
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalyzed aminocarbonylation of aryl halides using carbamoylsilane as an amide source†
Xue-Ping Wen,
Yu-Ling Han and
Jian-Xin Chen *
*
College of Chemistry and Materials Science, Shanxi Normal University, Linfen 041004, China. E-mail: jjxxcc2002@yahoo.com
First published on 21st September 2017
Abstract
The nickel-catalyzed aminocarbonylation of aryl halides using carbamoylsilane as an amide source is developed. The procedure can prepare both tertiary and secondary amides, and is applicable to various carbamoylsilanes and aryl halides containing different functional groups. The types and the relative position of substituents on the aryl ring impact the coupling efficiency.
Amides are very important functional groups in the synthesis of organic molecules, and play a significant role in biological systems, and the fields of natural products, polymers, and pharmaceuticals.1 In the past decades, a great number of methods for the synthesis of amides have been developed.2 Among the developed methods, the palladium-catalyzed aminocarbonylation is an important and the most frequently used procedure for the selective and direct synthesis of aryl amides starting from aryl halides, amines and carbon monoxide.3 Since the pioneering work of Heck in 1974,4 various improved protocols for amino-carbonylation have emerged using CO as a carbonyl source.5 However, the troublesome handling, transport, and storage of hazardous CO, the reaction setup requiring high-pressure reactors, high temperature and harsh reaction conditions restrict its applications in lab-scale synthesis.6 Therefore, a simple and newer CO-free procedure for the aminocarbonylation to amides using an easy to handle carbonyl source would obviously be of great interest to synthetic chemists.6b,7 Previously, various carbamoylsilanes had been synthesized in good yields,8 which can be used as amides source to introduce into an organic substrate, for example, addition of carbamoylsilane to the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of imines,9 addition of carbamoylsilane to the CO bond of α-ketoesters or α-ketoamides,10 and the aminocarbonylation of acid chlorides.11 Cunico et al. had reported aminocarbonylation of aryl halides with carbamoylsilane catalyzed by palladium complexes.12 Drawbacks of the method are the high cost and toxicity of palladium catalysts, and the need to dispose toxic byproducts. We have reported the direct functionalization of aromatics by carbamoylsilanes leading to formation of amides.13 However, the scope is limited to electron-deficient aromatics. Recently, we found that nickel complexe could be used as catalyst in the aminocarbonylation of aryl halides using carbamoylsilanes as amides source, which is inexpensive and low toxic compared to palladium catalyst. To the best of our knowledge, aminocarbonylation of aryl halides with carbamoylsilane catalyzed by nickel complexe have never been reported. One example of amides synthesized catalyzed by nickel complexe is the aminocarbonylation of aryl halides using formamide derivatives as amides source. This reaction must undergo in the presence of strong base.14 Herein, we report a general Ni-catalyzed process for the conversion of aryl halides to aryl amides that can be carried out under mild reaction conditions by using various carbamoylsilanes as amides source.
N bond of imines,9 addition of carbamoylsilane to the CO bond of α-ketoesters or α-ketoamides,10 and the aminocarbonylation of acid chlorides.11 Cunico et al. had reported aminocarbonylation of aryl halides with carbamoylsilane catalyzed by palladium complexes.12 Drawbacks of the method are the high cost and toxicity of palladium catalysts, and the need to dispose toxic byproducts. We have reported the direct functionalization of aromatics by carbamoylsilanes leading to formation of amides.13 However, the scope is limited to electron-deficient aromatics. Recently, we found that nickel complexe could be used as catalyst in the aminocarbonylation of aryl halides using carbamoylsilanes as amides source, which is inexpensive and low toxic compared to palladium catalyst. To the best of our knowledge, aminocarbonylation of aryl halides with carbamoylsilane catalyzed by nickel complexe have never been reported. One example of amides synthesized catalyzed by nickel complexe is the aminocarbonylation of aryl halides using formamide derivatives as amides source. This reaction must undergo in the presence of strong base.14 Herein, we report a general Ni-catalyzed process for the conversion of aryl halides to aryl amides that can be carried out under mild reaction conditions by using various carbamoylsilanes as amides source.
First, the aminocarbonylation of bromobenzene with N,N-dimethylcarbamoyl(trimethyl)silane catalyzed by [(Ph)3P]4Ni(0) was chosen as a model reaction to investigate the effects of solvents and temperature. When a range of solvents often used for cross-coupling reactions were tested, the reaction proceeded smoothly in tested solvents except dichloromethane. As shown in Table 1, we observed: when the reaction was carried out in toluene, the highest yields of product was achieved, and the reaction time was short (Table 1, entry 4), while in dichloromethane the reaction could not be conducted. Several other tested solvents, including benzene, THF, and acetonitrile, gave inferior results compared to toluene (Table 1, entries 2–5). The temperature study shows that 75 °C is the optimum temperature to obtain a higher yield of coupling product. Increase or decrease in reaction temperature led to reduce in yield of coupling product (Table 1, entries 6–9). Next, model reaction was run in toluene at 75 °C to examine the effect of catalyst loading, which reveled that 2 mol% of [(Ph)3P]4Ni (relative to bromobenzene) was essential for best results. When the amount of catalyst used was less than 2 mol%, low yields of the desired products were obtained, while increasing the catalyst loading results also in slightly lower yields (Table 1, entries 10–12).
