DOI:
10.1039/C7RA08683K
(Paper)
RSC Adv., 2017,
7, 50875-50879
Isolation, structure elucidation, and KDD study of (−)-Celosine, a new skeleton with potent anti-atherosclerosis activity†
Received
6th August 2017
, Accepted 25th October 2017
First published on 1st November 2017
Abstract
Natural (−)-Celosine was isolated as a novel endocyclicditerpene with an unprecedented skeleton from Celosia cristata L. Its structure was established by comprehensive 1D and 2D NMR spectroscopic analysis in combination with single-crystal X-ray crystallographic diffraction of synthesized (+)-Celosine. (−)-Celosine was expected to have an anti-atherosclerotic activity in vivo, and myeloperoxidase (MPO) expression may be the mechanism underlying this anti-atherosclerotic activity.
Introduction
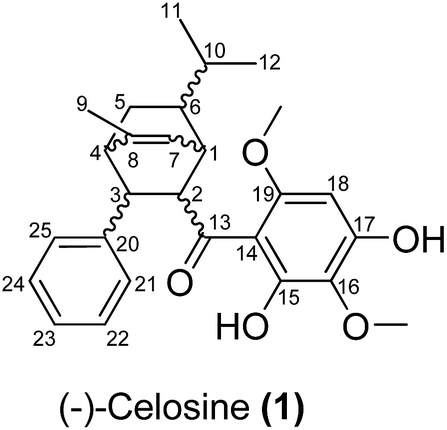
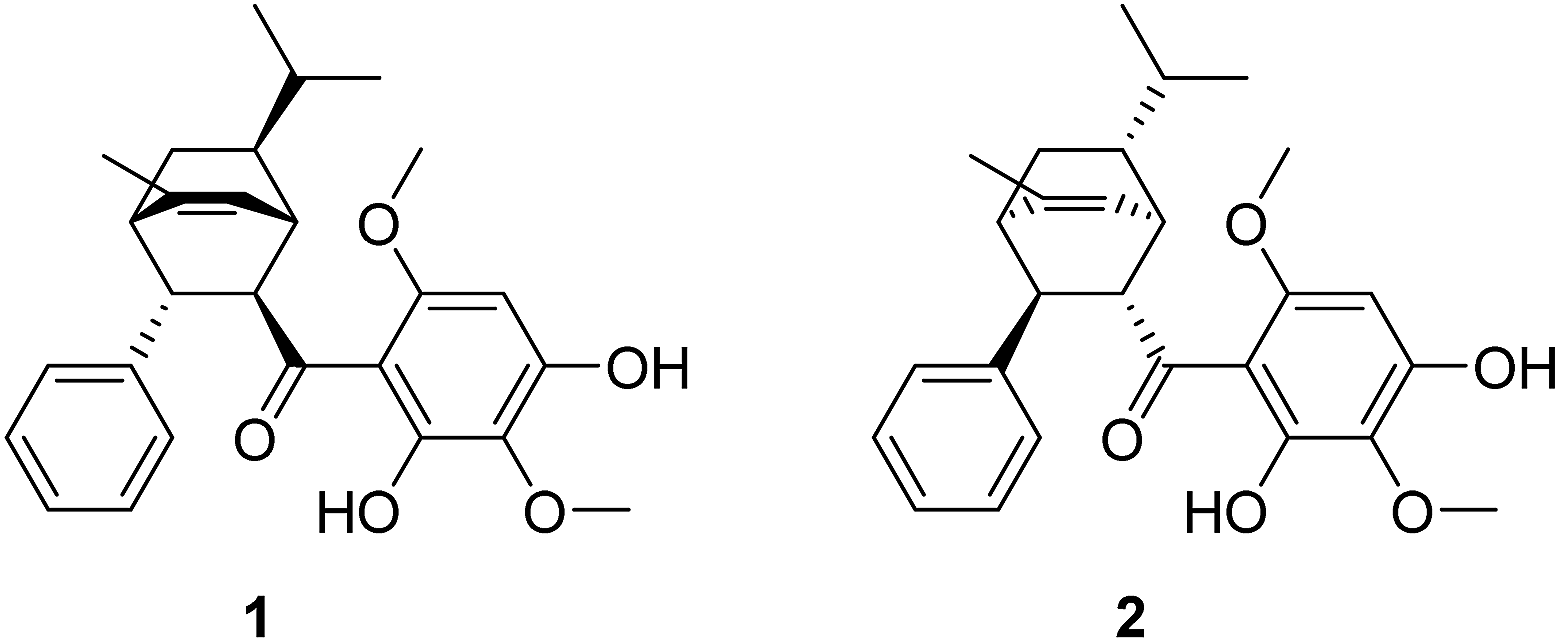
Celosia cristata L., known as Jiguanhua etc. in Chinese, has been long used in traditional Chinese medicine (TCM) for the treatment of hematischesis, infection, and hypertension.1–4 Previous studies5–7 on the whole dry plant of Celosia cristata L. have led to the isolation of many structurally novel compounds. The diversity of these novel compounds from Celosia cristata L. encouraged chemists to dig deep for new compounds, especially with a novel skeleton. In the present study, we selected Celosia cristata L. as the subject of our research and made a further investigation on their chemical constituents and pharmacological activities. By using various separation methods and chromatographic techniques, a chalcone–monoterpene complex was isolated, which represented a novel carbon skeleton of a natural endocyclic compound possessing 5 chiral centers (Fig. 1). This novel endocyclic skeleton was first obtained from natural source C. cristata of Celosia and named as “Celosine”.
