
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of novel hybrid POSS biomolecules via “Click” reactions†
Youssef El Aziz*a,
Nazia Mehrbanb,
Peter G. Taylor a,
Martin A. Birchallb,
James Bowena,
Alan R. Bassindalea,
Mateusz B. Pitakc and
Simon J. Colesc
a,
Martin A. Birchallb,
James Bowena,
Alan R. Bassindalea,
Mateusz B. Pitakc and
Simon J. Colesc
aThe Open University, Faculty of Science, Technology, Engineering & Mathematics, Walton Hall, Milton Keynes, MK7 6AA, UK. E-mail: youssef.elaziz@open.ac.uk; Fax: +44 (0)1908 858 327
bUniversity College London, Ear Institute, Brain Sciences, 332 Gray's Inn Rd, London WC1X 8EE, UK
cUK National Crystallography Service, Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
First published on 28th July 2017
Abstract
A novel alkyne-terminated cubic-octameric POSS was synthesised in high yield (82–90%). The X-ray crystal structure revealed intra- and intermolecular hydrogen bonding between the amide groups of the arms. Hybrid biomaterials were synthesised in nearly quantitative yields via a click reaction with (i) azido-N-Fmoc-norleucine and (ii) 3′-azido-3′-deoxythymidine.
Among the most commonly studied scaffolds for developing hybrid biomaterials1–4 is polyhedral oligomeric silsesquioxane (POSS). POSS units are symmetrical, three-dimensional cubic molecules, which are unique nanometer-sized hybrid inorganic–organic materials5,6 with the formula (RSiO3/2)8, known as T8. POSS contains an inorganic inner siloxane nanocore, with the possibility of chemical functionalisation at each of the eight corners of the cubic unit. POSS units have been used extensively as scaffolds for the development of liquid crystals,7,8 biocompatible materials,9 catalysts10,11 and dendrimers12 and can also be used in cross-linking polymers.13 Functionalisation of T8 with different substituents has usually been achieved by hydrosilylation,14,15 Heck,16–18 and cross-metathesis16,19 reactions.
Copper-catalyzed Azide–Alkyne Cycloaddition (CuAAC),20,21 ‘click’ chemistry, is a simple method for coupling organic molecules containing azide and alkyne functional groups in high yields and its use in the fields of peptide and protein biomedical and material sciences is accelerating.22,23 The click reaction has been used to synthesise POSS biomaterials such as hybrid POSS–PEG hydrogels24 that support chondrocyte attachment and proliferation. Only one synthetic approach towards peptidyl silsesquioxanes using click chemistry has been reported to date.25–30 focussing on the synthesis of octa(3-azidopropyl)polyhedral oligomeric silsesquioxane POSS-(N3)8 (Fig. 1) and its reaction with a variety of alkynes.25–36 The preparation of octa(3-azidopropyl)polyhedral oligomeric silsesquioxane POSS-(N3)8 requires a multi-step synthesis and suffers from a poor overall yield of 30–32%.32
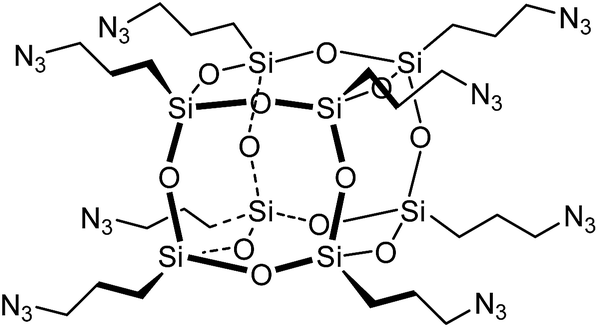 | ||
| Fig. 1 Octa(3-azidopropyl)POSS. | ||
The synthesis and click reaction of mono alkyne-heptaisobutyl POSS were first reported by Müller et al.37 and Wu et al.38 Müller et al.37 have used aminopropylheptaisobutyl polyhedral oligomeric silsesquioxane (POSS) to produce mono-alkyne-POSS in three steps and the click coupling occurred between the azido-terminated polystyrenes and alkyne-POSS to afford mono-, di-, and pentafunctional POSS-containing hybrid polystyrenes as star-shaped telechelic POSS-containing hybrid polymers. Wu et al.38 have prepared alkyne-functionalized-POSS in three steps from allyl-heptaisobutyl substituted-POSS and the product was linked to an azido-functionalized elastomer of poly(styrene-b-(ethylene-co-butylene)-b-styrene) (SEB–CH2N3) via a click coupling reaction to form a novel hybrid copolymer. These approaches focused mostly on the preparation of a hybrid copolymer based on mono-alkyne-heptaisobutyl and azido polystyrene. However, the preparation of octa-alkyne-terminated POSS and their click coupling reactions with azido-biomolecules to produce hybrid biomaterials has not been investigated yet. We believe that this approach will open a wide range of biomedical applications that were not accessible in the past.
