetime, easy preparation and low cost. Both the single fiber repeatability and fiber-to-fiber reproducibility were satisfactory. Based on these features, the proposed fiber has a great potential for widespread use as an effective and useful extraction tool.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 21677058), the Fundamental Research Funds for the Central Universities (Grants 20205170506) and China Scholarship Fund (201706775016).References
- P. Darnerud, Toxic effects of brominated flame retardants in man and in wild life, Environ. Int., 2003, 29, 841–853 CrossRef CAS PubMed.
- Stockholm Conference of Parties, Report of the Conference of the Parties of the Stockholm Convention on Persistent Organic Pollutants on the Work of Its Fourth Meeting: Stockholm Convention on Persistent Organic Pollutants, Geneva, 2009 Search PubMed.
- European Flame Retardants Association, Market Statistics, http://www.flameretardants.eu/DocShareNoFrame/docs/1/HGJJCNFBPCEOKPEPPIPEICDF53V443–HA4YW3PDB348BT/EFRA/docs/DLS/EFRA_web_11-2007_Market_statistics-1.pdf2007.
- L. V. Dishaw, C. M. Powes, I. T. Ryde, S. C. Roberts, F. J. Seidler, T. A. Slotkin and H. M. Stapleton, Is the PentaBDE replacement, tris (1,3-dichloro-2-propyl) phosphate (TDCPP), a developmental neurotoxicant? Studies in PC12 cells, Toxicol. Appl. Pharmacol., 2011, 256, 281–289 CrossRef CAS PubMed.
- N. Van den Eede, A. C. Dirtu, H. Neels and A. Covaci, Analytical developments and preliminary assessment of human exposure to organophosphate flame retardants from indoor dust, Environ. Int., 2011, 37, 454–461 CrossRef CAS PubMed.
- X. Liu, K. Ji and K. Choi, Endocrine disruption potentials of organophosphate flame retardants and related mechanisms in H295R and MVLN cell lines and in zebrafish, Aquat. Toxicol., 2012, 114, 173–181 CrossRef PubMed.
- J. D. Meeker, E. M. Copper, H. M. Stapleton and R. Hauser, Urinary metabolites of organophosphate flame retardants: temporal variability and correlations with house dust concentrations, Environ. Health Perspect., 2013, 121, 580–585 CrossRef PubMed.
- T. Reemtsma, J. B. Quintana, R. Rodil, M. G. Lopez and I. Rodriguez, Organophosphorus flame retardants and plasticizers in water and air I. Occurrence and fate, Trends Anal. Chem., 2008, 27, 727–737 CrossRef CAS.
- I. Rodríguez, F. Calvo, J. B. Quintana, E. Rubí, R. Rodil and R. Cela, Suitability of solid-phase microextraction for the determination of organophosphate flame retardants and plasticizers in water samples, J. Chromatogr. A, 2006, 1108, 158 CrossRef PubMed.
- Z. Q. Gao, Y. H. Deng, X. B. Hu, S. G. Yang, C. Sun and H. He, Determination of organophosphate esters in water samples using an ionic liquid-based sol-gel fiber for headspace solid-phase microextraction coupled to gas chromatography-flame photometric detector, J. Chromatogr. A, 2013, 1300, 141–150 CrossRef CAS PubMed.
- J. A. Andresen, A. Grundmann and K. Bester, Organophosphorus flame retardants and plasticisers in surface waters, Sci. Total Environ., 2004, 332, 155–166 CrossRef CAS PubMed.
- R. Rodil, J. B. Quintana and T. Reemtsma, Liquid chromatography-tandem mass spectrometry determination of nonionic organophosphorus flame retardants and plasticizers in wastewater Samples, Anal. Chem., 2005, 77, 3083–3089 CrossRef CAS PubMed.
- J. B. Quintana and T. Reemtsma, Potential of membrane-assisted solvent extraction for the determination of phosphoric acid triesters in wastewater samples by liquid chromatography-tandem mass spectrometry, J. Chromatogr. A, 2006, 1124, 22–28 CrossRef CAS PubMed.
