
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical monitoring of ROS generation by anticancer agents: the case of chartreusin†
Antonio Doménech-Carbó *a,
Gerardo Cebrián-Torrejón*ab,
Noemí Montoyaa,
Nico Ueberschaarc,
Marcus Tullius Scottid,
Zohra Benfoddab and
Christian Hertweckc
*a,
Gerardo Cebrián-Torrejón*ab,
Noemí Montoyaa,
Nico Ueberschaarc,
Marcus Tullius Scottid,
Zohra Benfoddab and
Christian Hertweckc
aDepartament de Química Analítica, Facultat de Química, Universitat de València, Dr Moliner 50, 46100 Burjassot, Valencia, Spain. E-mail: antonio.domenech@uv.es
bUniv. Nimes, EA7352 CHROME, Rue du Dr G. Salan, 30021, Nîmes Cedex 1, France. E-mail: gerardo.cebrian_torrejon@unimes.fr
cDepartment of Biomolecular Chemistry, Leibniz Institute for Natural Product Research and Infection Biology (HKI), Jena, Germany
dDepartment of Engineering and the Environment, Federal University of Paraíba, Rua da Mangueira s/n, Rio Tinto, PB 58297-000, Brazil
First published on 21st September 2017
Abstract
Solution phase and solid-sate electrochemical techniques centered in the voltammetry of microparticles approach are applied for testing the cytotoxic activity of anticancer drugs. The possibility of electrochemical generation of reactive oxygen species (ROS) is exploited for evaluating their contribution to cellular damage. The described methodology is applied to the case of chartreusin (Ch) whose electrochemistry in non-aqueous solutions and in the solid state in contact with aqueous electrolytes is described in the absence (experimental data were confirmed by theoretical calculations) and in the presence of double-stranded DNA (dsDNA). In parallel, scanning electrochemical microscopy (SECM) examination of dsDNA fibers was developed. Electrochemical data suggest that Ch-induced dsDNA interaction can operate by a two pathways: via intercalation and by mechanisms related with ROS generation. Although reduced Ch forms electrochemically generated can act as radical scavengers blocking the ROS generation chain, this property is interrupted upon binding to dsDNA.
Introduction
In order to develop new drugs against cancer, two key points should be considered, the exploration of the chemical diversity, increasing the pharmacological options to fight against drug resistance, and the study of correlation between modifications in genetic expression produced by the anticancer molecule and cytotoxicity. These aspects will allow a deeper understanding of the mechanism of action of drugs and will enhance the expectation for success in the research of new treatments.1,2As far as biological redox activity is recognized as playing a key role in a number of cytotoxic processes, including the triggering of cellular events that can be exploited for therapeutic uses,3 electrochemical studies in solution phase have been applied in medicinal chemistry typically for correlating biological activity with redox potentials.4 The biochemical significance of such correlations in regard to cytotoxic activity is modulated, however, by the difficulty in mimicking the biological environment in conventional electrochemical measurements, and the occurrence of undesired chemical reactions involving the electrochemically generated products, among other factors. On the other hand, it is uneasy to discriminate different possible cytotoxic actions. Typically, antitumoral activities can be exerted via DNA interaction as: groove binding, covalent linking or intercalation.5–7 Nevertheless endogenous DNA damage can also result from the action of intermediates of reactive oxygen species (ROS), which have the potential to interact with DNA bases or the deoxyribosyl backbone of DNA to produce damaged bases or strand breaks, and/or oxidize proteins or lipids generating intermediates reacting with DNA.8
In this context, a complementary electrochemical strategy, the voltammetry of microparticles (VMP), a solid-state technique yielding the electrochemical response of sparingly soluble materials,9,10 can be used in order to elucidate the mode of action of the cytotoxic agents. This methodology was previously used for screening the antimalarial activity of drugs11 and studying the dsDNA binding and damage by scorpiand-like polyamine receptors12a and natural β-carboline alkaloids.12b,c The use of VMP is potentially interesting in DNA studies, because solid-state electrochemical processes allow evaluating reactions that are inhibited in solution phase or obscured by faster electrochemical processes in conventional solution electrochemistry and permits the selection of hydrophilic or lipophilic environments via an appropriate electrolyte.12
Here, this methodology is extended upon exploiting the possibility of generating different ROS electrochemically13,14 in order to discriminate the action mode of cytotoxic compounds. Thus, the radical scavenging activity of plant extracts has been also studied electrochemically using chemical generation of ROS15–17 whereas the VMP technique has been applied to evaluate the antioxidant ability of samples from dry extracts of fruits and vegetables.17–19
The electrochemical signaling of cytotoxicity is applied to chartreusin (1, Ch), an aromatic polyketide glycoside isolated from Streptomyces chartreusis, that exhibits significant chemotherapeutic activity against bacteria20 and various tumor cell lines such as murine P388 and L1210 leukemia, and B16 melanoma cells,20–23 which has received increasingly growing attention.24–27 Chartreusin analogues are recognized as photoreactive DNA intercalators,28 but this compound and its derivatives could also exert their antitumor activities through binding to DNA,29 radical-mediated single-strand breaks, and inhibition of topoisomerase II.30 However the whole mode of action is not fully understood.
Chartreusin (1) structure (Fig. 1) consists of fucose, digitalose, and an unusual bislactone aglycone, named chartarin (Fig. 1),31 which include both potentially reducible and oxidizable moieties, thus providing an example of considerable structural and redox complexity where, as described by Goulart et al.,32 there is possibility of inner reorganization during the radical–biradical transitions. Electrochemical data are relevant as far as subtle structural changes can produce significant variations in the clastogenic potential as recently discussed by Saladino et al.33 The proposed electrochemical pathways, conducting to the different derivatives of chartreusin (1), were confirmed by theoretical calculations.
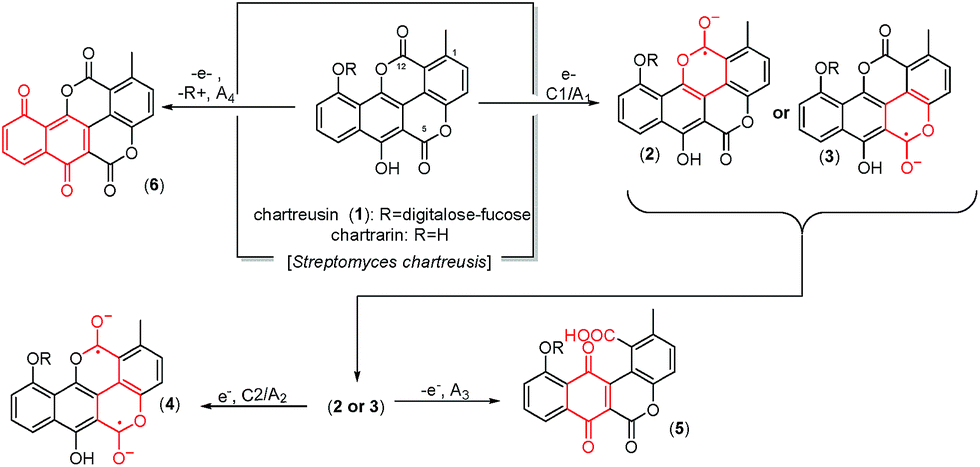 | ||
| Fig. 1 Structure of chartreusin and tentative scheme for electrochemical processes occurring in DMSO solution. Some equivalent processes could be proposed localized on the C12 unit rather than in the C5 one. | ||
In fact, it is known that chartreusin (1) and its related antibiotic elsamicin31,34 are able to bind to heterogeneous DNA, showing a clear preference for guanine/cytosine-rich DNA and that in the presence of a reducing environment and ferrous iron is able to degrade DNA.26,35 However, the DNA-cleaving activity appears to be partially blocked by the action of superoxide dismutase (SOD) and catalase. These results suggested that the bislactone aglycone moiety (chartarin) can be involved in the production of free radicals, so that chartreusin (1) could induce breakage of DNA through the generation of reactive oxygen-radical species.36 Quantum mechanical calculations suggested that only one of the “oxo groups” of the chartarin motif is reduced with no clear preference for either the C5 or the C12 group, the mono-hydroxylated species being subsequently oxidized by molecular oxygen to give the hydroperoxyl (HO2˙) radical.37
In order to provide some answers on the whole mode of action of chartreusin (1), the interaction between this compound and dsDNA has been monitored electrochemically in lipophilic and hydrophilic environments using conventional electrochemistry in DMSO solution, and solid-state electrochemistry respectively. Here, potential inputs are used in a two-fold function: provide the reducing environment required for the action of 1 on dsDNA26,35 and generating ROS species.