|
|||||
|---|---|---|---|---|---|
| Entry | Cata. (mol%) | Solvent | Temp (°C) | Timeb (h) | Yieldc,d (%) |
a Catalyst: tetrakis(triphenylphosphine)nickel(0).b To complete consumption of carbamoylsilane.c Isolated yield after chromatography on silica gel based on bromobenzene.d 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 mol ratio of bromobenzene and carbamoylsilane. :1.2 mol ratio of bromobenzene and carbamoylsilane. |
|||||
| 1 | 2 | Dichloromethane | 35 | 80 | 0 |
| 2 | 2 | Acetonitrile | 75 | 50 | 32 |
| 3 | 2 | THF | 65 | 44 | 36 |
| 4 | 2 | Toluene | 75 | 38 | 68 |
| 5 | 2 | Benzene | 75 | 41 | 58 |
| 6 | 2 | Toluene | 60 | 48 | 49 |
| 7 | 2 | Toluene | 90 | 34 | 64 |
| 8 | 2 | Toluene | 100 | 31 | 56 |
| 9 | 2 | Toluene | 110 | 70 | 20 |
| 10 | 1 | Toluene | 75 | 46 | 39 |
| 11 | 3 | Toluene | 75 | 37 | 60 |
| 12 | 5 | Toluene | 75 | 36 | 54 |
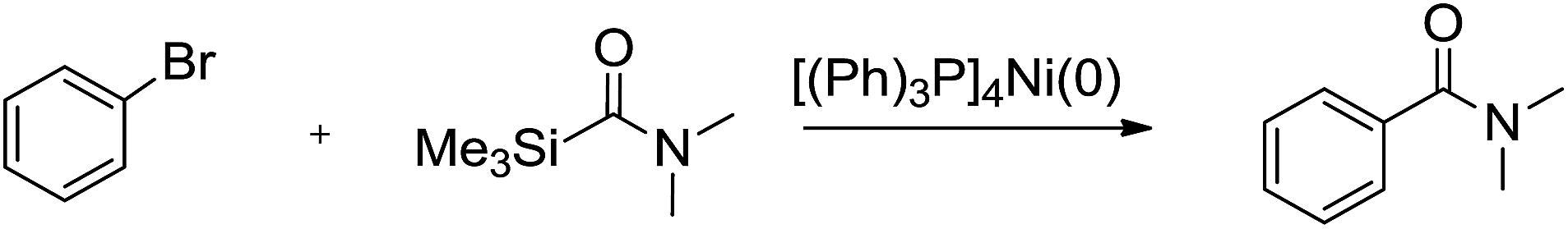
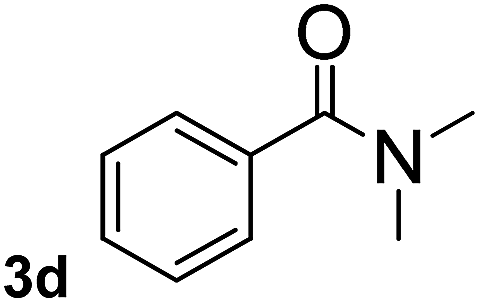
To explore the scope and limitation of the nickel-catalyzed aminocarbonylation of aryl halides using carbamoylsilane as an amide source, a series of aryl halides were screened to react with carbamoylsilane 2 in toluene solvent under 2 mol% of catalyst at 75 °C. Results are displayed in Table 2. Initially, aminocarbonylation of various halogen substituted benzene were performed. We found that chloro-, bromo- and iodo-benzene reacted with carbamoylsilane 2 smoothly in the presence of [(Ph)3P]4Ni(0), to give corresponding N,N-dimethylbenzamide directly, bromobenzene exhibited faster reaction rate and higher yield than others (Table 2, entries 1–3). However, fluorobenzene (1d) did not undergo the coupling reaction with carbamoylsilane 2 under the same conditions (Table 2, entry 4).