- M. García-López, I. Rodríguez and R. Cela, Development of a dispersive liquid-liquid microextraction method for organophosphorus flame retardants and plastizicers determination in water samples, J. Chromatogr. A, 2007, 1166, 9–15 CrossRef PubMed.
- M. García-López, I. Rodríguez and R. Cela, Evaluation of liquid-liquid microextraction using polypropylene microporous membranes for the determination of organophosphorus flame retardants and plasticizers in water samples, Anal. Chim. Acta, 2008, 625, 145–153 CrossRef PubMed.
- X. W. Wang, Y. Q. He, L. Lin, F. Zeng and T. G. Luan, Application of fully automatic hollow fiber liquid phase microextraction to assess the distribution of organophosphate esters in the Pearl River Estuaries, Sci. Total Environ., 2014, 470–471, 263–269 CrossRef CAS PubMed.
- C. L. Arthur and J. Pawliszyn, Solid phase microextraction with thermal desorption using fused silica optical fibers, Anal. Chem., 1990, 62, 2145–2148 CrossRef CAS.
- Z. Zhang, L. Zhu, Y. Ma, Y. Huang and G. Li, Preparation of polypyrrole composite solid-phase microextraction fiber coatings by sol-gel technique for the trace analysis of polar biological volatile organic compounds, Analyst, 2013, 138, 1156–1166 RSC.
- Y. Li, Y. Wang, J. Zhang and C. Sun, Determination of 16 polycyclic aromatic hydrocarbons in water using fluorinated polyaniline-based solid-phase microextraction coupled with gas chromatography, Environ. Monit. Assess., 2012, 184, 4345–4353 CrossRef CAS PubMed.
- J. Lopez-Darias, J. L. Anderson, V. Pino and A. M. Afonso, Developing qualitative extraction profiles of coffee aromas utilizing polymeric ionic liquid sorbent coatings in headspace solid-phase microextraction gas chromatography-mass spectrometry, Anal. Bioanal. Chem., 2011, 401, 2965–2976 CrossRef CAS PubMed.
- D. Djozan, Y. Assadi and S. H. Haddadi, Anodized aluminum wire as a solid-phase microextraction fiber, Anal. Chem., 2001, 73, 4054–4058 CrossRef CAS PubMed.
- N. Chang, Z. Gu, H. Wang and X. Yan, Metal-organic-framework-based tandem molecular sieves as a dual platform for selective microextraction and high-resolution gas chromatographic separation of n-alkanes in complex matrixes, Anal. Chem., 2011, 83, 7094–7101 CrossRef CAS PubMed.
- R. F. Jiang, F. Zhu, T. G. Luan, Y. X. Tong, H. Liu, G. F Ouyang and J. Pawliszyn, Carbon nanotube-coated solid-phase microextraction metal fiber based on sol-gel technique, J. Chromatogr. A, 2009, 1216, 4641–4647 CrossRef CAS PubMed.
- M. Sun, J. J. Feng, Y. N. Bu, X. J. Wang, H. M. Duan and C. N. Luo, Graphene coating bonded onto stainless steel wire as a solid-phase microextraction fiber, Talanta, 2015, 134, 200–205 CrossRef CAS PubMed.
- S. Y. Li, C. W. Lu, F. Zhu, R. F. Jiang and G. F. Ouyang, Preparation of C18 composite solid-phase microextraction fiber and its application to the determination of organochlorine pesticides in water samples, Anal. Chim. Acta, 2015, 873, 57–62 CrossRef CAS PubMed.
- Y. Y. Wen, Z. L. Niu, Y. L. Ma, J. P. Ma and L. X. Chen, Graphene oxide-based microspheres for the dispersive solid-phase extraction of non-steroidal estrogens from water samples, J. Chromatogr. A, 2014, 1368, 18–25 CrossRef CAS PubMed.