VMP has been combined with scanning electrochemical microscopy (SECM). This electrochemical technique provides information on electrochemical surface topography at the nanoscopic level38 and has been used to monitor dsDNA hybridization and to detect DNA lesions39–43 via using monolayers of DNA probes, their hybridization being monitored via guanine oxidation. One advantage of this technique over conventional bulk electrochemistry is that permits the sampling a local area of the immobilized substrate at the micrometer-level diameter of the SECM tip electrode minimizing contributions from imperfections on macroelectrodes. In such solutions, superoxide radical anion can be electrochemically generated in situ by applying a suitable potential to the substrate Pt electrode. The use of a redox competition based strategy44,45 permits to detect significant changes in the electrochemical topography of dsDNA fibers providing information on dsDNA damage activity of ROS and ferrocenium ion electrochemically generated in situ, as previously described for scorpiand-like macrocyclic receptors.12
Experimental
Chartreusin (1) was isolated from S. chartreusis. Double stranded deoxyribonucleic acid sodium salt from salmon testes (dsDNA) was purchased for Sigma-Aldrich and used as received. For experiments in aqueous media, the stock solution of DNA was prepared by dissolving in PBS pH 7.4.Electrochemical experiments were performed at 298 ± 1 K in a thermostated three-electrode cell with CH 660I equipment. A BAS MF2012 glassy carbon working electrode (GCE) (geometrical area 0.071 cm2), a platinum wire auxiliary electrode and an Ag/AgCl (3 M NaCl) reference electrode were used. Voltammetric measurements were performed at potential scan rates between 10 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mV s−1 in order to study the kinetics of the electrochemical processes. SECM experiments were performed with CH 920c equipment using a microdisk platinum electrode tip (CH 49, diameter 20 μm) and a Pt substrate electrode. The bipotentiostat mode was used to apply potentials to the tip (ET) and the electrode substrate (ES). In order to mimic physiological lipophilic environments, water-containing (1% v/v) DMSO (Carlo Erba, HPLC grade), optionally degassed by passing Ar during 15 min, was used as solvent incorporating Bu4NPF6 (Fluka) as a supporting electrolyte in concentration 0.10 M. In these experiments, a Pt-wire pseudoreference electrode was used and adding ferrocene (Carlo Erba) in concentration 0.1 mM for calibration, the potentials being passed to the Ag/AgCl scale using IUPAC conversion factors.46,47
000 mV s−1 in order to study the kinetics of the electrochemical processes. SECM experiments were performed with CH 920c equipment using a microdisk platinum electrode tip (CH 49, diameter 20 μm) and a Pt substrate electrode. The bipotentiostat mode was used to apply potentials to the tip (ET) and the electrode substrate (ES). In order to mimic physiological lipophilic environments, water-containing (1% v/v) DMSO (Carlo Erba, HPLC grade), optionally degassed by passing Ar during 15 min, was used as solvent incorporating Bu4NPF6 (Fluka) as a supporting electrolyte in concentration 0.10 M. In these experiments, a Pt-wire pseudoreference electrode was used and adding ferrocene (Carlo Erba) in concentration 0.1 mM for calibration, the potentials being passed to the Ag/AgCl scale using IUPAC conversion factors.46,47
Films of chartreusin (1) on glassy carbon electrode were prepared using the methodology previously described11,12 by pipetting 10 μL of 0.05 mM solution of 1 in ethanol and allowing the solvent to evaporate unto air. As a result, a uniform, fine coating was adhered to the base electrode. The voltammetric response of such modified electrodes was studied upon immersion into aqueous 0.25 M potassium phosphate buffer and/or PBS at pH 7.4, previously degassed by passing Ar during 15 min. Microparticulate deposits of chartreusin were examined before and after application of potential inputs using a Jeol JSM 6300 scanning electron microscope operating with a Link-Oxford-Isis X-ray microanalysis system. The analytical conditions were: accelerating voltage 20 kV, beam current 2 × 10−9 A, and, working distance 15 mm. Field-emission scanning electron microscope operating with X-ray microanalysis system (FESEM/EDX).
The structure of compounds chartreusin (1) and derivatives (2–7) due structural changes from electrochemical process occurring in DMSO solution (Fig. 1) were drawn using Marvin 17.3.3.0, 2016, ChemAxon (http://www.chemaxon.com). The software, Standardizer, JChem 17.4.3.0, 2016, ChemAxon (http://www.chemaxon.com), was used to canonize the structure. This process converts an arbitrarily chosen chemical structure to a unique notation, adds hydrogens, and cleans the molecular graph in three dimensions. The process uses a divide-and-conquer approach. The structure is split into small fragments which are organized into a tree using connectivity information. Conformers generated for the initial structure (represented by the root node in the tree) are optimized. The tree building process uses a proprietary extended version of the Dreiding force field.48
Geometric optimizations and conformational searches were performed using Spartan'16.49 The geometry of the chemical structure of the compound was initially optimized with a Merck Molecular Force Field (MMFF) force field,50 and a new geometric optimization was then performed based on the semi-empirical method, Austin Model 1 (AM1).51 A systematic search method was used which analyzed conformers and selected the conformers with the lowest minimum energy using AM1 and a Monte-Carlo algorithm.52 After that, the lowest minimum energies were selected and optimized based on a vibrational mode calculation using DFT (density functional theory).53
DFT calculations were performed using Spartan 10 for Windows (Wavefunction, Irvine, CA, USA).53,54 Each structure was examined at the B3LYP/6-311G* level and the lowest energy structures were selected for the calculations. The global minimum on the potential energy surface was used for the determination of each geometry.
Results and discussion
Electrochemistry in lipophilic environment
In order to mimic the redox behavior of chartreusin (1) in lipophilic environments, the electrochemical response of 1 solutions in DMSO was investigated.Fig. 2 shows cyclic voltammograms at glassy carbon electrode of a 0.03 mM solution of chartreusin in 0.10 M Bu4NPF6/DMSO at a potential scan rate of 50 mV s−1. In initial cathodic scan voltammograms (Fig. 2a), reduction peaks at −0.18 (C1) and −0.75 V (C2) vs. Ag/AgCl appear, followed by a shoulder at ca. −1.5 V (C3). The peaks C1, C2 are coupled, in the subsequent anodic scan, by weak oxidation counterparts at −0.58 (A2) and +0.10 V (A1). On prolonging this scan, anodic peaks at +0.59 (A3) and +0.90 V (A4) are recorded. In initial anodic scan voltammograms (Fig. 2b), only the peak A4 remains well-defined, thus denoting that this electrochemical process corresponds to the oxidation of the pristine chartreusin (1). This peak does not exhibit any cathodic counterpart at high scan rates, thus suggesting that the process A4 corresponds to an essentially irreversible process. In contrast, the peak A3 is accompanied by a cathodic one (C3) at scan rates above 500 mV s−1, thus suggesting that the electron transfer process is coupled with relatively slow chemical reactions. The peak A3 also appears when the potential is switched few mV past the peak C1, thus denoting that the species resulting from this cathodic process yields two competing oxidative pathways via processes A1 and A3.
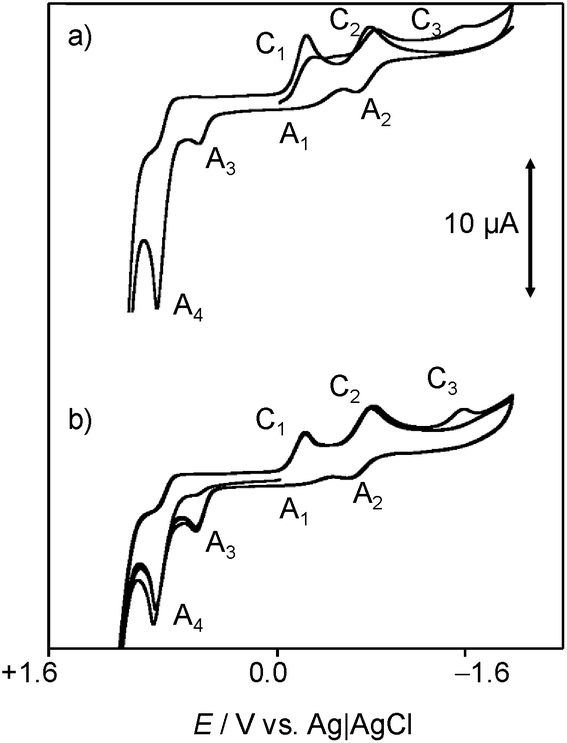 | ||
| Fig. 2 Cyclic voltammograms at glassy carbon electrode of a 0.03 mM chartreusin solution in 0.10 M Bu4NPF6/DMSO (1% water v/v). (a) Initial cathodic scan; (b) initial anodic scan. Potential scan rate 50 mV s−1. | ||
This voltammetry can be directly related with that extensively studied for quinones (Q) which, in aprotic solvents are reduced successively to the corresponding semiquinone anion radical (Q˙−) and the dianion (Q2−).55 On the basis of thermochemical calculations from Párraga et al.37,56 on elsamicin, a chartreusin analogue harboring identical bislactone aglycone (chartarin), the most favored reductive pathway involves one of the oxo groups but with no clear preference for either the C5 or the C12 group. The above one-electron processes would be responsible for peaks C1 and C2, respectively, for which a reasonable representation can be seen in Fig. 1. Here, inner reorganizations associated to the radical anion to biradical dianion conversion would be conceivable similarly to that described for a nor-β-lapachone derivative possessing two redox centers.32 The process A3 can tentatively be attributed, based on comparable processes described for quercetin,19 to the oxidation of the anion radical generated in the cathodic process C1 to a different quinone form involving, as represented in Fig. 1, ring opening. The process A4, occurring at clearly positive potentials with high degree of irreversibility can be attributed to the oxidation of the –OH and –OR motifs (yielding compound 6, see Fig. 1), in agreement with the electrochemistry of anisols.57
The above voltammetry changes drastically in the presence of dsDNA. Fig. 3 shows the (a) initial cathodic and (b) initial anodic scan voltammograms for a 0.03 mM of 1 incubated with 0.03 mM DNA solution in 0.10 M Bu4NPF6/DMSO. Here, a first cathodic signal consisting of overlapping peaks between −0.30 and −0.65 V (C1D/C2D), both without defined anodic counterparts, is followed by an irreversible cathodic process at ca. −1.5 V (C3D). In the anodic region of the voltammogram, only a well-defined oxidation wave at +0.72 V (A4D) appears preceding a shoulder at ca. +1.10 V. The intensity of all signals is lower than for native chartreusin (1) at the same concentration. Cyclic voltammograms, after semi-derivative convolution to increase peak resolution, for dsDNA plus chartreusin (1) (Fig. 3c) and 1 alone (Fig. 3d), show clearly that the chartreusin cathodic peak C1 becomes resolved into several peaks (C1D) in the presence of dsDNA. Such reduction processes, in contrast with process C1 for native chartreusin, behaves highly irreversible at the tested scan rates. These features can be interpreted on assuming that 1 is strongly coordinated to dsDNA so that the potential of the voltammetric signals become significantly shifted relative to the uncoordinated chartreusin, a well-known feature in electrochemical studies on cytotoxic reagents.4 The considerable peak splitting appearing in the initial reduction process (C1D) of 1 and, to a lower extent, in the initial oxidation signal (A4D), suggests that chartreusin binds to dsDNA for more than one site. These data suggest that, in lipophilic biological environments, effective intercalation occur modulating the redox chemistry of chartreusin.