|
||||
|---|---|---|---|---|
| Entry | Aryl halide | Product | Time (h) | Yieldb,c (%) |
| a 2 mol% of tetrakis(triphenylphosphine)nickel(0).b Isolated yield after chromatography on silica gel based on aryl halides.c 1:1.2 mol ratio of aryl halides and carbamoylsilane. |
||||
| 1 | 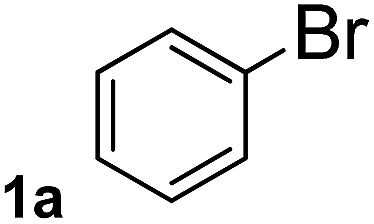 |
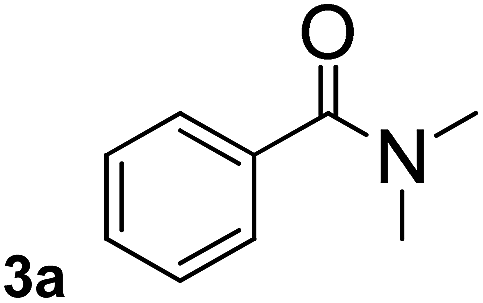 |
38 | 68 |
| 2 |  |
 |
40 | 58 |
| 3 |  |
 |
47 | 35 |
| 4 |  |
 |
50 | 0 |
| 5 | 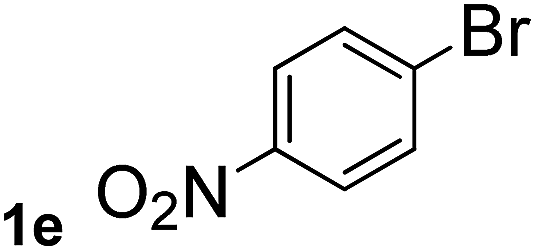 |
 |
27 | 75 |
| 6 | 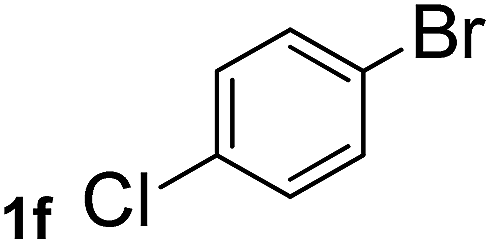 |
 |
36 | 71 |
| 7 | 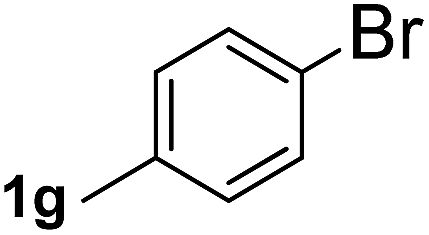 |
 |
48 | 58 |
| 8 | 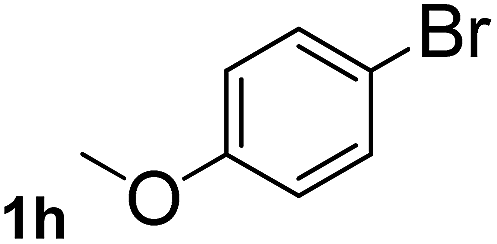 |
 |
39 | 69 |
| 9 | 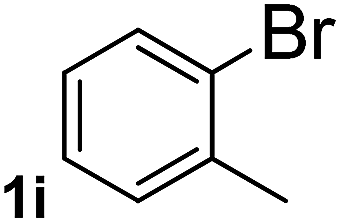 |
 |
46 | 42 |
| 10 | 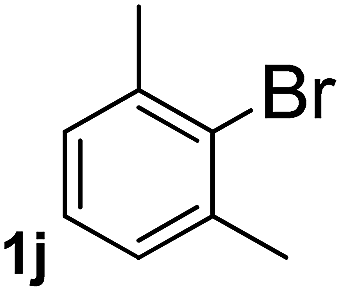 |
 |
52 | 0 |
| 11 | 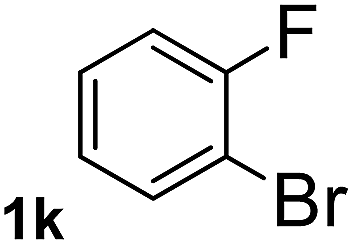 |
 |
39 | 57 |
| 12 | 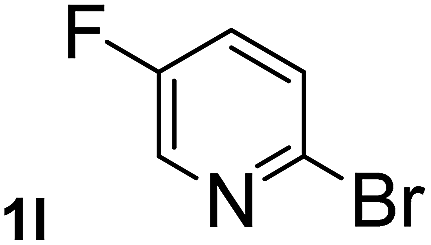 |
 |
28 | 62 |
| 13 | 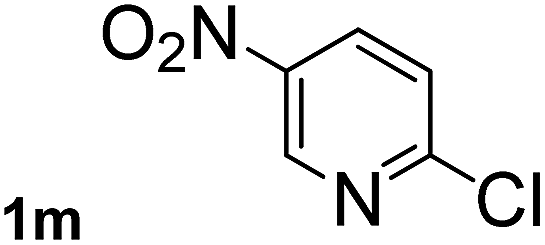 |
 |
15 | 67 |
| 14 | 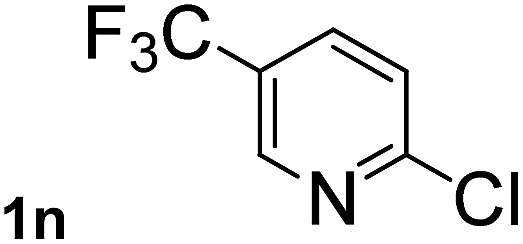 |
 |
28 | 78 |
| 15 | 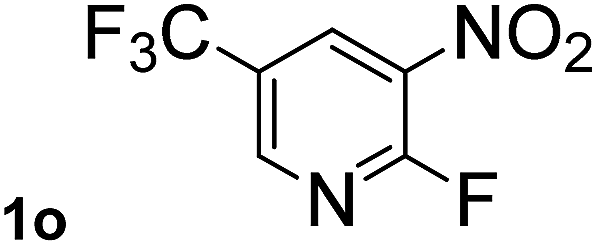 |
 |
72 | 0 |
| 16 | 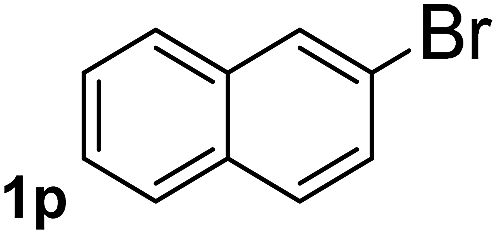 |
 |
36 | 68 |
| 17 | 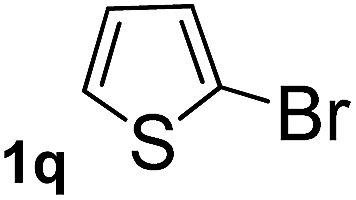 |
 |
28 | 62 |
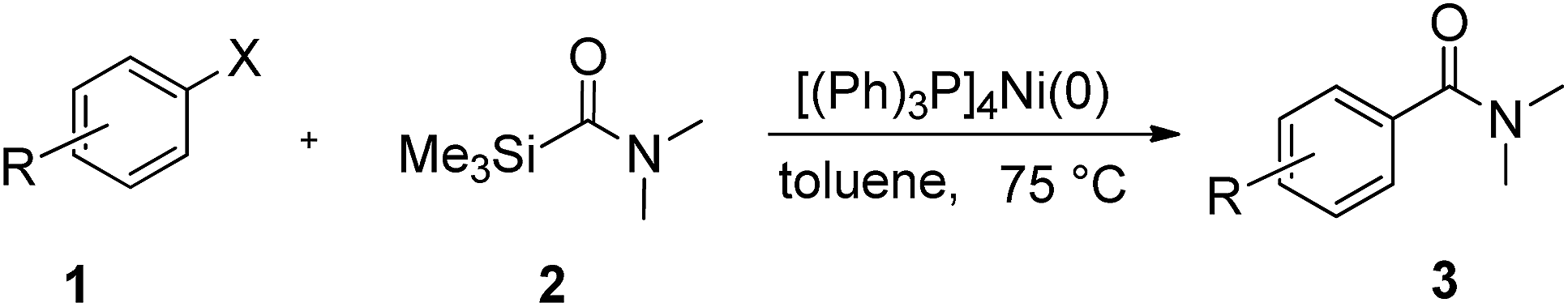
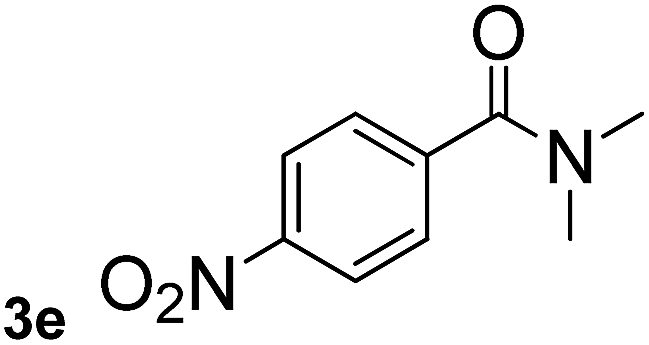
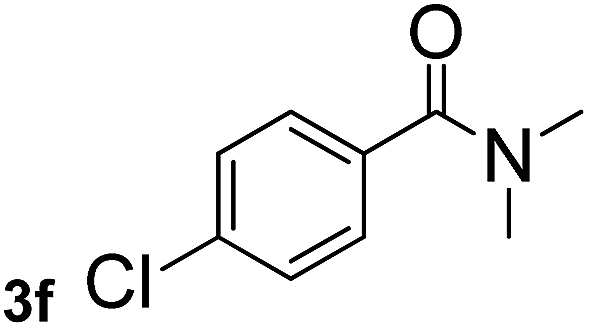
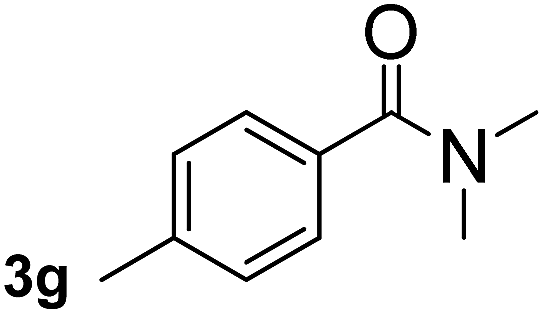
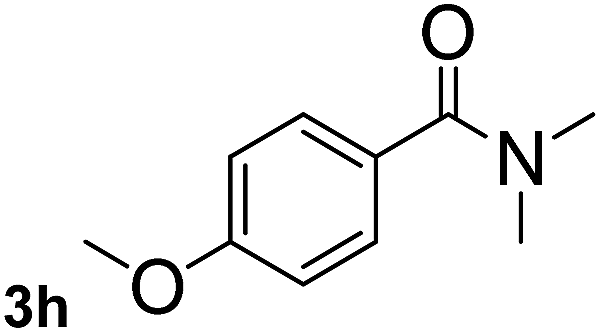
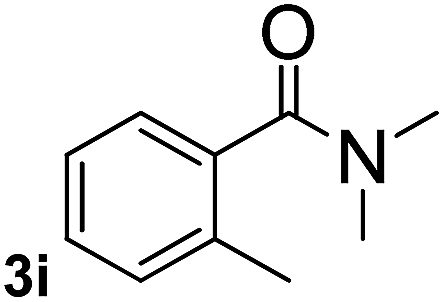
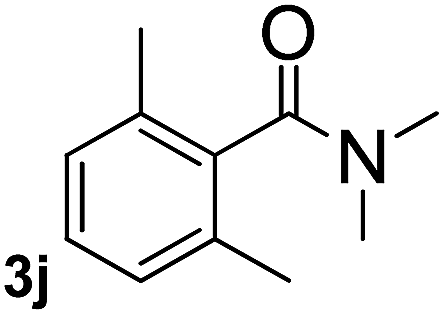
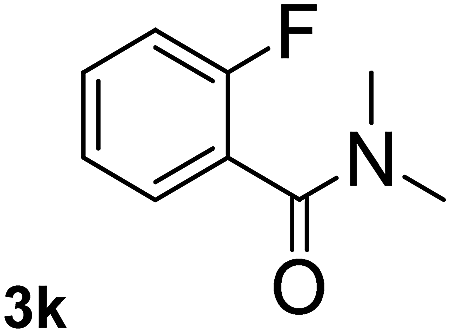
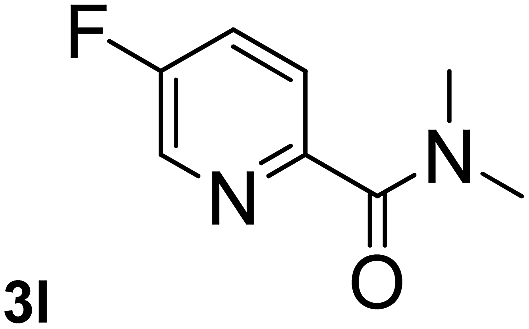
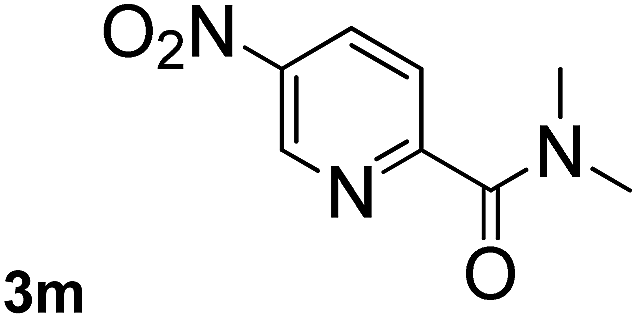