Herein we report a novel synthetic approach to hybrid biomaterials based on octa-alkyne-terminated POSS and their reaction with (i) azido-N-Fmoc-L-norleucine and (ii) 3′-azido-3′-deoxythymidine, using the CuAAC reaction. Our approach presents a particularly versatile route which provides a facile and convenient way to functionalise a cubic silsesquioxane core with biomolecules that are more readily available as their azido derivative than their alkyne derivative.
Compound 2 was prepared in one step from commercially available materials; octa(3-aminopropyl)octasilsesquioxane (1) and 5-hexynoic acid (Scheme 1), in 82–90% yield. Product 2 was isolated and purified by column chromatography, followed by characterisation using standard techniques (see ESI†). The crystal structure determined by X-ray crystallography (Fig. 2) suggests that intra- and inter-molecular hydrogen bonding between the arms were a fundamental driving force for the formation of a well-defined crystal structure.‡
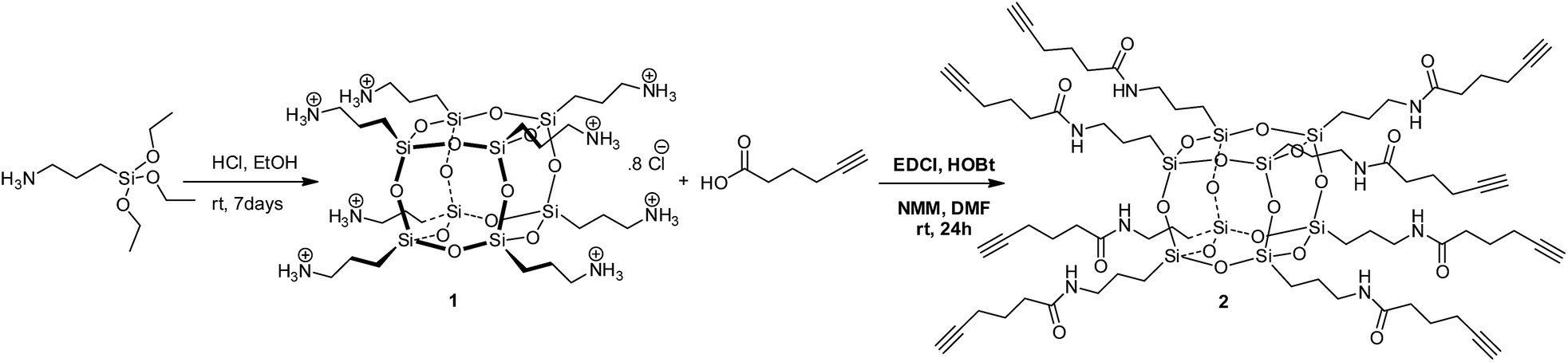 | ||
| Scheme 1 Synthesis of octa-alkyne-terminated POSS (2). | ||
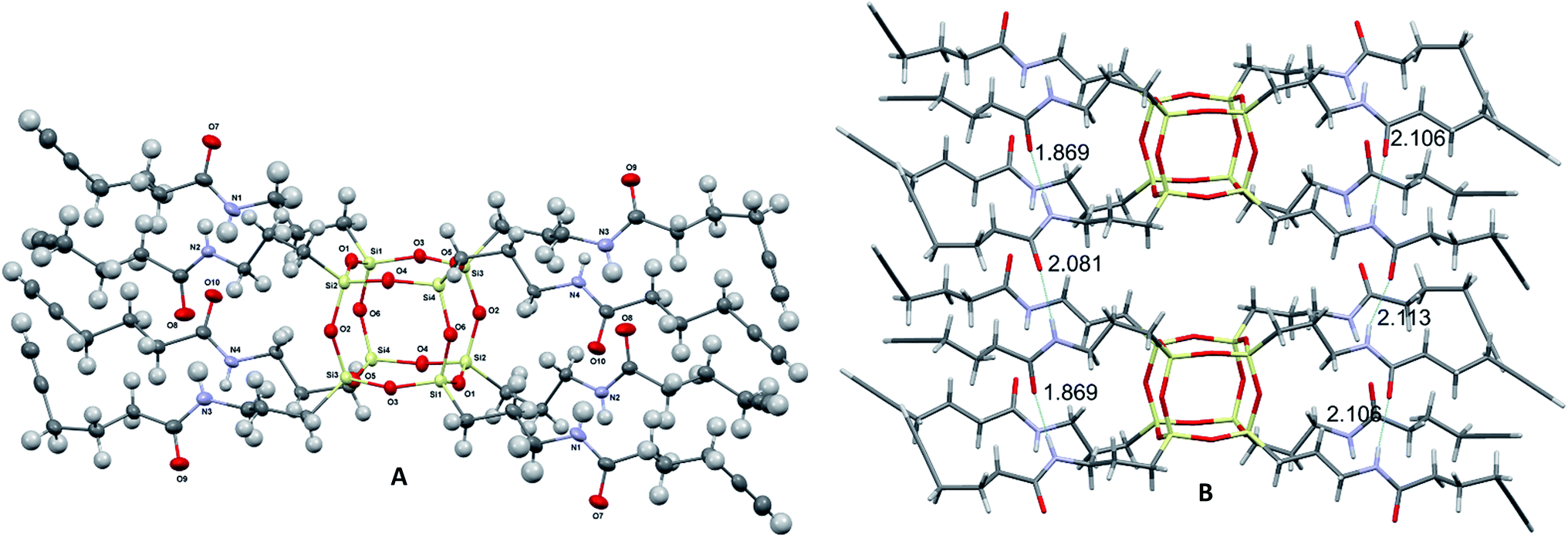 | ||
| Fig. 2 Molecular structure of 2 grown fragment (thermal ellipsoids drawn with 50% probability) (A) and the font intra- and inter- H-bonding distances (B). | ||
The length of intramolecular nitrogen–hydrogen (N⋯H) bonds varies between 2.09(3) and 2.12(3) Å, whereas for an intermolecular bond the distance is 1.87(3) Å.