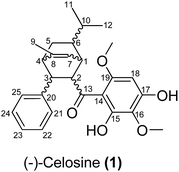 |
| | Fig. 1 2D structure of (−)-Celosine (1). | |
Results and discussion
Celosine was obtained as a white amorphous powder. The HR-ESI-MS showed a quasi-molecular ion peak at m/z 437.2314 [M + H]+ (calcd for C27H33O5, 437.2328), suggesting a molecular formula C27H32O5 with 12 double-bond equivalents (DBEs). The IR spectrum displayed absorptions for hydroxyls (3394 cm−1) and double bonds (1622 and 1589 cm−1). The 1H NMR spectrum (Table 1) showed typical signals of one hydrogen-bonded hydroxyl group at δH 14.22 (s), six aromatic protons, one olefinic proton at δH 5.49 (1H, br. d, J = 6.4 Hz), two methoxyl groups (δH 3.90, 3.87, each 3H, s) and three methyl groups at δH 1.93 (3H, d, J = 1.7 Hz), 0.90 (3H, d, J = 6.6 Hz) and 0.84 (3H, d, J = 6.5 Hz). The five typical signals at δH 7.29 (2H, m), 7.26 (2H, m), 7.18 (1H, br. t, J = 7.2 Hz) indicated the presence of mono-substituted benzene while the remaining single aromatic proton signal at δH 6.03 (1H, s) indicated a penta-substituted benzene. The 13C NMR and DEPT spectra data (Table 1), with the aid of HSQC experiments, further revealed the presence of one mono-substituted benzene, one penta-substituted benzene, one double bond, one carbonyl, three methyl groups, two methoxyl groups, one sp3 methylene and six sp3 methines. The proton resonance at δH 14.22 showed no correlation with any carbons in the HSQC spectrum and were assigned to the hydroxyl. The above-mentioned functionalities accounted for 10 DBEs, and the remaining 2 DBEs require the existence of 2 additional rings in the molecule.
Table 1 The 1H, 13C NMR data of (−)-Celosine (1) (CDCl3; δ, ppm; J, Hz)
| No. |
δH (J in Hz) |
δC |
DEPT |
| 1 |
2.97 d (6.4) |
39.0 |
CH |
| 2 |
4.09 dd (6.8, 1.5) |
56.2 |
CH |
| 3 |
3.61 br. d (7.1) |
42.6 |
CH |
| 4 |
2.45 t (2.3) |
43.4 |
CH |
| 5α |
1.92 (overlapped) |
25.8 |
CH2 |
| 5β |
0.78–0.82 (overlapped) |
| 6 |
1.52 tdd (9.5, 4.7, 2.0) |
49.0 |
CH |
| 7 |
5.49 br. d (6.4) |
120.0 |
CH |
| 8 |
— |
145.8 |
C |
| 9 |
1.93 d (1.7) |
19.9 |
CH3 |
| 10 |
1.14 dp (9.4, 6.4) |
33.4 |
CH |
| 11 |
0.90 d (6) |
21.3 |
CH3 |
| 12 |
0.84 d (6.5) |
20.8 |
CH3 |
| 13 |
— |
206.2 |
C |
| 14 |
— |
105.6 |
C |
| 15 |
— |
159.1 |
C |
| 16 |
— |
128.6 |
C |
| 17 |
— |
154.6 |
C |
| 18 |
6.03 s |
89.3 |
CH |
| 19 |
— |
158.5 |
C |
| 20 |
— |
143.8 |
C |
| 21 |
7.26 (overlapped) |
128.1 |
CH2 |
| 22 |
7.29 (overlapped) |
128.3 |
CH2 |
| 23 |
7.18 br. t (7.2) |
125.9 |
CH2 |
| 24 |
7.29 (overlapped) |
128.3 |
CH2 |
| 25 |
7.26 (overlapped) |
128.1 |
CH2 |
| 15-OH |
14.22 s |
— |
— |
| 16-OCH3 |
3.87 s |
16.5 |
CH3 |
| 19-OCH3 |
3.90 s |
17.5 |
CH3 |
The planar structure of Celosine was established by interpretation of 2D NMR spectra (1H–1H COSY, HSQC and HMBC). The 1H–1H COSY spectrum (Fig. 2) displayed four coupled spin systems of H-1 (δH 2.97)/H-2 (δH 4.09)/H-3 (δH 3.61)/H-4 (δH 2.45)/H-5 (δH 1.92, 0.80)/H-6 (δH 1.52)/H-1 (δH 2.97); H-1 (δH 2.97)/H-7 (δH 5.49); H-6 (δH 1.52)/H-10 (δH 1.14)/H-11 (δH 0.90) (/H-12 (δH 0.84)) and H-21 or 25 (δH 7.26)/H-22 or 24 (δH 7.29)/H-23 (δH 7.18). The coupled spin systems from H-1 to H-6 indicated the existence of a ring with six carbons.