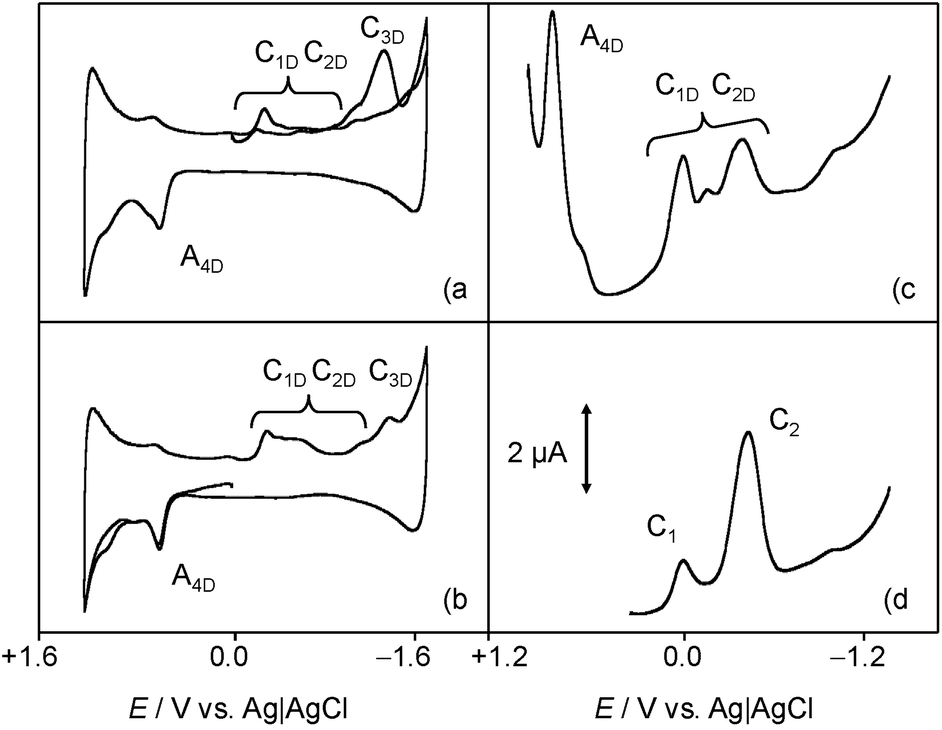 | ||
| Fig. 3 (a, b) Cyclic and (c, d) square wave voltammograms at glassy carbon electrode of (a–c) 0.03 mM chartreusin plus 0.03 mM dsDNA and (d) 0.03 mM chartreusin solutions in 0.10 M Bu4NPF6/DMSO. (a) Initial cathodic and (b) anodic scan voltammograms at a potential scan rate of 50 mV s−1. (c, d) Potential scan initiated at +1.05 V in the negative direction; potential step increment 4 mV; square wave amplitude 25 mV, frequency 5 Hz. Semi-derivative convolution of cyclic voltammograms was performed to increase peak resolution. | ||
Electrochemistry in hydrophilic environment
Due to the low water solubility of chartreusin (1) (logD: 2.46 at pH 7.4; ChemAxon prediction), the electrochemistry of this compound in a hydrophilic environment was studied using 1 films on glassy carbon electrode in contact with aqueous phosphate buffer and/or PBS buffer. The films remained stable during voltammetric experiments and allowed to at least 10–20 repeated measurements without significant changes for each freshly prepared electrode.
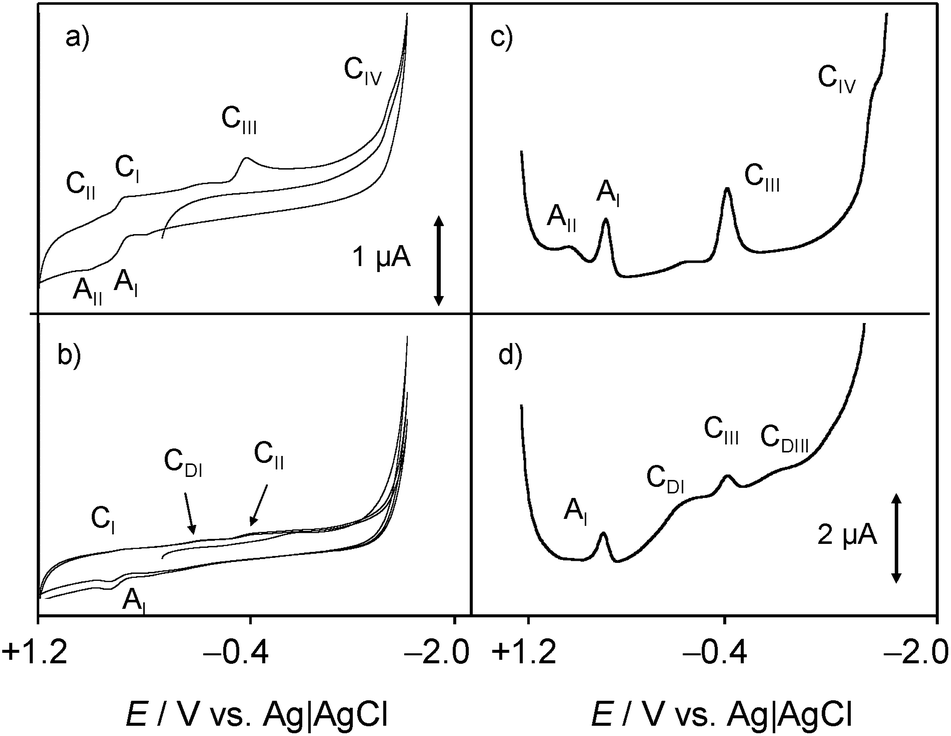
Fig. 4 shows the (a, b) cyclic and (c, d) square wave voltammograms of 1-modified glassy carbon electrodes immersed into 0.25 M aqueous potassium phosphate buffer at pH 7.4 (a, c) in the absence and (b, d) in the presence of dsDNA. The voltammetric response chartreusin (1) differs from that recorded in the lipophilic environment, being dominated by two successive essentially reversible one-electron couples at midpeak potentials of +0.57 V (AI/CI) and +0.80 V (AII/CII), accompanied by an apparently irreversible reduction process at −0.40 V (CIII) and a cathodic shoulder (CIV) near the solvent discharge.