The completion of the cycloaddition reaction was confirmed by MALDI-TOF and the reaction progress was monitored by observing the disappearance of the azide asymmetric stretch at 2093 cm−1 and the triple bond C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C asymmetric stretch of T8[propylhex-5-ynamide]8 (2) at 2100 cm−1 by FT-IR spectroscopy together with monitoring the disappearance in the 13C-NMR spectrum of the two peaks (89.20 and 76.56 ppm) representing the triple bond of 2.
C asymmetric stretch of T8[propylhex-5-ynamide]8 (2) at 2100 cm−1 by FT-IR spectroscopy together with monitoring the disappearance in the 13C-NMR spectrum of the two peaks (89.20 and 76.56 ppm) representing the triple bond of 2.
Compounds 3 and 4 have been analysed and characterised using NMR (1H, 13C and 29Si) spectroscopy, infrared and MALDI-TOF mass spectrometry in positive ion mode with a DHB matrix.
Trastoy et al.32 have reported an efficient preparation of highly functionalised cubic-octameric POSS frameworks by click chemistry and the highest yield (96%) was obtained with the CuSO4·H2O/sodium ascorbate precatalyst system20 using a biphasic organic solvent/water mixture at room temperature for 24 hours. We have used these reaction conditions for the functionalisation of the octa-alkyne-terminated POSS with azido-N-Fmoc-norleucine and 3′-azido-3′-deoxythymidine (Scheme 2).