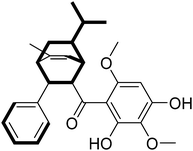 |
| | Fig. 2 Key 1H–1H COSY correlations (bold bonds) of (−)-Celosine. | |
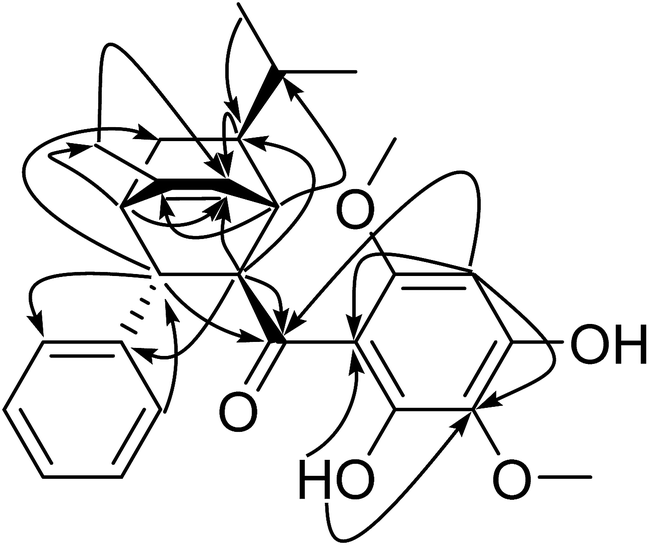
The downfield chemical shift of hydroxyl (δH 14.22) suggested its double-bonding position (thus adjacent) to the carbonyl group. The multiple HMBC correlations (Fig. 3) of 15-OH/C-14 and C-16; H-16/C-16; H-18/C-13, C-14, C-16, C-17 and C-19; 19-OCH3/C-19 established the 2,4-dihydroxy-3,6-dimethoxybenzoyl group. The HMBC correlations of H-2/C-20, H-3/C-21, H-21/C-3 suggested the location of mono-substituted benzyl moiety at C-3. The HMBC correlations of H-11/C-6, H-11/C-12, H-1/C-10, H-2/C-6 together with 1H–1H COSY correlations of H-6/H-10/H-11(H-12) revealed the presence of the isopropyl at C-6. The HMBC correlations of H-18/C-13, H-1/C-13, H-2/C-13 suggested the location of carbonyl group at C-2 and C-14. After counting the above six membered ring as one DBEs, the last DBE in the molecule was solved by revealing the double bond bridge between C-1 and C-4 forming a second ring with the HMBC correlations of H-1/C-8, H-2/C-7, H-4/C-7 and 1H–1H COSY correlation of H-1/H-7. The HMBC correlations of H-4/C-9 and H-9/C-7 indicated a methyl locating at C-8 position. The planar structure of Celosine was thus established.
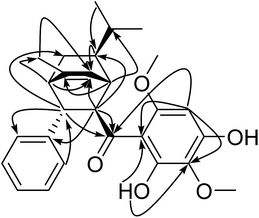 |
| | Fig. 3 Key HMBC correlations (arrows) of (−)-Celosine. | |
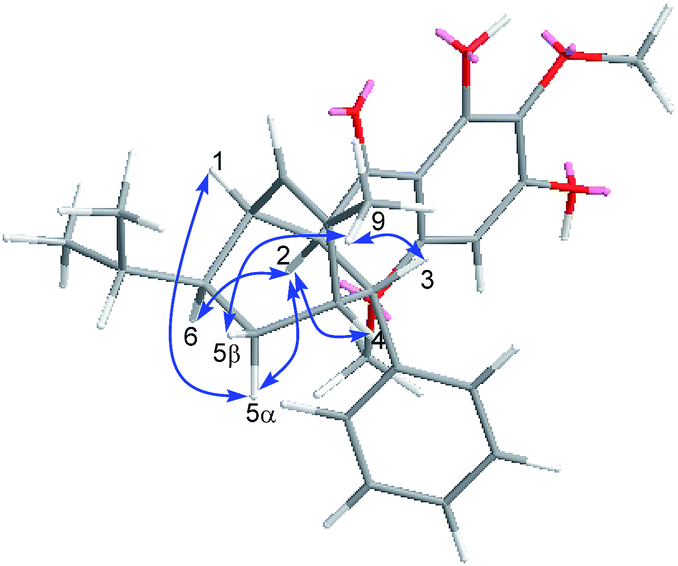
The relative configuration of Celosine was assigned by ROESY (Fig. 4) spectrum. The correlations of H-5α with H-1, H-2 and H-2 with H-4, H-6 revealed that H-1, H-2, H-4, H-5α and H-6 were cofacial and randomly assigned to be α-oriented. The correlations of 9-CH3 with H-5β and H-3 revealed that H-5β, H-3 and 9-CH3 were cofacial and β-oriented. The two possible configurations were shown in Fig. 5.