 | ||
| Fig. 4 (a, b) Cyclic and (c, d) square wave voltammograms of chartreusin-modified glassy carbon electrodes immersed into: (a, c) 0.25 M aqueous potassium phosphate buffer at pH 7.4; (b, d) the same solution containing 0.5 mM dsDNA. (a, b) Potential scan rate 50 mV s−1. (c, d) Potential scan initiated at +1.05 V in the negative direction; potential step increment 4 mV; square wave amplitude 25 mV, frequency 5 Hz. | ||
This voltammetry can be described on the basis of quinone/hydroquinone electrochemistry4,37,55 in terms of two rounds of electron-transfer (E) coupled with proton receipt (chemical reaction (C)) processes (ECEC-type mechanisms). The repetition of the recorded voltammetric pattern in successive potential scans denoted that the observed redox processes can be ascribed to solid-to-solid transformations. Such processes can be described in terms of the Lovric and Scholz model on the electrochemistry of ion-insertion solids.58–60 In the case of organic compounds in contact with aqueous electrolytes, solid state electrochemical reactions are initiated at the solid particle/base electrode/electrolyte three-phase junction and propagate through the solid via proton insertion coupled with electron hopping between immobile redox centers in the solid. Generically, the reductive processes can be represented as solid state transformations of the type:
| Chsolid + nHaq+ + ne− → H2Chsolid | (1) |
The solid-state nature of the electrochemical processes was assessed by FESEM images recorded on microparticulate deposits of 1 in contact with phosphate buffer before and after application of reductive potential inputs. As can be seen in Fig. 5a, the parent chartreusin crystals exhibit a smooth surface which, after application of a potential of −0.50 V during 5 min, becomes corrugated and with irregular profiles. This pattern is consistent with the aforementioned modeling58–60 but suggests that, possibly, the reduced species have a certain solubility, thus resulting in some erosion of the original crystals.
 | ||
| Fig. 5 FESEM images for a microparticulate deposit of chartreusin on a graphite plate immersed into 0.10 M potassium phosphate buffer solution at pH 7.4, (a) before, and (b) after application of a potential step at +0.50 V during 5 min. | ||
In agreement with previous results for dyes,61 the AI and AII processes can be attributed to the oxidation of the parent chartreusin form to a quinone involving the issue of protons from the solid 1 to the solution phase, while the processes CIII, CIV would correspond to successive reductions of the parent chartreusin coupled to protonation.
In the presence of dsDNA, the voltammetric response again changes. Now, the AII/CII couple disappears whereas the process CIII is accompanied by cathodic peaks at −0.10 (CDI) and −0.78 V (CDII) which can be assigned to the reduction of chartreusin–dsDNA adducts. Since, in view of the solid state of chartreusin forms, no possibility of (at least complete) intercalation occurs, one can conclude that under the electrochemical conditions studied here, chartreusin binds to dsDNA and that the formed adduct species can experience one-electron reduction processes.
SECM imaging
To gain deeper insights into chartreusin–dsDNA interaction, SECM experiments were performed on dsDNA fibers attached to a Pt substrate electrode with the help of Paraloid B72 polymer in contact with DMSO solutions of 1. In these experiments, both generation/collection38 and redox competition44,45 methods were applied using ferrocene as the redox probe. This approach parallels those recently proposed for determining dsDNA interactions with scorpiand-like macrocyclic receptors.12 Under the used experimental conditions, dsDNA fibers emerge through the polymer pores and do not exhibited appreciable dissolution, as depicted in Fig. 6a. Different potentials were applied to the tip and the substrate electrodes taking into account that the voltammetric wave for the well-known one-electron oxidation of ferrocene appears at +0.55 V, accompanied, in air-saturated solutions, by the cathodic wave at ca. −1.0 V corresponding to the reduction of O2 to radical anion superoxide.62,63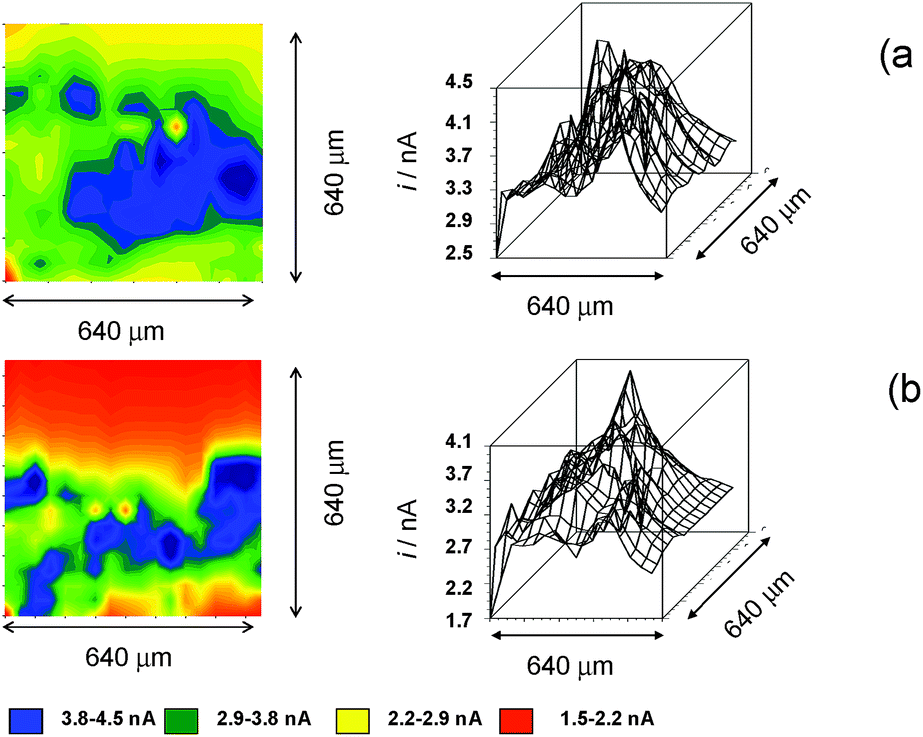 | ||
| Fig. 6 SECM topographic images of a deposit of dsDNA fibers attached to Pt substrate electrode immersed into a 0.10 M Bu4NPF6/DMSO solution containing: (a) 0.01 mM ferrocene; (b) 0.01 mM ferrocene plus 0.03 mM chartreusin. (a) ES = 0.00 V; ET = +0.60 V. Insets depict the corresponding color maps at the resolution of the tip diameter (20 μm). | ||
Under application of a tip potential slightly more positive than that for which the ferrocene oxidation signal appears, oxidation of ferrocene becomes diffusion-controlled and leads to enhanced/decreased tip current (referred to as SECM positive/negative feedback) depending on the distance to the substrate and the conductivity of the same so that the tip current is enhanced on approaching the substrate electrode when the substrate surface is conducting. In these conditions, the tip exhibits positive feedback in the regions where the dsDNA fibers occupy partially the gap between the tip and the substrate electrode, so that the tip current is enhanced, relative to the surrounding regions, where it is dominated by the insulating polymer blocking effect.
As a result, a peaked profile is recorded in the immediacy of dsDNA fibers. Under these conditions, the addition of 1 to the solution produces a modification of the profile. This observation is again consistent with the recognized binding ability of chartreusin to dsDNA. As far as ferrocene and ferrocenyl derivatives can be conjugated to dsDNA oligonucleotides through unsaturated bonds at the nucleobase,64,65 there was competition between ferrocene and chartreusin for binding with suitable dsDNA sites.
Accordingly, the concentration of ferrocene increases in the regions where is displaced from the dsDNA fibers by (1); i.e., peaked regions in Fig. 6b would correspond to those where chartreusin is preferentially anchored to de dsDNA fibers. This result was consistent with SECM imaging recorded using chartreusin itself as the redox probe (vide infra).
Interaction with ROS species
A second series of voltammetric and SECM experiments were performed in air-saturated, water-containing DMSO solutions in order to test the interaction of chartreusin with electrochemically generated ROS species.Fig. 7 shows the cyclic voltammogram at glassy carbon electrode of an air-saturated solution of 1 in 0.10 M Bu4NPF6/DMSO. In the absence of chartreusin (1), the reduction of dissolved oxygen occurs via a reversible one-electron transfer yielding superoxide ion resulting in a couple of peaks (Cox/Aox) at an equilibrium potential of −0.80 V.53,54 This process can be represented as:
| O2 + e− → O2˙− | (2) |
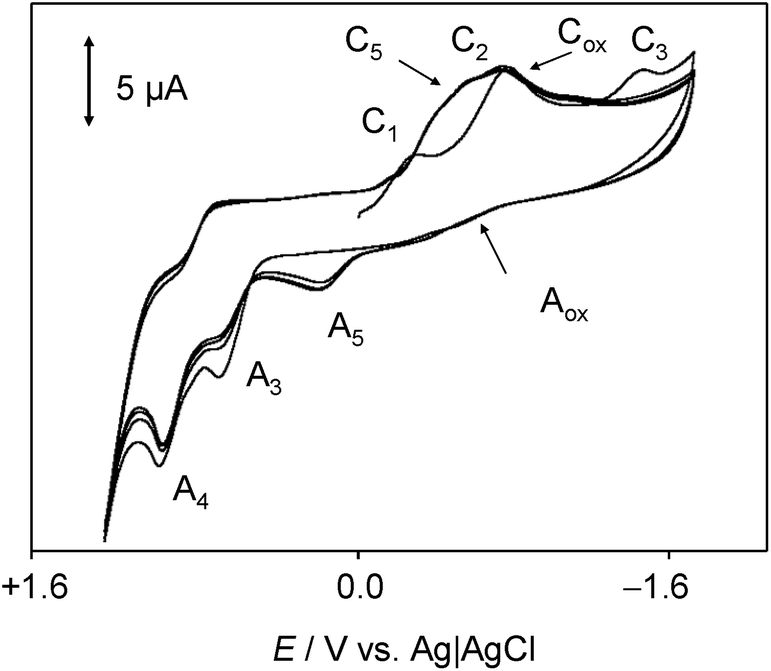 | ||
| Fig. 7 Cyclic voltammogram at glassy carbon electrode of air-saturated 0.03 mM chartreusin solution in 0.10 M Bu4NPF6/DMSO. Potential scan rate 50 mV s−1. | ||
In the presence of 1, the initial cathodic scan voltammograms show peak C1, followed by a series of overlapping signals between −0.4 and −0.8 V which can be attributed to the reduction process C2 accompanied by a new cathodic signal (C5) and the peak for the reduction of dissolved oxygen, Cox. In the reverse scan, all possible anodic signals coupled with the above peaks are almost entirely absent. In contrast, the signals A3 and A4 remain well-defined, now being accompanied by an additional oxidation peak at +0.15 V (A5). Upon repetitive cycling the potential scan, the peak A5 increases progressively at the expense of peaks A3 and A4.