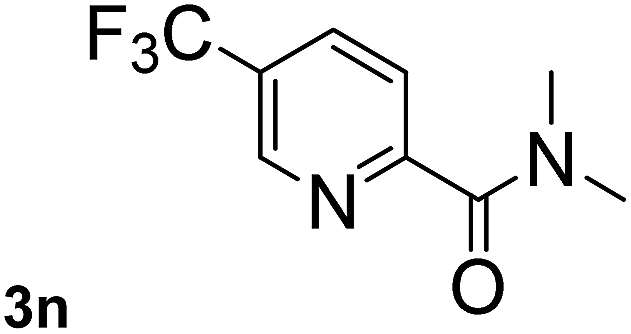
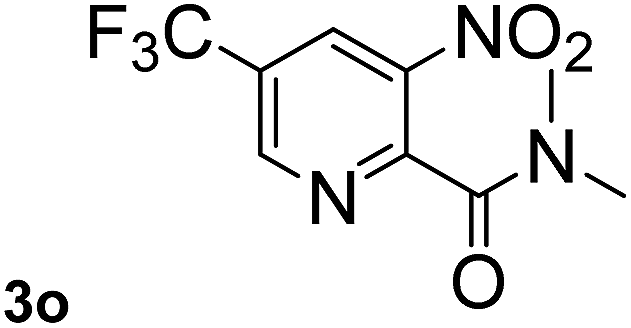
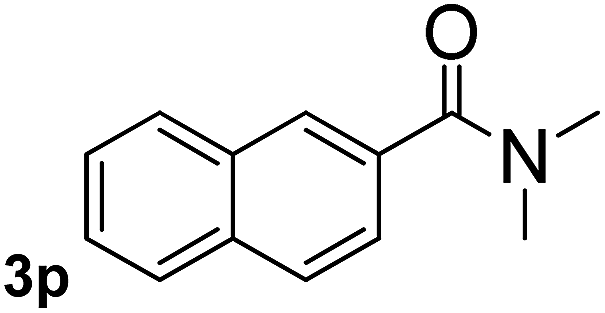
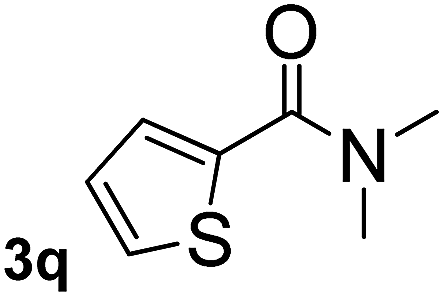
Next, aryl halides containing different functional groups were tested to investigate the effect of electronic property and relative position of substituents on the aryl ring. A comparison of the results obtained from 1e–h indicates that the electronic properties of the substituent in the phenyl ring influenced both the reaction time and yields of aminocarbonylation. Aryl bromides possessing an electron-withdrawing group, such as a nitro or chloro, reacted faster and gave higher isolated yields than that possessing electron-donating group, such as a methyl or methoxy group. The stronger electron-withdrawing the substituent, the faster the process and the higher the yield was (Table 2, entries 5–8). For aryl halides containing Br and Cl simultaneously, the aminocarbonylation only selectively occurred at the bromo-terminal, such as 4-bromochlorobenzene (1f) gave exclusively 4-chloro aryl amide 3f. The reaction results of aryl bromides 1i and 1j indicate that the coupling reaction is highly sensitive to the relative position of substituent on the phenyl ring. Substrate 1i, the relatively minor steric impediment of an ortho methyl inhibiting the coupling reaction, afforded product 3i in low yield, whereas ortho dimethyl completely inhibits the coupling reaction, 1j gave no desired product due to big steric hindrance (Table 2, entries 9 and 10). Fluoro substituted aryl bromide at ortho (an electron-withdrawing group) also led to low yield of product (Table 2, entries 11). As expected, an electron-deficient heteroaryl (pyridyl) bromides and chlorides possessing fluoro, trifluoromethyl or nitro (1l, 1m or 1n) gave the coupling reaction products in good yield. However, fluoropyridine 1o bearing two electron-withdrawing groups proved to be totally inert even at 75 °C for 3 days. 2-Bromonaphthalene (1p) provided a moderate yield of desired product 3p. Aryl bromide 1q containing an electron-rich heteroaryl was also reactivity with carbamoylsilane 2 to give product 3q in good yield.
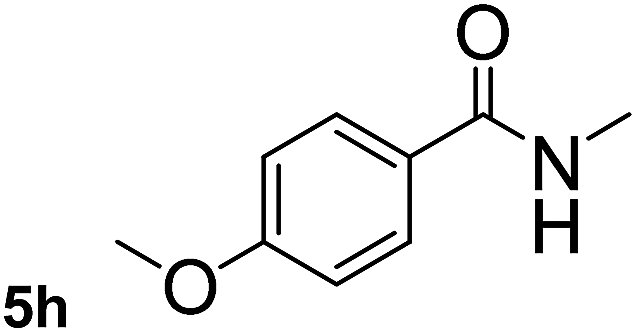
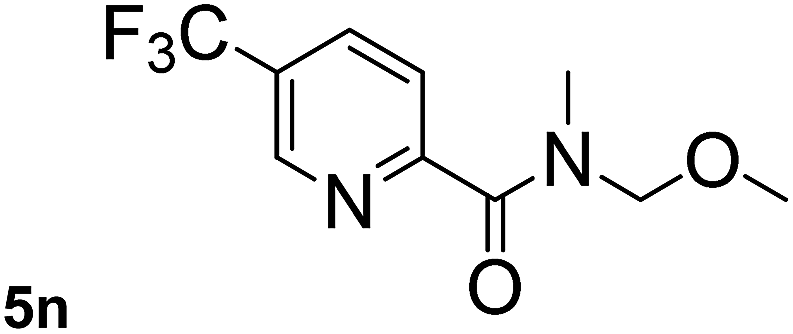
This approach is an easy and practical method for the aminocarbonylation of aryl halides, suitable for the formation of tertiary amides. For more application within different areas, secondary amides need to be synthesized, for example, the synthesis of heterocyclic compounds.15 We must further develop this method towards the preparation of secondary amides. Therefore, carbamoylsilanes containing an amino protecting group are expected to solve this problem. Considering this, the N-methoxymethyl-N-methylcarbamoyl(trimethyl)silane was selected as an amide source to synthesize secondary amides under same reaction conditions, wherein methoxymethyl group was used as an amino protecting group. To our surprise, all reactions proceeded smoothly and most of reactions afforded secondary amides directly in good yields, the methoxymethyl group could be hydrolyzed in the separation process. As shown in Table 3, we observed that aryl halides 1a, 1e, 1g, and 1h, bearing different functional groups, gave the corresponding secondary amides in good yields, while hetero aryl halide 1n afforded the methoxymethyl-protected amide 5n in 73% yield (Table 3, entry 5).