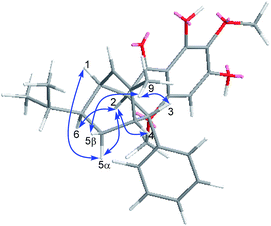 |
| | Fig. 4 Key ROESY correlations (blue arrows) of (−)-Celosine. | |
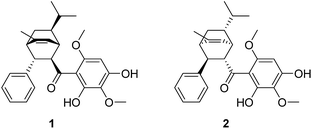 |
| | Fig. 5 The two possible configurations of (−)-Celosine. | |
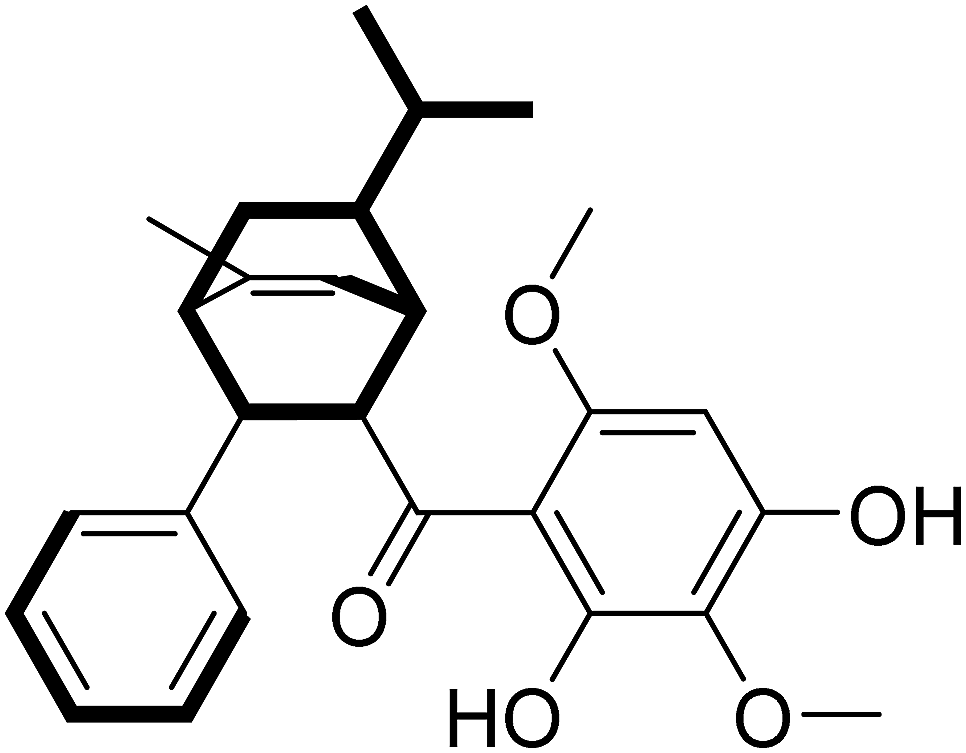
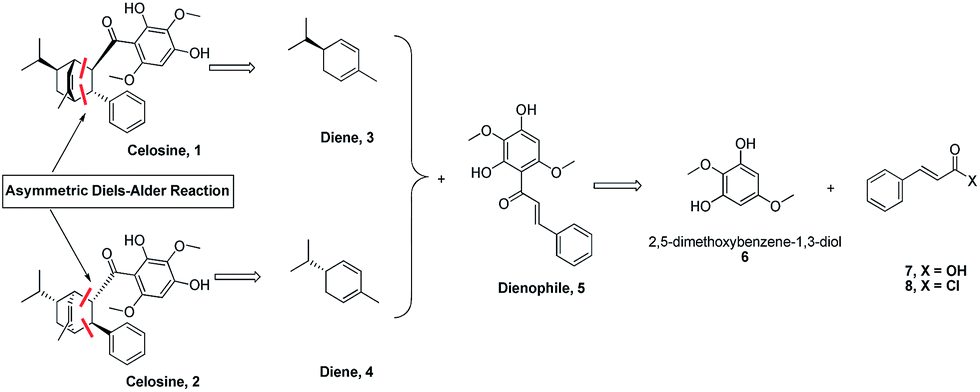
Mp 59.5–60.2 °C, IR (KBr) Vmax: 3394, 1621, 1589 and 1427 cm−1, [α]25D = −139.4 (c = 0.1, MeOH). The suitable single-crystal could not be obtained from the natural source of Celosine due to the less amount of the sample. Therefore, the total synthesis of Celosine was carried out to determine the absolute configuration of Celosine.
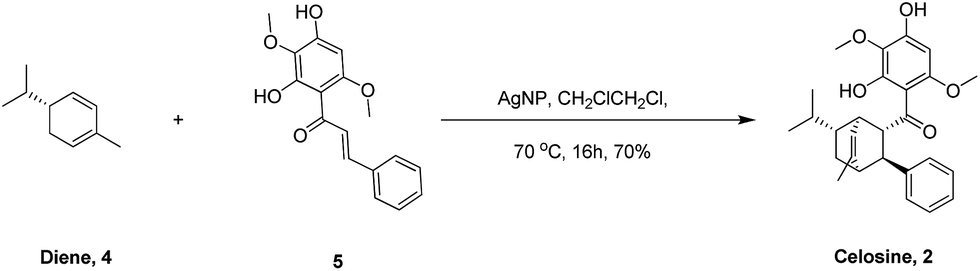
Based on the skeleton of Celosine, Diels–Alder cycloaddition8–15 would be the best choice to complete the total synthesis (Scheme 1). Diene compound 3 (or compound 4) and dienophile compound 5,16 which was also isolated from Celosia cristata L. and other CTMs,17,18 are the two key intermediates to construct Celosine 1 (or Celosine 2) through asymmetric endo Diels–Alder cycloaddition. α-Phellandrene, named after Eucalyptus phellandra, is also a natural product,18 and the only commercial available isomer (−)-α-phellandrene (diene compound 4) was used for the synthesis of Celosine 2. Compound 5 could be generated through Friedel Crafts acylation reaction from 2,5-dimethoxybenzene-1,3-diol (compound 6) and cinnamoyl acid or chloride (compound 7 or 8).