These features can be interpreted on assuming that the electrochemically generated chartreusin anion radical (Ch˙−) reacts with oxygen species via different competing pathways. One first possibility would be the reaction with O2 forming the superoxide radical anion and regenerating the parent chartreusin:
| Ch˙− + O2 → Ch + O2˙− | (3) |
In view of the electrode potentials for the Ch/Ch˙− and O2/O2˙− couples (see Fig. 7), however, this possibility would be thermochemically disfavored. In hydrophilic media, the chartreusin and peroxide anion radicals would be protonated and may react to form HO˙ radicals, thus regenerating the parent chartreusin (1) and activating the formation of subsequent ROS. A possible sequence of reactions would be:
| O2˙− + H+ → HO2˙ | (4) |
| Ch˙− + H+ → ChH | (5) |
| ChH˙ + HO2˙ → 2HO˙ + Ch | (6) |
In alternative pathway, however, a peroxo adduct deactivating the ROS formation chain, could be formed. The produced species would be electrochemically oxidized via the process A5. Although attempts to isolate such products were unsuccessful, on the basis of literature data on the reaction of quercetin with ROS64–67 and the electrochemical response of this system,19 one plausible hypothesis in this context would be the formation of peroxo-bridged adducts (8 and 9).
Fig. 8 depicts a tentative scheme for explaining the possible abstraction of the hydroperoxo radical. This is indirectly supported by voltammetric data in air-saturated phosphate buffer at relatively high potential scan rates. The reduction of dissolved oxygen yields as a first step superoxide radical anion, subsequently undergoing protonation and reduction competing with disproportionation, resulting in the generation of HO˙ and HO2˙ radicals displaying an anodic signal at ca. +0.8 V.68–71 In the presence of chartreusin films, this signal disappears (see ESI, Fig. S1 and S2†). In particular, Kano et al.65 indicated that the reaction of quercetin with the superoxide anion radical is triggered by proton abstraction from quercetin to yield dismutated products of superoxide and deprotonated quercetin, which allows quercetinase-like dioxygenation to give the corresponding depside in a high yield. In spite of structural differences with chartreusin (1), in view of the similarity of the electrochemical response of chartreusin with quercetin,19 it is conceivable that peroxo-bridged chartreusin-based forms (9) could be tentatively considered as responsible of the C5/A5 signals.
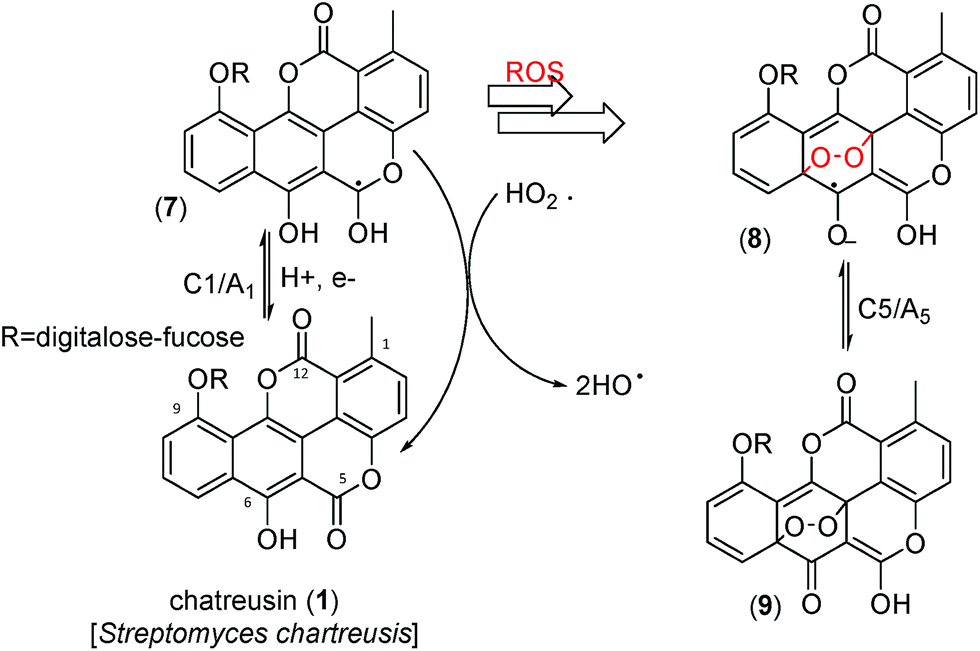 | ||
| Fig. 8 Tentative scheme able to describe electrochemical processes occurring at glassy carbon electrode of air-saturated 0.03 mM chartreusin solution in 0.10 M Bu4NPF6/DMSO. | ||
Analyzing the energy of the chartreusin (1) and modified structures (2–9) from the electrochemical processes occurring in DMSO solution, one can verify that the energy all structures are similar (1–4), excepting for the structures A3 (5), A4 (6) and A5 (9) (Table 1, Fig. 1 and 8). Structure 5 (A3) shows energy value little bit lower than (1), A1 (2 and 3) and A2 (4). On the other hand, the structure 6 (A4) presents the highest energy of all. The structure of 4 (A2) and its higher energy than 5 (A3) suggest not only the deprotonation of the hydrogen at position C6 as the opening of the ring at position C12 (ester group), leading to the structure 5 (A3), with lower energy. The structure 9 (A5) (Fig. 8) shows the lowest energy among the compounds.
| Compound | Potential electrostatic surface | Energy (au) | Energy (kJ mol−1) |
|---|---|---|---|
| Chartreusin (1) |  |
−2291.81 | −6017160.62 |
| C2/A2 (4) | 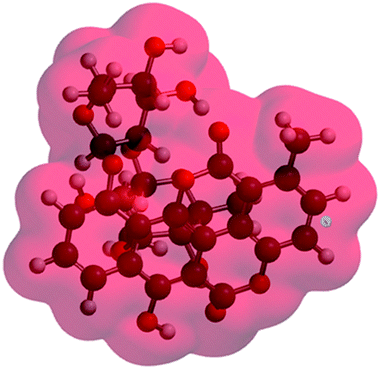 |
−2291.70 | −6016856.90 |
| C1/A1_1 (2) | 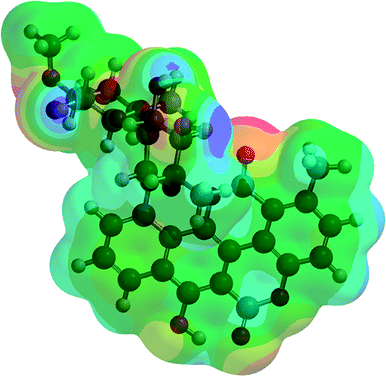 |
−2291.75 | −6016996.60 |
| C1/A1_2 (3) | 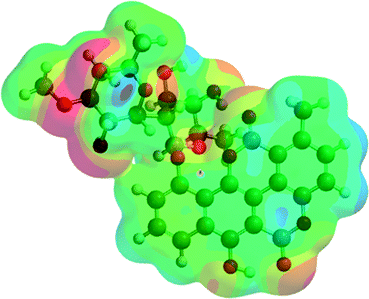 |
−2291.77 | −6017052.13 |
| A3 (5) | 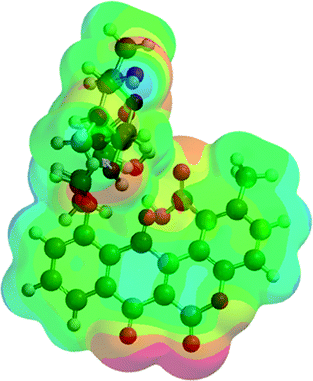 |
−2367.00 | −6214557.02 |
| A4 (6) | 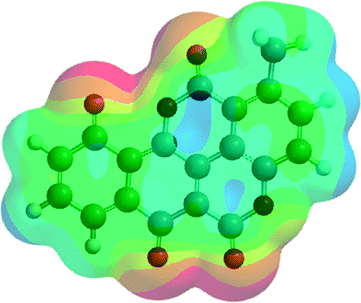 |
−1180.18 | −3098562.99 |
| A5 (9) | 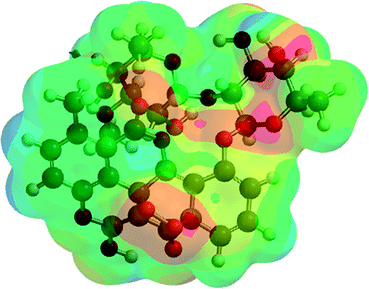 |
−2441.98 | −6411426.50 |
Consistently with quercetin data,19 such species would produce oxidation signals at less positive potentials than the parent chartreusin (1). Fig. 9 depicts the 2nd scan voltammograms of air-saturated DMSO solutions of 1 (a) and 1 plus dsDNA in the potential region around the Cox/Aox couple at a relatively high potential scan rate (500 mV s−1). Under these conditions, the voltammetric signals associated to reactive species can be more clearly seen.