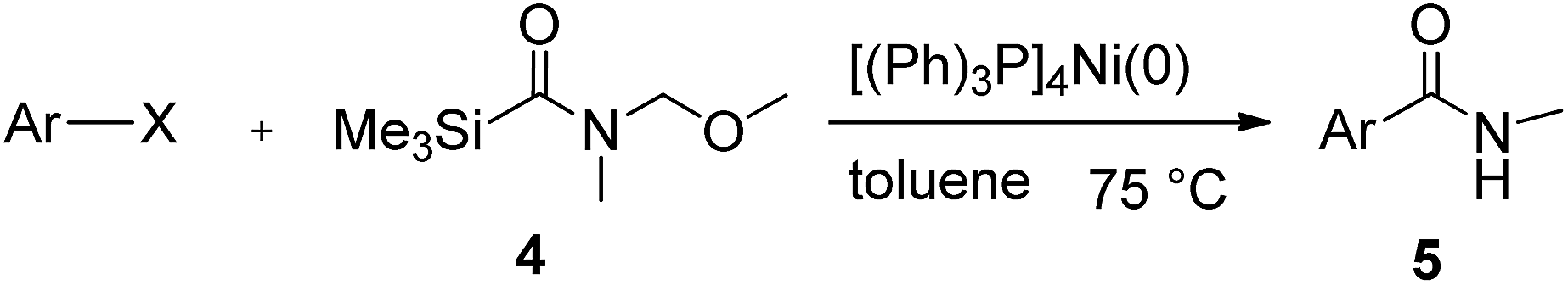
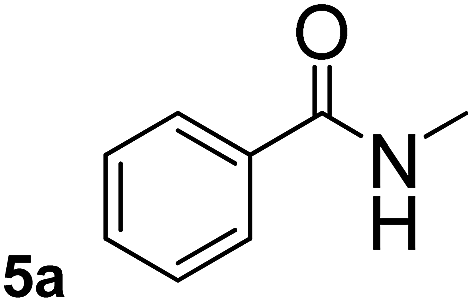
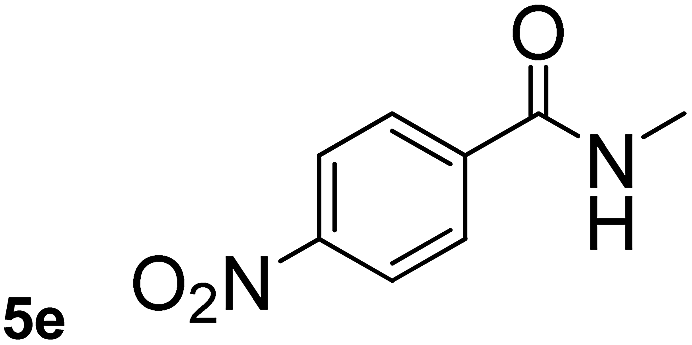
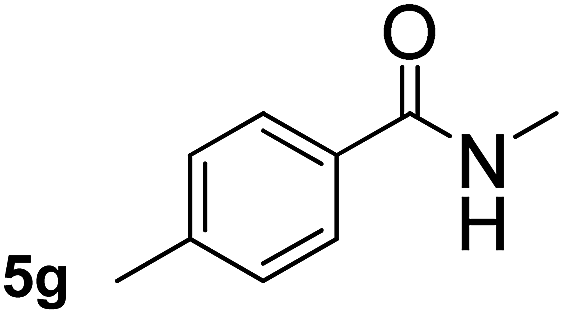
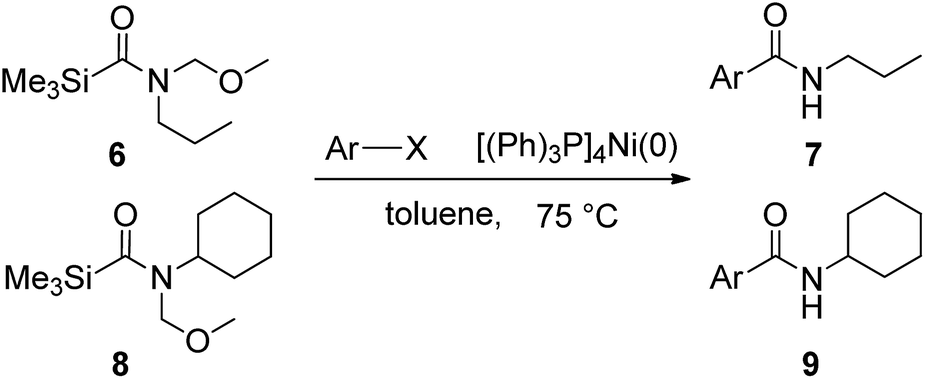
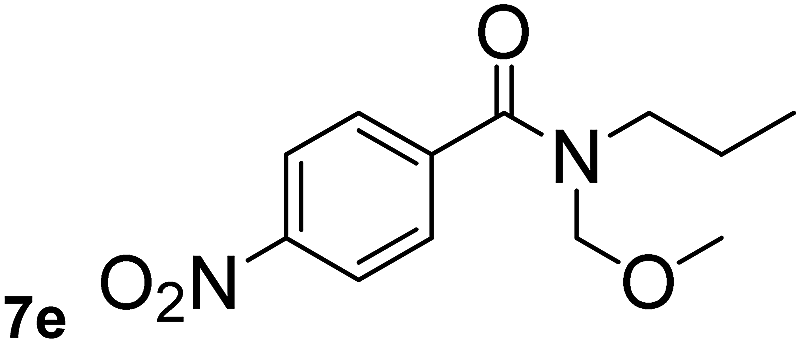
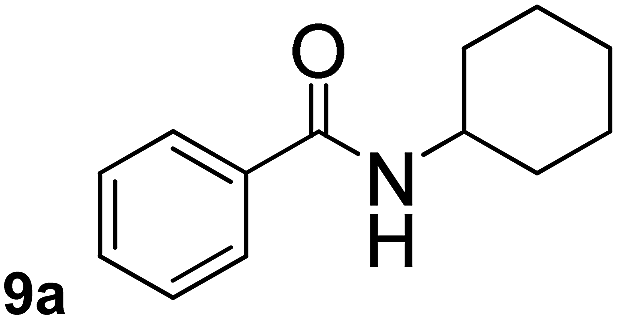
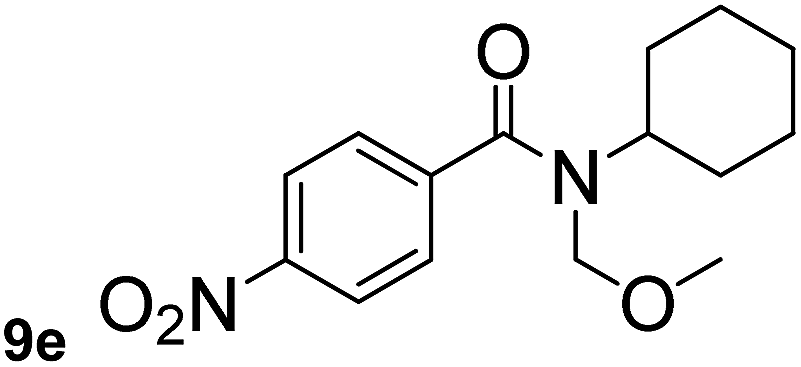
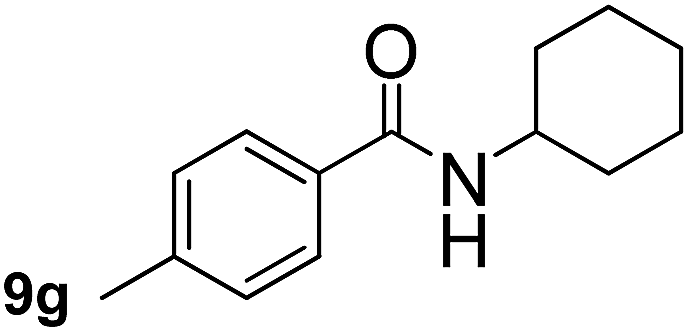
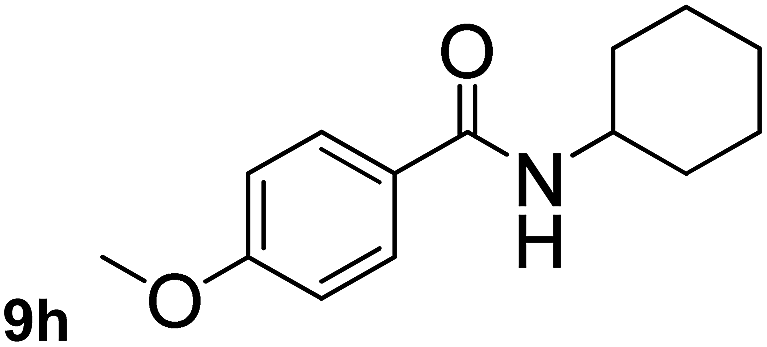
In order to study the synthetic potential and general applicability of developed methodology, carbamoylsilanes having different alkyls were selected as amides source, such as N-methoxymethyl-N-propylcarbamoyl(trimethyl)silane (6) and N-methoxymethyl-N-cyclohexylcarbamoyl(trimethyl)silane (8), to react with aryl halides bearing different functional groups (1a, 1e, 1g, 1h and 1n) under same reaction conditions. Experimental results are displayed in Table 4. We observed that both carbamoylsilanes reacted smoothly and afforded the corresponding amides in moderate yields. Aryl halides bearing an electron-withdrawing and electron-donating group were tolerated. Aryl halides 1e and 1n gave the methoxymethyl-protected amides 7e, 9e and 9n in 56%, 60% and 62% yields respectively (Table 4, entries 2, 7 and 10), while others directly afforded secondary amides in good yields.
In summary, a novel nickel-catalyzed aminocarbonylation method of aryl halides using carbamoylsilanes as amides source has been developed. The protocol could prepare both tertiary and secondary amides, and tolerate a broad range of aryl halides bearing different functional groups to provide