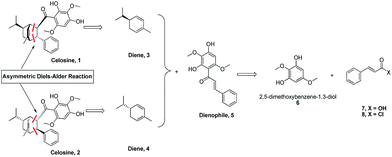 |
| | Scheme 1 Retro synthesis analysis of Celosine. | |
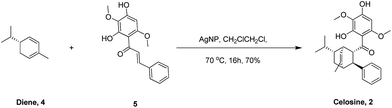
After preparation of the key intermediate 5 (ESI S7†), attention was turned to the asymmetric Diels–Alder cycloaddition. Huan and his coworkers19 published a robust Diels–Alder cycloaddition method via the catalysis of silver nanoparticles supported by silica. As listed in Table 2, the reaction parameters shown in entry 1 and entry 2 are totally the same with Huan's, but the reaction results could not be accepted even though some product could be detected. From Huan's another paper,20 we learned that the cycloaddition reaction was affected severely by the substituent groups on dienes and dienophiles. But we found that the multi-substituent groups on (−)-α-phellandrene (diene, compound 4) and chalcone (compound 5) made some obstacles for cycloaddition, and therefore, dichloroethane (DCE) was chosen for a higher reaction temperature. As shown in entry 4 and 5, the yields increased to 75% at 70 °C for 16 h after the change of solvent. What interests us most is that the cycloaddition could occur without participate of any catalyst as shown in entry 6 and 7 and only one regioisomer was detected, suggesting that the configuration of compound 2 was the most stable one based on thermodynamic theory.
Table 2 Optimization of asymmetric Diels–Alder cycloaddition condition
|
| Entry |
Catalyst |
Solvent |
Temp (°C) |
Time (h) |
Yield (%) |
Endo/exo |
| Silver nanoparticles. TLC semi quantitive. Not detected. Determined by chiral HPLC. No catalyst. |
| 1 |
AgNPa |
DCM |
25 |
16 |
10b |
NDc |
| 2 |
AgNP |
DCM |
40 |
16 |
25–30 |
ND |
| 3 |
AgNP |
DCE |
40 |
16 |
20–25 |
ND |
| 4 |
AgNP |
DCE |
50 |
16 |
50 |
Endo onlyd |
| 5 |
AgNP |
DCE |
70 |
16 |
75 |
Endo only |
| 6 |
—e |
DCE |
70 |
16 |
15 |
ND |
| 7 |
— |
DCE |
70 |
48 |
45 |
Endo only |
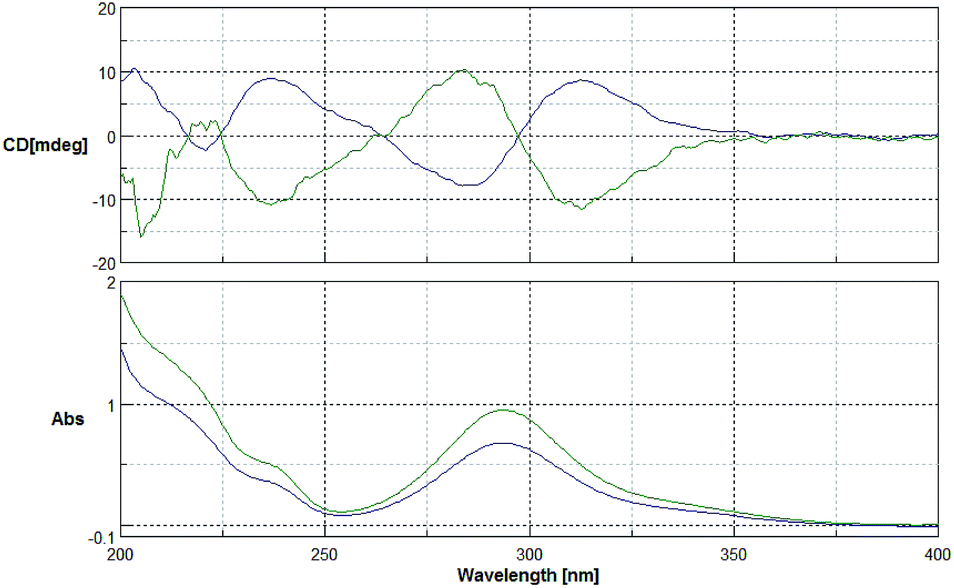
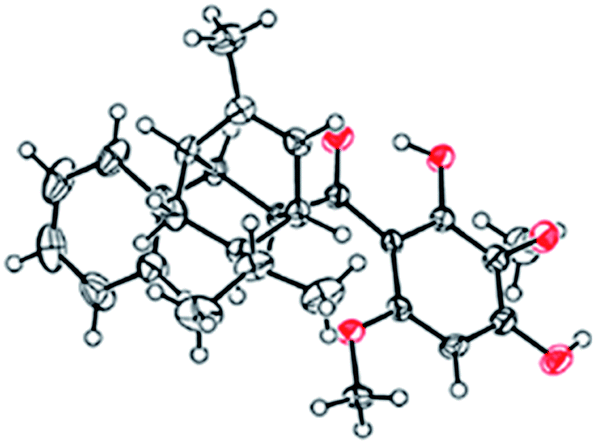
The 1H- and 13C-NMR spectra showed that the natural source Celosine and the synthesized Celosine possessed the same 2D structure (ESI S15†). However, the specific optical rotation ([α]25D = +142.6 (c = 0.1, MeOH)) of the synthesized Celosine was opposite to that of the natural one, demonstrating that the structure of Celosine 1 was the exact structure of the natural Celosine. To further search the correlation between the two Celosines, CD spectroscopy was collected as depicted in Fig. 6. Totally opposite signals had been recorded either, confirming that the two compounds were