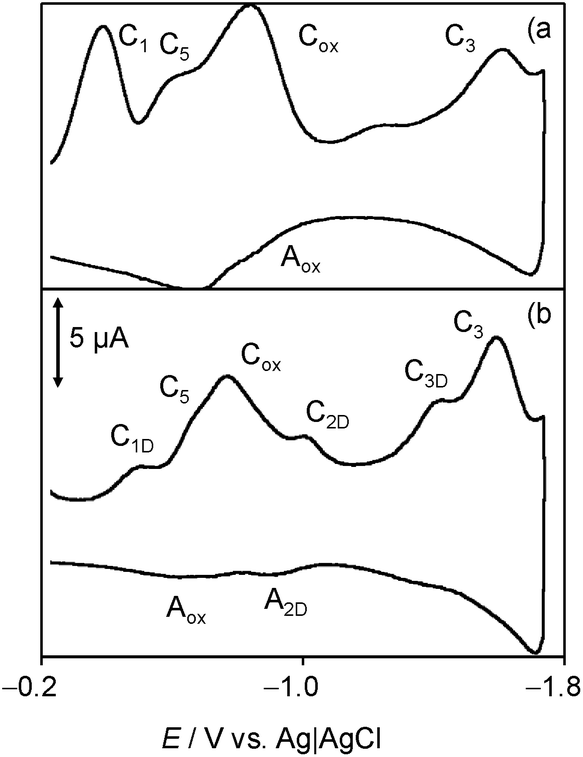 | ||
| Fig. 9 Cyclic voltammograms at glassy carbon electrode, after semi-derivative convolution, of: (a) 0.03 mM chartreusin and (b) 0.03 mM chartreusin plus 0.03 mM dsDNA air-saturated solutions in 0.10 M Bu4NPF6/DMSO. Detail of the 2nd scan. Potential scan rate 500 mV s−1. | ||
On comparing the voltammograms in Fig. 8, one can see that the presence of dsDNA enhances the couple C5/A5 and lowers the peak Aox while the Cox signal is accompanied by cathodic peaks attributable to the reduction of chartreusin (1) bound to dsDNA (C1D, C2D), as judged upon comparing peak potential values with those for chartreusin and chartreusin plus dsDNA deoxygenated solutions.
These results can be rationalized on considering pharmacological studies revealing that 1 and its derivatives (1–9) exert their antitumor activities through binding to dsDNA, promoting strand scission via ROS generation in the presence of a suitable reducing agent, and inhibition of topoisomerase II.28,31 Interestingly, in the absence of dsDNA, the aforementioned Ch˙− deactivating pathway operates partially blocking the ROS generation chain yielding HO2˙ and HO˙, ultimately responsible of dsDNA strand scission.28 Notice that the electrochemical reduction of Ch in our experiments plays the role to the synergistic reducing agent for the chartreusin (1) damage of dsDNA.26,35 The presence of dsDNA, however, inhibits the deactivation of Ch˙− thus facilitating the advance of the ROS generation chain. A simplified scheme of these competing pathways is depicted in Fig. 8.
Fig. 10 compares the SECM color maps of dsDNA fiber on Pt substrate electrode in contact with air-saturated 0.10 M Bu4NPF6/DMSO on the SECM cell in the absence (a, b) and in the presence (c, d) of 1. The experimental arrangement differs from that used in Fig. 5 in which here the dsDNA fibbers were fixed only by one of their edges to the substrate electrode. Then, when ES = 0.00 V and the tip is held at a potential where the reduction of dissolved oxygen (Cox process) occurs (ET = −0.80 V), the fibbers appear as a negative feedback features contrasting with the positive feedback due to the O2 reduction at the uncovered regions of the Pt base electrode (see Fig. 10a). When 1 is added, the negative feedback features appears much more concentrated in several regions of the fibbers (Fig. 10b). As far as the tip current reflects the superposition of the reductions of O2 and 1, the above feature can be attributed to the non-homogeneous accumulation of chartreusin (1) in the dsDNA fibers.
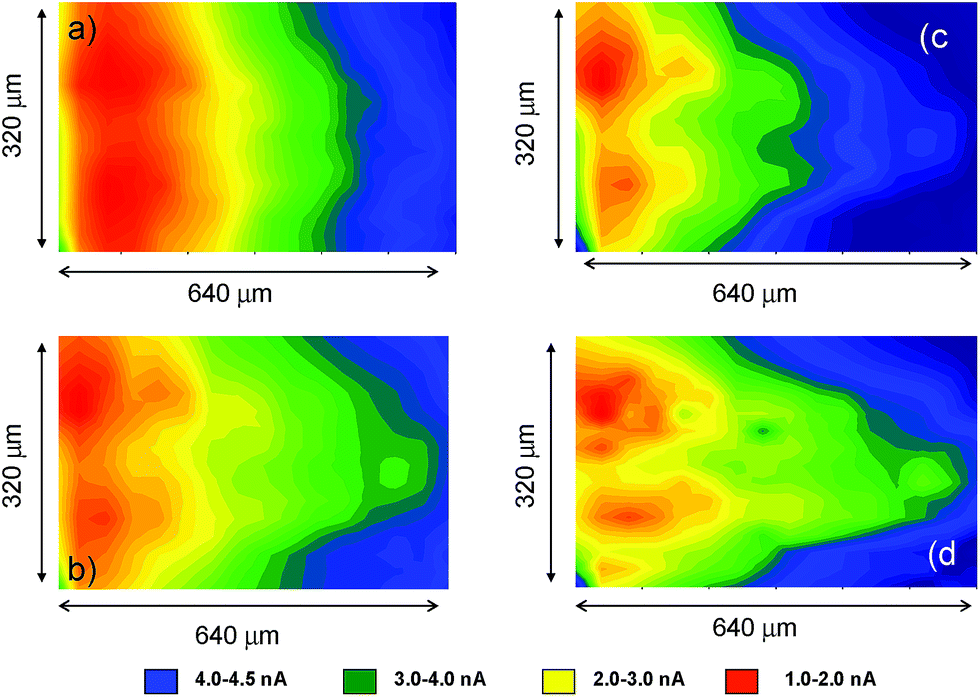 | ||
| Fig. 10 SECM color maps for a deposit of dsDNA fibers attached to Pt substrate electrode immersed into air-saturated: (a, b) 0.10 M Bu4NPF6/DMSO solution: (c, d) id. plus 0.03 mM chartreusin. (a, c) ES = 0.00 V; ET = −0.70 V; (b, d) ES = −1.00 V; ET = −0.70 V. | ||
When a potential negative enough to oxidize O2 and 1 is applied to the substrate whereas the tip is held at a potential near that of the signal Aox, the tip current should reflect the current for the reoxidation of the superoxide radical anion electrochemically generated in the base Pt electrode. The result in the absence of 1 is that positive feedback region becomes sharply contrasted (Fig. 10c). In the presence of chartreusin (1) (Fig. 10d), the map color approaches to some extent that in Fig. 10c: these features would be in agreement with the aforementioned reaction scheme: in the regions where 1 was not bound to dsDNA, the previously proposed deactivating pathway and hence the tip current become lowered.