good yields of the products under mild reaction conditions. The mild and simple procedure, the easy availability of aryl halides and the low toxity of reagents provide a valuable method for the preparation of various aryl amides. We believe that the current methodology will prove to be an attractive alternative to the reported methods for aminocarbonylation reactions due to utilizing carbamoylsilanes used as amides source instead of a toxic CO source.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Shanxi Province Foundation for Returness (No. 0713), the Natural Science Foundation of Shanxi Province (No. 2012011046-9) and Foundation of Shanxi Normal University (No. SD2015CXXM-83), China.Notes and references
- (a) J. J. Luszczki, M. J. Swiader, K. Swiader, R. Paruszewski, W. A. Turski and S. J. Czuczwar, Fundam. Clin. Pharmacol., 2008, 22, 69 CrossRef CAS PubMed; (b) J. W. Bode and S. S. Sohn, J. Am. Chem. Soc., 2007, 129, 13798 CrossRef CAS PubMed; (c) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 790 CrossRef CAS PubMed; (d) I. Kobayashi, H. Muraoka, M. Hasegawa, T. Saika, M. Nishida, M. Kawamura and R. Ando, J. Antimicrob. Chemother., 2002, 50, 129 CrossRef CAS PubMed; (e) A. Graul and J. Castaner, Drugs Future, 1997, 22, 956 CrossRef CAS; (f) K. R. Reddy, C. U. Maheswari, M. Venkateshwar and M. L. Kantam, Eur. J. Org. Chem., 2008, 3619 CrossRef CAS; (g) M. D. Reddy and E. B. Watkins, J. Org. Chem., 2015, 80, 11447 CrossRef CAS PubMed; (h) M. D. Reddy, F. R. Fronczek and E. B. Watkins, Org. Lett., 2016, 18, 5620 CrossRef CAS PubMed.