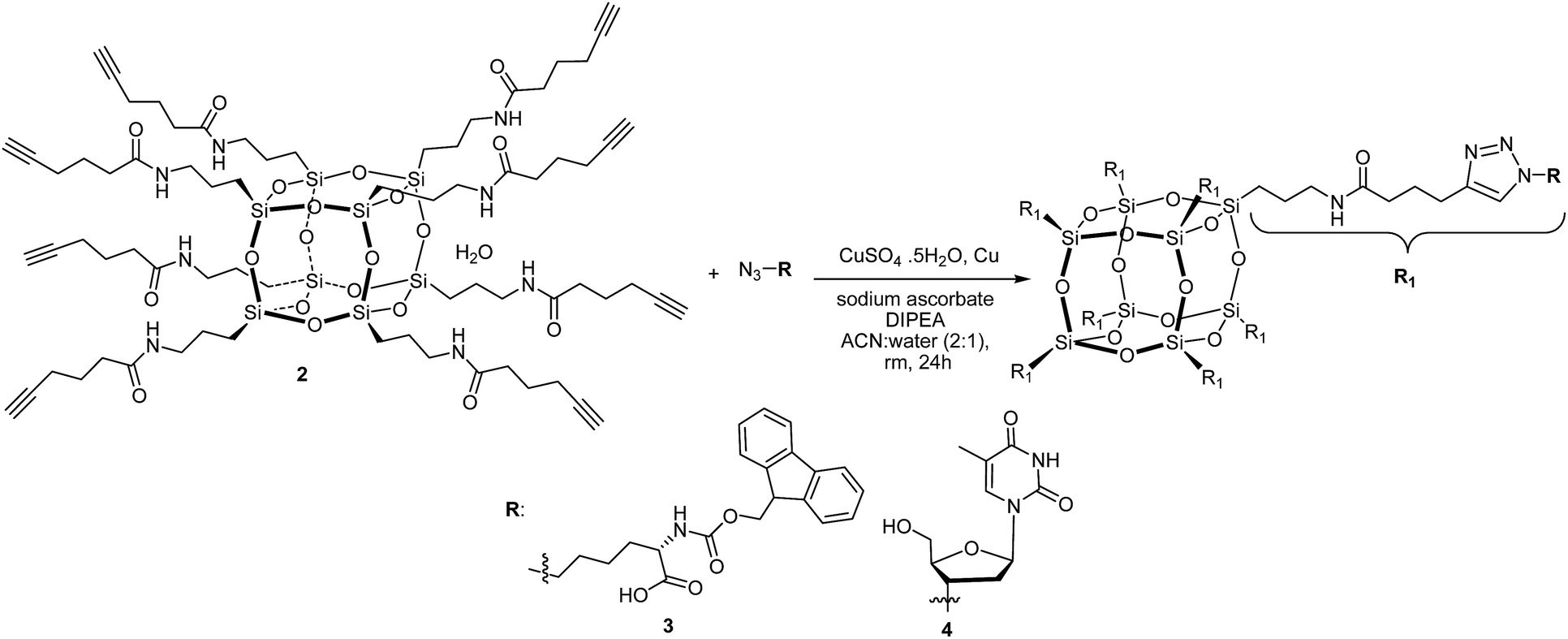 | ||
| Scheme 2 Synthesis of 3 and 4 using CuAAC approach. | ||
The MALDI-TOF MS of compound 3 and 4 revealed that the octa-alkyne-terminated POSS has been fully functionalised with azido-N-Fmoc-norleucine for 3 and 3′-azido-3′-deoxythymidine for 4. The molecular ion peak of 3 observed at found 4787 Da is attributed to [M + H]+ and 4 observed at 3835.3 Da is attributed to [M + Cu]+.
Conclusions
In this study we have described a novel, efficient method for the synthesis of 3D radially symmetrical biomolecule-POSS hybrids. We have developed a one-step synthesis of 2 from commercially available octakis(3-aminopropyl)octa-silsesquioxane (1) with high yield (82–90%). The X-ray crystal structure shows that compound 2 exhibits plane-to-plane stacking with an intra- and inter-molecular hydrogen bond network. The octa-alkyne-terminated POSS was efficiently and regioselectively octafunctionalised with two azido-R species (where R are Fmoc-Leu and thymidine) by copper(I)-catalysed 1,3-dipolar azide–alkyne cycloaddition (CuAAC) under biphasic conditions. This led to new hybrid biofunctional nanocages 3 and 4 in high yield. This new strategy of functionalisation of terminated alkyne-POSS via CuAAC opens many possibilities for the efficient and controlled assembly of new hybrid biomaterials with a high degree of symmetry and with carefully tailored functional properties.Acknowledgements
We thank the EPSRC National Mass Spectrometry Service Centre (NMSSC) at Swansea and MEDAC Ltd. of Brunel University for elemental analysis. We thank the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data.Notes and references
- S. Fabritz, S. Horner, O. Avrutina and H. Kolmar, Org. Biomol. Chem., 2013, 11, 2224–2236 CAS.