Voltammetric data at chartreusin-modified electrodes in air-saturated aqueous solutions were also consistent with the proposed reaction scheme. Fig. 11 compares cyclic voltammograms of 1-modified glassy carbon electrodes immersed into air-saturated aqueous potassium phosphate buffer at pH 7.4 in the absence and in the presence of dsDNA. The mechanism of O2 reduction on carbon electrodes can be described in terms of a cascade of processes72 initiated by a one-electron reduction followed by protonation (eqn (1) and (3)), competing with disproportionation:
| 2O2˙− (ads) + H2O → HO2− + O2 + OH− | (7) |
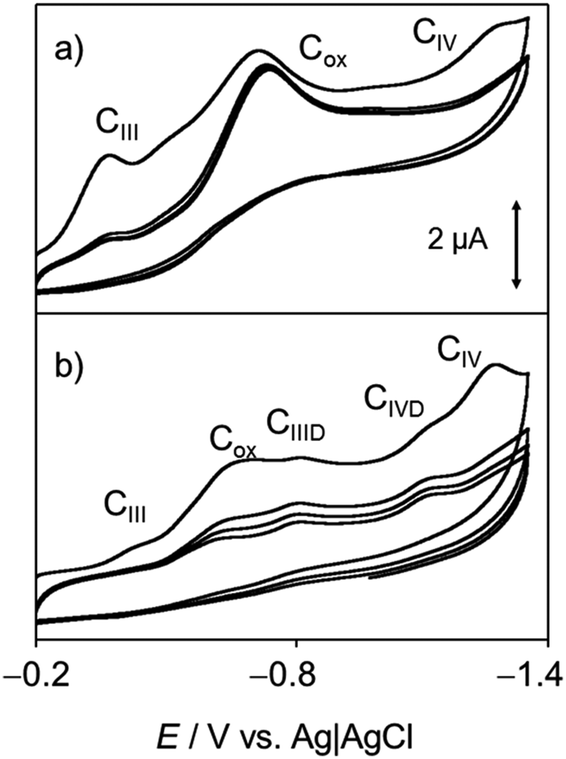 | ||
| Fig. 11 Cyclic voltammograms of chartreusin-modified glassy carbon electrodes immersed into air-saturated 0.25 M aqueous potassium phosphate buffer at pH 7.4 (a) in the absence and (b) in the presence of 0.5 mM dsDNA. Potential scan rate 50 mV s−1. | ||
Accompanied by a subsequent electrochemical reduction:
| HO2˙ (ads) + e− → HO2− | (8) |
The process described by eqn (3) being the rate-determining step. As a result, the reduction of O2 looks like an apparently irreversible voltammetric wave at ca. −0.5 V. At chartreusin-modified electrodes (Fig. 11a), the 1 reduction signals CIII, CIV (see Fig. 4) appear superimposed to the O2-reduction (Cox) with no apparent modification of the voltammetric profiles obtained for O2 and 1 independently. In the presence of dsDNA, however (Fig. 11b), the peak Cox becomes lowered while the CIII, CIV peaks are accompanied by additional signals attributable to the reduction of dsDNA-bound chartreusin, CIIID, CIVD. Such features suggest that, consistently with the foregoing set of considerations, in hydrophilic environment the reaction scheme leading to the development of the ROS generation chain is promoted, possibly following the reaction scheme proposed for the surface redox catalysis of O2 reduction on quinone-modified electrodes:73
| Ch (surf) + H+ + e− → HChrd (surf) | (9) |
| HChrd (surf) + O2 → Ch (surf) + HO2 | (10) |
Whose effectiveness requires that the rate of O2 interception was faster than the subsequent reduction of the reduced quinone form (here, HChrd) or the redox potentials of the first and second reductions of the parent quinone (processes CIII and CIV) were clearly separated, as is the case of chartreusin (see Fig. 4 and 11).
It is pertinent to note, however, that the observed voltammetric and SECM results correspond to ‘electrochemical conditions’, where superoxide radical anion and chartreusin reduced/oxidized forms can be generated upon applying the adequate potential inputs. Such conditions, obviously, differ from those existing in biological media. In spite of this limitation, solid state confinement permits to minimize the effect of competing chemical and electrochemical reactions occurring in conventional solution electrochemistry so that the reported data offer information on the reactivity of dsDNA under severe structural and conformational constraints to some extent similar to those often involved in biological processes. The application of the described electrochemical methodology would be potentially interesting for complementing existing assays for studying dsDNA damage.
Conclusions
Electrochemistry of chartreusin was studied and the data confirmed by theoretical calculations. Voltammetric experiments in both DMSO solutions as well as at chartreusin-modified film electrodes immersed into aqueous biomimetic buffer revealed that there is a significant interaction between chartreusin and dsDNA. SECM examination of dsDNA fibers denoted the existence of local concentrations of chartreusin. SECM imaging upon in situ electrochemical generation of reactive oxygen species (ROS) were in agreement with the idea that chartreusin-induced dsDNA damage can operate via intercalation and ROS generation mechanisms. Electrochemical generation of reactive oxygen species in DMSO solution revealed that, under electrochemical conditions, the one-electron reduced form of chartreusin can react with superoxide radical anion resulting in deactivation of the ROS generation chain but, in the presence of dsDNA, radical scavenging is blocked allowing the progression of the ROS generation chain. Reported data can be considered as indicative of the capabilities of electrochemical techniques for studying the processes involved in DNA damage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support is gratefully acknowledged from the MEC Project CTQ2014-53736-C3-2-P which is also supported with ERDF funds.Notes and references
- B. C. Baguley and E. S. Marshall, Eur. J. Cancer, 2004, 40, 794 CrossRef CAS PubMed.
- J. Portugal, BMC Pharmacol., 2009, 9, 11 CrossRef PubMed.
- J. Hrbac and R. Kohen, Drug Dev. Res., 2000, 50, 516 CrossRef CAS.
- (a) M. Albrecht, O. Schneider and A. Schmidt, Org. Biomol. Chem., 2009, 7, 1445 RSC; (b) M. F. Jacobsen, E. E. Ferapontova and K. V. Gothelf, Org. Biomol. Chem., 2009, 7, 905 RSC; (c) B. Chailap and T. Tuntulani, Org. Biomol. Chem., 2012, 10, 3617 RSC; (d) D. A. Lanfranchini, D. Belorgev, T. Müller, H. Vezin, M. Lanzer and E. Davioud-Charvet, Org. Biomol. Chem., 2012, 10, 4795 RSC.
- E. C. Long and J. K. Barton, Acc. Chem. Res., 1990, 23, 271 CrossRef CAS.
- M. J. Hannon, Chem. Soc. Rev., 2007, 36, 280 RSC.
- H. K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 44, 349 CrossRef CAS PubMed.
- M. Valko, M. Izakovic, M. Mazur, C. J. Rhodes and J. Telser, Mol. Cell. Biochem., 2004, 266, 37 CrossRef CAS PubMed.
- F. Scholz, U. Schröder, R. Gulabowski and A. Doménech-Carbó, Electrochemistry of Immobilized Particles and Droplets, Springer, Berlin-Heidelberg, 2nd edn, 2014 Search PubMed.
- A. Doménech-Carbó, J. Labuda and F. Scholz, IUPAC Technical Report, Pure Appl. Chem., 2013, 85, 609 Search PubMed.
- A. Doménech-Carbó, A. Maciuk, B. Figadère, E. Poupon and G. Cebrián-Torrejón, Anal. Chem., 2013, 85, 4014 CrossRef PubMed.
- (a) M. Martini, M. T. Albelda, M. Inclán, F. M. Valle-Algarra, E. García-España and A. Doménech-Carbó, J. Electroanal. Chem., 2014, 720 Search PubMed; (b) A. Doménech-Carbó, G. Cebrián-Torrejón, L. de Miguel, V. Tordera, D. Rodrigues-Furtado, S. Assad-Kahn, A. Fournet, B. Figadère, R. P. Váquez-Manrique and E. Poupon, Electrochim. Acta, 2014, 115, 546 CrossRef; (c) G. Cebrián-Torrejón, A. Doménech-Carbó, M. T. Scotti, A. Fournet, B. Figadère and E. Poupon, J. Mol. Struct., 2015, 1102, 242 CrossRef.
- B. Marselli, J. Garcia-Gomez, P.-A. Michaud, M. A. Rodrigo and C. Comninellis, J. Electrochem. Soc., 2003, 150, D79 CrossRef CAS.
- A. Kapałka, G. Foti and C. Comninellis, Electrochim. Acta, 2009, 54, 2018 CrossRef.
- F. Scholz, G. L. L. González, L. M. de Carvalho, M. Hilgemann, K. Z. Brainina, H. Kahlert, R. S. Jack and D. T. Minh, Angew. Chem., Int. Ed., 2007, 46, 8079 CrossRef CAS PubMed.
- L. M. de Carvalho, M. Hilgemann, F. Scholz, H. Kahlert, M. B. da Rosa, M. Wuster, U. Lindequist, P. C. do Nascimento and D. Bohrer, Electroanalysis, 2010, 22, 406 CrossRef.
- Š. Komorsky-Lovrić and I. Novak, Chem. Commun., 2009, 1467 Search PubMed.
- Š. Komorsky-Lovrić and I. Novak, J. Food Sci., 2011, 76, C919 CrossRef PubMed.
- A. Doménech-Carbó, R. Gavara, P. Hernández and I. Domínguez, Talanta, 2015, 114, 1207 CrossRef PubMed.