enantiomers. Fortunately, the crystal structure of Celosine 2 was obtained and lead to a successful single crystal X-ray diffraction with anomalous dispersion of Cu Kα radiation (Fig. 7), which confirmed the planar structure and unambiguously determined the absolute configuration of Celosine 2. As the mirror image of Celosine 2, the planar structure and absolute configuration of Celosine 1 was also confirmed by comparing their CD data and the specific optical rotation data. Thus the natural source Celosine and the synthetic Celosine were renamed as (−)-Celosine and (+)-Celosine respectively based on their opposite specific optical rotation.
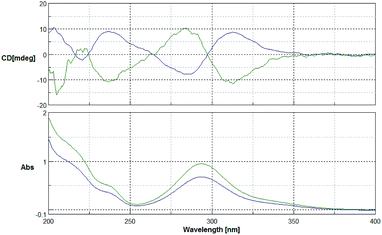 |
| | Fig. 6 CD spectrum of the two Celosines (green line for natural source Celosine, and the blue line for synthetic Celosine). | |
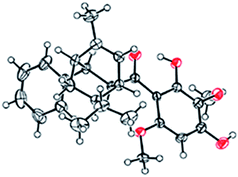 |
| | Fig. 7 The crystal structure of Celosine 2. | |
Knowledge discovery in database (KDD) is a new information processing techniques quickly emerging in recent years with the aim to discover useful information hidden in databases currently available by making use of various information processing tools.21–24 KDD was initially used in the field of TCM to predict the pharmacological target by searching DNP and MDDR3D databases. Myeloperoxidase (MPO), also known as peroxidase, is a heme protease with a heme prosthetic group. This leukocytic ferment mainly secreted by neutrophils plays an important role in the formation of atherosclerosis, and the increased expression and activity of MPO can promote the formation of atherosclerosis. As the classical target of anti-atherosclerosis, MPO was chosen as a predicted target for further study (ESI S17†).
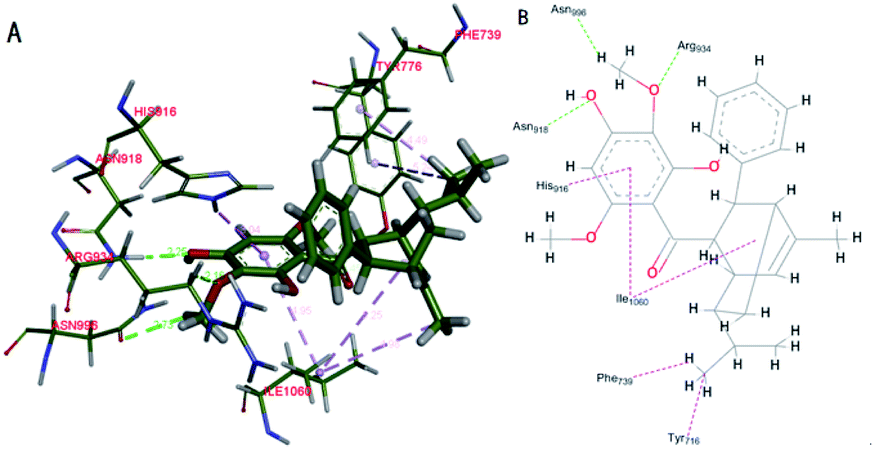
Based on the docking study, the (−)-Celosine molecule could form multiple interactions with the residues around the docking pocket of MPO. MD simulation proved that the result of complex docking is stable and reliable (ESI S17†). The interaction between the (−)-Celosine molecule and important residues including HIE916, ASN918, ARG934, ASN996, TYR776, PHE739 and ILE1060 indicated the orientation of further optimization (Fig. 8). It was predicted by the KDD and MPO docking study that (−)-Celosine should possess an anti-atherosclerosis activity. To validate the prediction, (−)-Celosine was used to treat atherosclerosis in rat model established by feeding of high-fat diet and clamping of the carotid artery. The expression of MPO, MMP-9 and Lp-PLA2 was detected by using atorvastatin as the positive control.