- C. P. Teng, K. Y. Mya, K. Y. Win, C. C. Yeo, M. Low, C. He and M.-Y. Han, NPG Asia Mater., 2014, 6, e142 CrossRef.
- Y.-C. Lin and S.-W. Kuo, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2127–2137 CrossRef CAS.
- H. Ghanbari, B. G. Cousins and A. M. Seifalian, Macromol. Rapid Commun., 2011, 32, 1032–1046 CrossRef CAS PubMed.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- P. D. Lickiss and F. Rataboul, in Adv. Organomet. Chem., ed. F. H. Anthony and J. F. Mark, Academic Press, 2008, vol. 57, pp. 1–116 Search PubMed.
- I. M. Saez and J. W. Goodby, Liq. Cryst., 1999, 26, 1101–1105 CrossRef CAS.
- Elsa, G. H. Mehl, J. W. Goodby and D. J. Photinos, Chem. Commun., 2000, 851–852 Search PubMed.
- F. J. Feher, K. D. Wyndham and M. A. Scialdone, Chem. Commun., 1998, 1469–1470 RSC.
- L. A. Bivona, O. Fichera, L. Fusaro, F. Giacalone, M. Buaki-Sogo, M. Gruttadauria and C. Aprile, Catal. Sci. Technol., 2015, 5, 5000–5007 CAS.
- C.-H. Lu and F.-C. Chang, ACS Catal., 2011, 1, 481–488 CrossRef CAS.
- I. M. Saez, J. W. Goodby and R. M. Richardson, Chem.–Eur. J., 2001, 7, 2758–2764 CrossRef CAS.
- I. A. Zucchi, M. J. Galante and R. J. J. Williams, Eur. Polym. J., 2009, 45, 325–331 CrossRef CAS.
- L. Zheng, R. J. Farris and E. B. Coughlin, Macromolecules, 2001, 34, 8034–8039 CrossRef CAS.
- M. F. Roll, M. Z. Asuncion, J. Kampf and R. M. Laine, ACS Nano, 2008, 2, 320–326 CrossRef CAS PubMed.
- G. Cheng, N. R. Vautravers, R. E. Morris and D. J. Cole-Hamilton, Org. Biomol. Chem., 2008, 6, 4662–4667 CAS.
- C. M. Brick, R. Tamaki, S. G. Kim, M. Z. Asuncion, M. Roll, T. Nemoto, Y. Ouchi, Y. Chujo and R. M. Laine, Macromolecules, 2005, 38, 4655–4660 CrossRef CAS.
- A. Sellinger, R. Tamaki, R. M. Laine, K. Ueno, H. Tanabe, E. Williams and G. E. Jabbour, Chem. Commun., 2005, 3700–3702 RSC.
- S. Sulaiman, A. Bhaskar, J. Zhang, R. Guda, T. Goodson and R. M. Laine, Chem. Mater., 2008, 20, 5563–5573 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- K. Nwe and M. W. Brechbiel, Cancer Biother. Radiopharm., 2009, 24, 289–302 CrossRef CAS PubMed.
- J.-F. Lutz and Z. Zarafshani, Adv. Drug Delivery Rev., 2008, 60, 958–970 CrossRef CAS PubMed.
- X. Pan, H. Gao, G. Fu, Y. Gao and W. Zhang, RSC Adv., 2016, 6, 23471–23478 RSC.
- Z. Wang, Y. Li, X.-H. Dong, X. Yu, K. Guo, H. Su, K. Yue, C. Wesdemiotis, S. Z. D. Cheng and W.-B. Zhang, Chem. Sci., 2013, 4, 1345–1352 RSC.