- (a) B. E. Leach, K. M. Calhoun, L. E. Johnson, C. M. Teeters and W. G. Jackson, J. Am. Chem. Soc., 1953, 75, 4011 CrossRef CAS; (b) E. Simonitsch, W. Eisenhuth, O. A. Stamm and H. Schmid, Helv. Chim. Acta, 1964, 47, 1459 CrossRef CAS; (c) J. P. McGovren, G. L. Neil, S. L. Crampton, M. I. Robinson and J. D. Douros, Cancer Res., 1977, 37, 1666 CAS.
- M. Uramoto, T. Kusano, T. Nishio, K. Isono, K. Shishido and T. Ando, FEBS Lett., 1983, 153, 325 CrossRef CAS PubMed.
- J. Portugal, Curr. Med. Chem., 2003, 3, 411 CAS.
- T. Tashiro, K. Kon, M. Yamamoto, N. Yamada, T. Tsuruo and S. Tsukagoshi, Cancer Chemother. Pharmacol., 1994, 34, 287 CrossRef CAS PubMed.
- N. Ueberschaar, Z. Xu, K. Scherlach, M. Metsä-Ketelä, T. Bretschneider, H. M. Dahse, H. Goerls and C. Hertweck, J. Am. Chem. Soc., 2013, 135, 17408 CrossRef CAS PubMed.
- Z. Xu, K. Jakobi, K. Welzel and C. Hertweck, Chem. Biol., 2005, 12, 579 CrossRef CAS PubMed.
- P. Kirubakaran, R. Kothapalli, D. Singh Kh, S. Nagamani, S. Arjunan and K. Muthusamy, Bioinformation, 2011, 6, 100 CrossRef PubMed.
- K. Knop, S. Stumpf and U. S. Schubert, Rapid Commun. Mass Spectrom., 2013, 27, 2201 CrossRef CAS PubMed.
- N. Ueberschaar, H.-M. Dahse, T. Bretschneider and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 6185 CrossRef CAS PubMed.
- X. Salas and J. Portugal, FEBS Lett., 1991, 292, 223 CrossRef CAS PubMed.
- A. Lorico and B. H. Long, Eur. J. Cancer, 1993, 29A, 1985 CrossRef CAS PubMed.
- F. Barceló, D. Capó and J. Portugal, Nucleic Acids Res., 2002, 30, 4567 CrossRef.
- D. M. Hernández, M. A. B. F. de Moura, D. P. Valencia, F. J. González, I. González, F. C. de Abreu, E. N. da Silva Junior, V. F. Ferreira, A. Ventura Pinto, M. P. F. Goulart and C. Frontana, Org. Biomol. Chem., 2008, 6, 3414 Search PubMed.
- R. Bernini, M. Barontini, P. Mosesso, G. Pepe, S. M. Willför, R. E. Sjöholm, P. C. Eklund and R. Saladino, Org. Biomol. Chem., 2009, 7, 2367 CAS.
- W. C. Krueger, L. M. Pschigoda and A. Moscowitz, J. Antibiot., 1986, 39, 1298 CrossRef CAS PubMed.
- M. Yagi, T. Nishimura, H. Suzuki and N. Tanaka, Biochem. Biophys. Res. Commun., 1981, 98, 642 CrossRef CAS PubMed.
- M. Uesugi, T. Sekida, S. Matsuki and Y. Sugiura, Biochemistry, 1991, 30, 6711 CrossRef CAS PubMed.
- A. Párraga, M. Orozco and J. Portugal, Eur. J. Biochem., 1992, 208, 227 CrossRef.
- Electrochemical Microscopy, ed. A. J. Bard and M. V. Mirkin, Taylor & Francis, Boca Raton, 2001 Search PubMed.
- L. Stoica, S. Neugebauer and W. Schuhmann, Adv. Biochem. Eng./Biotechnol., 2008, 109, 455 CrossRef CAS PubMed.
- J. Wang and F. Zho, J. Electroanal. Chem., 2002, 537, 95 CrossRef CAS.
- M. Komatsu, K. Yamashita, K. Uchida, H. Kondo, S. Takenaka, M. Komatsu, K. Yamashita, K. Uchida, H. Kondo and S. Takenaka, Electrochim. Acta, 2006, 51, 2023 CrossRef CAS.
- L. H. Lie, M. V. Mirkin, S. Hakkarainen, A. Houlten and B. R. Horrocks, J. Electroanal. Chem., 2007, 603, 67 CrossRef CAS.
- I. Palchetti, S. Laschi, G. Marrazza and M. Mascini, Anal. Chem., 2007, 79, 7206 CrossRef CAS PubMed.
- L. Guadagnini, A. Maljusch, X. Chen, S. Neugebauer, D. Tonelli and W. Schumann, Electrochim. Acta, 2009, 54, 3753 CrossRef CAS.
- D. A. Walsh, L. E. Li, M. S. Bakare and K. T. Voisey, Electrochim. Acta, 2009, 54, 4647 CrossRef CAS.
- G. Gritzner, Pure Appl. Chem., 1990, 62, 1839 CrossRef CAS.
- A. Doménech-Carbó, J. Phys. Chem. C, 2012, 116, 25977 Search PubMed.
- G. Imre, G. Veressc, A. Volfordd and Ö. Farkas, J. Mol. Struct.: THEOCHEM, 2003, 666, 51 CrossRef.
- Spartan model homepage for windows, http://www.wavefun.com/products/windows/SpartanModel/win_model.html, access in April 2016.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490 CrossRef CAS.
- M. J. S. E. Dewar, G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902 CrossRef CAS.
- N. Metropolis and S. Ulam, J. Am. Stat. Assoc., 1949, 44, 335 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- L. Vereecken, K. Pierloot and J. Peeters, J. Chem. Phys., 1998, 108, 1068 CrossRef CAS.
- J. Q. Chambers, in The Chemistry of Quinonoid Compounds, ed. S. Patai and Z. Rapport, John Wiley & Sons Ltd, N. York, 1988, pp. 719–757 Search PubMed.
- C. A. Wraight, Front. Biosci., 2004, 9, 309 CrossRef CAS.
- C. de la Fuente, J. A. Acuna, M. D. Vazquez, M. L. Tascon and P. S. Batanero, Talanta, 1999, 49, 441 CrossRef CAS PubMed.
- M. Lovric and F. Scholz, J. Solid State Electrochem., 1997, 1, 108–113 CrossRef CAS.
- M. Lovric and F. Scholz, J. Solid State Electrochem., 1999, 3, 172 CrossRef CAS.
- U. Schröder, K. B. Oldham, J. C. Myland, P. J. Mahon and F. Scholz, J. Solid State Electrochem., 2000, 4, 314 CrossRef.
- A. Doménech-Carbó and M. T. Doménech-Carbó, J. Solid State Electrochem., 2006, 10, 949 CrossRef.
- D. T. Sawyer, G. Chiericato, C. T. Angelis, E. J. Nanni and T. Tsuchiya, Anal. Chem., 1982, 54, 1720 CrossRef CAS.
- M. E. Ortiz, L. J. Núñez-Vergara and J. A. Squella, J. Electroanal. Chem., 2003, 549, 157 CrossRef CAS.
- V. Krishnamachari, L. H. Levine, P. W. Paré and J. Agric, Food Chem., 2002, 50, 4357 CrossRef CAS.
- K. Kano, T. Mabuchi, B. Uno, Y. Esaka, T. Tanaka and M. Iinuma, J. Chem. Soc., Chem. Commun., 1994, 5, 593 RSC.
- I. G. Zenkevich, A. Y. Eshchenko, S. V. Makarova, A. G. Vitenberg, Y. G. Dobryakov and V. A. Utsal, Molecules, 2007, 12, 654 CrossRef CAS PubMed.
- Q. Y. Wei, B. Zhou, Y. J. Cai, L. Yang and Z. L. Liu, Food Chem., 2006, 96, 90 CrossRef CAS.
- T. Yano, D. A. Tryk, K. Hashimoto and A. Fujishima, J. Electrochem. Soc., 1998, 145, 1870 CrossRef CAS.
- B. Marselli, J. Garcia-Gomez, P. A. Michaud, M. A. Rodrigo and C. Comninellis, J. Electrochem. Soc., 2003, 150, D79 CrossRef CAS.
- A. Kapałka, G. Foti and C. Comninellis, Electrochim. Acta, 2009, 54, 2018 CrossRef.
- P. Cofré and D. T. Sawyer, Anal. Chem., 1986, 58, 1057 CrossRef.
- (a) J. Xu, W. Huang and R. L. McCreery, J. Electroanal. Chem., 1996, 410, 235 CrossRef; (b) F. Kuang, D. Zhang, Y. Li, Y. Wan and B. Hou, J. Solid State Electrochem., 2009, 13, 385 CrossRef CAS.
- (a) K. Tammeveski, K. Kontturi, R. J. Nichols, R. J. Potter and D. J. Schiffrin, J. Electroanal. Chem., 2001, 515, 101 CrossRef CAS; (b) A. Sarapu, K. Vaik, D. J. Schiffrin and K. Tammeveski, J. Electroanal. Chem., 2003, 541, 23 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08238j |
| This journal is © The Royal Society of Chemistry 2017 |