- S. H. Zhang, Z. Li, C. Wang and Z. Wang, Cyclodextrin-functionalized reduced graphene oxide as a fiber coating material for the solid-phase microextraction of some volatile aromatic compounds, J. Sep. Sci., 2015, 38, 1711–1720 CrossRef CAS PubMed.
- Y. Guo, S. Guo, J. Ren, Y. Zhai, S. Dong and E. Wang, Cyclodextrin functionalized graphene nanosheets with high supramolecular recognition capability: synthesis and host_guest inclusion for enhanced electrochemical performance, ACS Nano, 2010, 4, 4001–4010 CrossRef CAS PubMed.
- C. Xu, J. Wang, L. Wan, J. Lin and X. Wang, Microwave-assisted covalent modification of graphene nanosheets with hydroxypropyl-β-cyclodextrin and its electrochemical detection of phenolic organic pollutants, J. Mater. Chem., 2011, 21, 10463–10471 RSC.
- G. Crini, Kinetic and equilibrium studies on the removal of cationic dyes from aqueous solution by adsorption onto a cyclodextrin polymer, Dyes Pigm., 2008, 77, 415–426 CrossRef CAS.
- J. Zhang, X. Shen and Q. Chen, Separation processes in the presence of cyclodextrins using molecular imprinting technology and ionic liquid cooperating approach, Curr. Org. Chem., 2011, 15, 74–85 CrossRef CAS.
- Y. P. Chin, S. Mohamad and M. R. B. Abas, Removal of parabens from aqueous solution using β-cyclodextrin cross-linked polymer, Int. J. Mol. Sci., 2010, 11, 3459–3471 CrossRef CAS PubMed.
- M. Raoov, S. Mohamad, M. R. Abas and H. Surikumaran, New macroporous β-cyclodextrin functionalized ionic liquid polymer as an adsorbent for solid phase extraction with phenols, Talanta, 2014, 130, 155–163 CrossRef CAS PubMed.
- C. López-de-Dicastillo, M. Gallur, R. Catalá, R. Gavara and P. Hernandez-Muñoz, Immobilization of β-cyclodextrin in ethylene-vinyl alcohol copolymer for active food packaging applications, J. Membr. Sci., 2010, 353, 184–191 CrossRef.
- L. Qian, Y. Guan and H. Xiao, Preparation and characterization of inclusion complexes of a cationic β-cyclodextrin polymer with butylparaben or triclosan, Int. J. Pharm., 2008, 357, 244–251 CrossRef CAS PubMed.
- T. Sun, J. Jia, N. Fang and Y. Wang, Application of novel activated carbon fiber solid-phase microextraction to the analysis of chlorinated hydrocarbons in water by gas chromatography-mass spectrometry, Anal. Chim. Acta, 2005, 530, 33–40 CrossRef CAS.
- Y. Liu, Y. F. Shen and M. L. Lee, Porous layer solid phase microextraction using silica bonded phases, Anal. Chem., 1997, 69, 190–195 CrossRef CAS.
- J. X. Wang, D. Q. Jiang, Z. Y. Gu and X. P. Yan, Multiwalled carbon nanotubes coated fibers for solid-phase microextraction of polybrominated diphenyl ethers in water and milk samples before gas chromatography with electron-capture detection, J. Chromatogr. A, 2006, 1137, 8–14 CrossRef CAS PubMed.
- A. Medinia, M. F. Mousavi and M. Shamsipur, Nano-structured lead dioxide as a novel stationary phase for solid-phase microextraction, J. Chromatogr. A, 2006, 1134, 24–31 CrossRef PubMed.
- J. X. Yu, L. Dong, C. Wu, L. Wu and J. Xing, Hydroxyfullerene as a novel coating for solid-phase microextraction fiber with sol-gel technology, J. Chromatogr. A, 2002, 978, 37–48 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07903f |
| This journal is © The Royal Society of Chemistry 2017 |