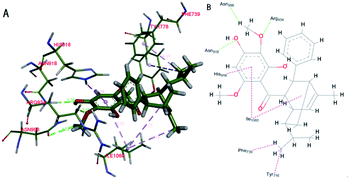 |
| | Fig. 8 Docking of (−)-Celosine into the binding site of MPO. Hydrogen bonds are shown as green dotted line. Hydrophobic bonds are shown as pink dotted line ((A) schematic perspective (B) plan sketch). | |
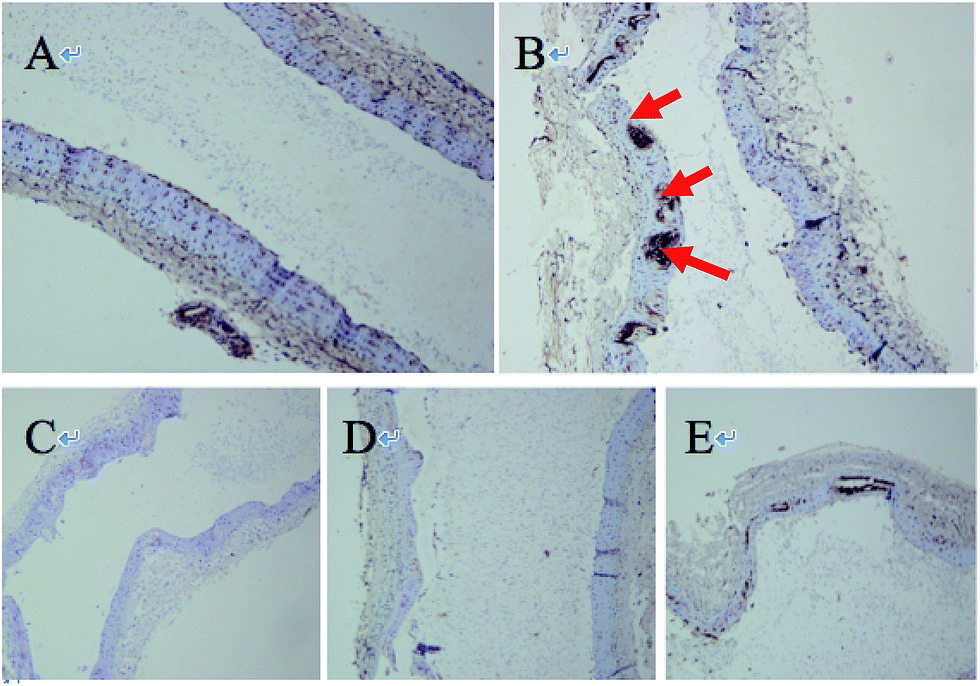
As listed in Table 3, the expression of MPO and MMP-9 was increased significantly in modeled group and high dose (−)-Celosine treatment improved the vascular function of the carotid artery significantly in a dose dependent manner. However, no significant effect was observed on the expression of Lp-PLA2. HE staining showed significant atherosclerotic changes in the modeled group, as represented by endothelial detachment, rupture, intimal thickening and inward bulge, and obvious plaque formation. The degree of intimal thickening and uplift was greatly improved after (−)-Celosine treatment (ESI S22†). Immunohistochemistry showed high MPO expression in large number of endothelial cells in modeled group. In contrast, the MPO expression was decreased significantly in (−)-Celosine treatment group in a dose dependent manner (Fig. 9).
Table 3 Expression of MPO, MMP-9 and Lp-PLA2 in different animal groups
| |
MPO |
MMP-9 |
Lp-PLA2 |
| p > 0.05. |
| Norm group |
11.19 ± 3.53 |
4.78 ± 0.36 |
19.98 ± 2.62 |
| Disease group |
19.69 ± 5.21a |
9.22 ± 0.90a |
22.17 ± 4.56 |
| Atorvastatin group |
16.62 ± 3.66 |
5.56 ± 0.33 |
21.76 ± 1.63 |
| (−)-Celosine group |
High dose |
15.07 ± 2.98 |
5.73 ± 0.66 |
21.55 ± 1.01 |
| Low dose |
15.36 ± 2.86 |
6.08 ± 0.28 |
21.91 ± 3.04 |
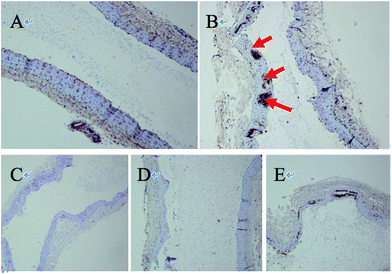 |
| | Fig. 9 Immunohistochemical picture of vascular protein MPO ((A) normal group, (B) disease group, (C) atorvastatin group, (D and E) (−)-Celosine high/low dose group). | |
Conclusions
In summary, (−)-Celosine with a new carbon skeleton, was isolated from Celosia cristata L., whose structure and relative configuration was confirmed by the spectroscopic and chemical methods. The total synthesis and the single crystal structure of the (+)-Celosine unfolded the true picture of the two compounds. Based on the KDD technique, (−)-Celosine was predicted as having an anti-atherosclerosis activity, and MPO, the classical target of anti-atherosclerosis, was chosen for docking and MD simulation study. MD simulation demonstrated that the result of complex docking was stable and reliable. The anti-atherosclerosis tests in vivo showed that the expression of MPO was decreased significantly after (−)-Celosine treatment, indicating the prediction was precise and rational. This finding has provided a new structure class for the treatment of atherosclerosis and related diseases.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by The seed fund program of Shanghai university of medicine & health Sciences (HSMF-17-22-031), Excellent Young Medical Expert of Shanghai (2017YQ048), China Postdoctoral Science Foundation (184279) and research project of Shanghai municipal health and Family Planning Commission (201540027), Shanghai Rising-Star Program (16QB1403800).