- H. Su, J. Zheng, Z. Wang, F. Lin, X. Feng, X.-H. Dong, M. L. Becker, S. Z. D. Cheng, W.-B. Zhang and Y. Li, ACS Macro Lett., 2013, 2, 645–650 CrossRef CAS.
- H. B. Tinmaz, I. Arslan and M. A. Tasdelen, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1687–1695 CrossRef CAS.
- B. Trastoy, D. A. Bonsor, M. E. Pérez-Ojeda, M. L. Jimeno, A. Méndez-Ardoy, J. M. García Fernández, E. J. Sundberg and J. L. Chiara, Adv. Funct. Mater., 2012, 22, 3191–3201 CrossRef CAS.
- X. Feng, S. Zhu, K. Yue, H. Su, K. Guo, C. Wesdemiotis, W.-B. Zhang, S. Z. D. Cheng and Y. Li, ACS Macro Lett., 2014, 3, 900–905 CrossRef CAS.
- S. Fabritz, D. Heyl, V. Bagutski, M. Empting, E. Rikowski, H. Frauendorf, I. Balog, W.-D. Fessner, J. J. Schneider, O. Avrutina and H. Kolmar, Org. Biomol. Chem., 2010, 8, 2212–2218 CAS.
- G. M. Ziarani, M. S. Nahad, N. Lashgari and A. Badiei, Acta Chim. Slov., 2015, 62, 709–715 CrossRef CAS.
- B. Trastoy, M. E. Pérez-Ojeda, R. Sastre and J. L. Chiara, Chem.–Eur. J., 2010, 16, 3833–3841 CrossRef CAS PubMed.
- V. Ervithayasuporn, X. Wang and Y. Kawakami, Chem. Commun., 2009, 5130–5132 RSC.
- Z. Ge, D. Wang, Y. Zhou, H. Liu and S. Liu, Macromolecules, 2009, 42, 2903–2910 CrossRef CAS.
- A. Blazquez-Moraleja, M. Eugenia Perez-Ojeda, J. R. Suarez, M. Luisa Jimeno and J. L. Chiara, Chem. Commun., 2016, 52, 5792–5795 RSC.
- W. Zhang and A. H. E. Müller, Polymer, 2010, 51, 2133–2139 CrossRef CAS.
- W. Zhang and A. H. E. Müller, Macromolecules, 2010, 43, 3148–3152 CrossRef CAS.
- M. Niu, R. Xu, P. Dai and Y. Wu, Polymer, 2013, 54, 2658–2667 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, 1H, 13C, 19F, 29Si NMR, IR, MALDI-TOF MS, elemental analysis, and analytical data for all new compounds. For ESI and crystallographic data in CIF or other electronic format. CCDC 1514595. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra07915j |
‡ Crystal data of compound 2: C72H112N8O20Si8 (M = 1634.41 g mol−1): triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 9.6202(3) Å, b = 14.1254(3) Å, c = 17.6565(6) Å, α = 71.392(2)°, β = 74.675(3)°, γ = 70.560(2)°, V = 2110.47(12) Å3, Z = 1, T = 100.15 K, μ(Mo Kα) = 0.198 mm−1, Dcalc = 1.286 g cm−3, 28 (no. 2), a = 9.6202(3) Å, b = 14.1254(3) Å, c = 17.6565(6) Å, α = 71.392(2)°, β = 74.675(3)°, γ = 70.560(2)°, V = 2110.47(12) Å3, Z = 1, T = 100.15 K, μ(Mo Kα) = 0.198 mm−1, Dcalc = 1.286 g cm−3, 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 211 reflections measured (6.088° ≤ 2Θ ≤ 50.054°), 7434 unique (Rint = 0.0375, Rsigma = 0.0368) which were used in all calculations. The final R1 was 0.0459 (I > 2σ(I)) and wR2 was 0.1280 (all data). 211 reflections measured (6.088° ≤ 2Θ ≤ 50.054°), 7434 unique (Rint = 0.0375, Rsigma = 0.0368) which were used in all calculations. The final R1 was 0.0459 (I > 2σ(I)) and wR2 was 0.1280 (all data). |
| This journal is © The Royal Society of Chemistry 2017 |