- (a) H. Dong and M.-F. Hou, Chin. J. Org. Chem., 2017, 37, 267 CrossRef CAS; (b) R. W. Foster, E. N. Lenz, N. S. Simpkins and D. Stead, Chem.–Eur. J., 2017, 23, 8810 CrossRef CAS PubMed; (c) Y.-H. Yang, J.-J. Gu, Z. Fang, Z. Yang, P. Wei and K. Guo, RSC Adv., 2017, 7, 22797 RSC; (d) T. Tang, L. Zhang, H. Dong, Z.-X. Fang, W.-Q. Fu, Q.-Y. Yu and T.-D. Tang, RSC Adv., 2017, 7, 7711 RSC; (e) Y. S. Seo, D. S. Kim and C. H. Jun, Chem.–Asian J., 2016, 11, 3508 CrossRef CAS PubMed; (f) S.-Y. Guo, Z. Fang, Z. Yang, C.-K. Liu, Z.-X. Dai, L.-H. Zho and K. Guo, RSC Adv., 2016, 6, 1503 RSC.
- (a) B. C. Soderberg, in Comprehensive Organometallic Chemistry II, ed. L. S. Hegedus, E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon, Oxford, UK, 1995, vol. 12, p. 249 Search PubMed; (b) H. M. Colguhoun, D. J. Thompson and M. V. Twigg, Carbonylation, Plenum, New York, 1991, p. 145 Search PubMed.
- (a) A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3318 CrossRef CAS; (b) A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327 CrossRef CAS.
- (a) R. S. Mane and B. M. Bhanage, J. Org. Chem., 2016, 81, 1223 CrossRef CAS PubMed; (b) Y. Li, D. Xue, C. Wang, Z.-T. Liu and J. Xiao, Chem. Commun., 2016, 52, 2960 RSC; (c) J. Cheng, X.-X. Qi, M. Li, P.-H. Chen and G.-S. Liu, J. Am. Chem. Soc., 2015, 137, 2480 CrossRef CAS PubMed; (d) S. T. Gadge and B. M. Bhanage, RSC Adv., 2014, 4, 10367 RSC; (e) W.-W. Fang, Q.-Y. Deng, M.-Z. Xu and T. Tu, Org. Lett., 2013, 15, 3678 CrossRef CAS PubMed; (f) T.-T. Dang, Y.-H. Zhu, J. S. Y. Ngiam, S. C. Ghosh, A. Chen and A. M. Seayad, ACS Catal., 2013, 3, 1406 CrossRef CAS; (g) C. Csajagi, B. Borcsek, K. Niesz, I. Kovacs, Z. Szekelyhidi, Z. Bajko, L. Urg and F. Darvas, Org. Lett., 2008, 10, 1589 CrossRef CAS PubMed; (h) J. R. Martinelli, D. M. M. Freckmann and S. L. Buchwald, Org. Lett., 2006, 8, 4843 CrossRef CAS PubMed; (i) A. Schnyder, M. Beller, G. Mehltretter, T. Nsenda, M. Studer and A. F. Indolese, J. Org. Chem., 2001, 66, 4311 CrossRef CAS PubMed; (j) W.-F. Xiong, C.-R. Qi, T.-Z. Guo, M. Zhang, K. Chen and H.-F. Jiang, Green Chem., 2017, 19, 1642 RSC.
- (a) P. G. Alsabeh, M. Stradiotto, H. Neumann and M. Beller, Adv. Synth. Catal., 2012, 354, 3065 CrossRef CAS; (b) D. N. Sawant, Y. S. Wagh, K. D. Bhatte and B. M. Bhanage, J. Org. Chem., 2011, 76, 5489 CrossRef CAS PubMed.
- (a) M. A. P. Martins, C. P. Frizzo, D. N. Moreira, L. Buriol and P. Machado, Chem. Rev., 2009, 109, 4140 CrossRef CAS PubMed; (b) T. Morimsoto and K. Kakiuchi, Angew. Chem., Int. Ed., 2004, 43, 5580 CrossRef PubMed.
- R. F. Cunico and J.-X. Chen, Synth. Commun., 2003, 33, 1963 CrossRef CAS.
- (a) W.-T. Tong, H. Liu and J.-X. Chen, Tetrahedron Lett., 2015, 56, 1335 CrossRef CAS; (b) H. Liu, Q.-L. Guo and J.-X. Chen, Tetrahedron Lett., 2015, 56, 5747 CrossRef CAS; (c) Q.-L. Guo, X.-P. Wen and J.-X. Chen, Tetrahedron, 2016, 72, 8117 CrossRef CAS.
- (a) W.-D. Li, Y.-H. Liu and J.-X. Chen, Tetrahedron Lett., 2015, 56, 4328 CrossRef CAS; (b) X.-J. Chen and J.-X. Chen, Mendeleev Commun., 2013, 23, 106 CrossRef CAS.
- (a) F. Ma, H. Liu and J.-X. Chen, Tetrahedron Lett., 2016, 57, 5246 CrossRef CAS; (b) J.-X. Chen and R. F. Cunico, J. Org. Chem., 2004, 69, 5509 CrossRef CAS PubMed.
- R. F. Cunico and B. C. Maity, Org. Lett., 2002, 4, 4357 CrossRef CAS PubMed.
- Y. H. Liu, P. Cao and J. X. Chen, Tetrahedron Lett., 2016, 57, 937 CrossRef CAS.
- (a) J. Ju, M. Jeong, J. Moon, H. M. Jung and S. Lee, Org. Lett., 2007, 9, 4615 CrossRef CAS PubMed; (b) Y. Jo, J. Ju, J. Choe, K. H. Song and S. Lee, J. Org. Chem., 2009, 74, 6358 CrossRef CAS PubMed.
- (a) P. Hasan, B. Aneja, M. M. Masood, M. B. Ahmad, U. Yadava, C. G. Daniliuc and M. Abid, RSC Adv., 2017, 7, 11367 RSC; (b) M. Muselli, C. Baudequin, C. Perrio, C. Hoarau and L. Bischoff, Chem.–Eur. J., 2016, 22, 5520 CrossRef CAS PubMed; (c) N. S. Upadhyay, V. H. Thorat, T. R. Sato, P. Annamalai, S.-C. Chuang and C.-H. Cheng, Green Chem., 2017, 19, 3219–3224 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental information, characterization data for new compounds. See DOI: 10.1039/c7ra08009c |
| This journal is © The Royal Society of Chemistry 2017 |