Notes and references
- J.-J. Zhang, L.-Y. Xu and H. Shi, Chin. J. Pharm., 2006, 15, 25–26 CAS.
- J.-F. Chen and Y. Yan, Chin. J. Patho. Bio., 2010, 5, 720–724 Search PubMed.
- D.-B. Weng, H.-F. Wang and J.-Y. Weng, Chin. Bull. Bot., 2000, 17, 565–568 Search PubMed.
- M. Begam, S. Narwal, S. Roy, S. Kumar, M.-L. Lodha and H.-C. Kapoor, Biochemistry, 2006, 71, S44–S48 CAS.
- J.-J. Wang, X.-M. Zhang and Z.-W. Huang, Northwest Pharm. J., 2008, 06, 354–356 Search PubMed.
- Y. Wang, Z.-Y. Lou, Q.-B. Wu and M.-L. Guo, Fitoterapia, 2010, 81, 1246–1252 CrossRef CAS PubMed.
- X. Pang, H. X. Yan, Z.-F. Wang, M.-X. Fan, Y. Zhao, X.-T. Fu, C.-Q. Xiong, J. Zhang, B.-P. Ma and H.-Z. Guo, J. Asian Nat. Prod. Res., 2014, 16, 240–247 CrossRef CAS PubMed.
- C.-F. Chang, C.-F. Li, C.-C. Tsai and T.-H. Chuang, Org. Lett., 2016, 18, 638–641 CrossRef CAS PubMed.
- G. Yang, Q.-F. Jia, L. Chen, Z.-Y. Du and J. Wang, Chem. inform., 2016, 47, 76759–76763 Search PubMed.
- J. Zhang, Y.-L. Xiao, K. Chen, W.-Q. Wu, H.-F. Jiang and S.-F. Zhu, Adv. Synth. Catal., 2016, 358, 2684–2691 CrossRef CAS.
- A.-C. Aragonès, N.-L. Haworth, N. Darwish, S. Ciampi, N.-J. Bloomfield, G.-G. Wallace and I. Diez-Perez, Nature, 2016, 531, 88–91 CrossRef PubMed.
- M.-J. Umerani, D.-J. Dibble, A.-G. Wardrip, A. Mazaheripour, E. Vargas, J.-W. Ziller and A.-A. Gorodetsky, J. Mater. Chem. C, 2016, 4, 4060–4066 RSC.
- J.-E. Sears and D.-L. Boger, Acc. Chem. Res., 2016, 49, 241–251 CrossRef CAS PubMed.
- S. Gupta, M.-I. Alam, T.-S. Khan, N. Sinha and M.-A. Haider, RSC Adv., 2016, 6, 60433–60445 RSC.
- R. Huang, X. Chang, J. Li and C.-J. Wang, J. Am. Chem. Soc., 2016, 138, 3998–4001 CrossRef CAS PubMed.
- M. Ahmed, M. Khaleduzzaman and M.-S. Islam, Phytochemistry, 1990, 29, 2009–2011 CrossRef CAS.
- Y. Hua, L. He and H.-Q. Wang, China J. Chin. Mater. Med., 2003, 28, 530–533 CAS.
- Z.-L. Liu and S.-S. Du, E-J. Chem., 2011, 8, 1937–1943 CrossRef CAS.
- H. Cong, C.-F. Becker, S.-J. Elliott, M.-W. Grinstaff and J. A. Porco, J. Am. Chem. Soc., 2010, 132, 7514–7518 CrossRef CAS PubMed.
- H. Cong, D. Ledbetter, G.-T. Rowe, J.-P. Caradonna and J.-A. Porco, J. Am. Chem. Soc., 2008, 130, 9214–9215 CrossRef CAS PubMed.
- J.-S. Fang, A.-L. Liu and G.-H. Du, Acta Pharmacol. Sin., 2014, 49, 1357–1364 CAS.
- M. Whittle, P. Willett, W. Klaffke and P. van Noort, J. Chem. Inf. Comput. Sci., 2003, 43, 449–457 CrossRef CAS PubMed.
- M. Xue, M.-Y. Zheng, B. Xiong, Y.-L. Li, H.-L. Jiang and J. K. Shen, J. Chem. Inf. Model., 2010, 50, 1378–1386 CrossRef CAS PubMed.
- E.-J. Gardiner and V.-J. Gillet, J. Chem. Inf. Model., 2015, 55, 1781–1803 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, product characterizations, and 1H and 13C NMR spectra. CCDC 1488037. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08683k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Bin Hud,
Jifa Zhanga,
Peng Dub,
Man Wanga,
Linlin Xiaoa and
Peiming Yang*c
*b,
Bin Hud,
Jifa Zhanga,
Peng Dub,
Man Wanga,
Linlin Xiaoa and
Peiming Yang*c