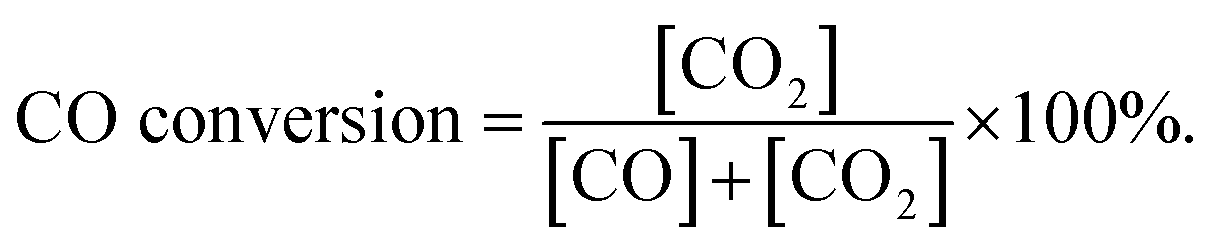DOI:
10.1039/C7RA08781K
(Paper)
RSC Adv., 2017,
7, 45420-45431
High efficiency and stability of Au–Cu/hydroxyapatite catalyst for the oxidation of carbon monoxide
Received
9th August 2017
, Accepted 18th September 2017
First published on 22nd September 2017
Abstract
A highly efficient and stable Au–Cu/hydroxyapatite (HAP, Ca10(PO4)6(OH)2) catalyst was reported. HAP was prepared through a deposition–precipitation method. Au–Cu/HAP catalysts were obtained by a two-step impregnation approach. The samples were characterized by XRD, TEM, SEM, UV-Vis, ICP, XPS, H2-TPR, and O2-TPD. CO oxidation reaction was carried out to evaluate the catalytic performance of samples. TEM and UV-Vis results showed that the metallic particles supported on Au–Cu/HAP were smaller than those on Au/HAP, and they were highly dispersed on the HAP support. Patterns of XPS revealed that CuO nanoparticles were formed in the Cu/HAP catalyst, while CuO and Cu2O species coexisted in Au–Cu/HAP catalyst. Based on the O2-TPD data, the addition of copper to Au/HAP gave rise to more new O2 adsorption sites and bigger O2 adsorption capacity. The catalysis results indicated that the Au–Cu/HAP was capable of obtaining higher efficiency and stability compared with Au/HAP and Cu/HAP for CO oxidation. It was likely that the synergistic interaction between gold and CuOx phase created the most active sites on Au–Cu/HAP and was responsible for the enhanced activity and stability in comparison with Au/HAP and Cu/HAP.
1. Introduction
In the late 1980s, Haruta et al.1 first found that supported gold catalysts in which gold particles were below 5 nm in size had surprisingly high activity for low temperature CO oxidation. Since then, gold catalysts have been applied a variety of important reactions, including CO oxidation, water–gas shift and the water pollutant removal.2–5 Metal oxides used as gold supports have been classified into active (Fe2O3, MnO2, CeO2 and TiO2) and inert ones (SiO2, MgO and Al2O3) according to their reducibility.6,7 The former group exhibited exceptionally efficient activity for CO oxidation. For example, on Au/TiO2 catalysts, the CO oxidation reaction was observed even at the temperature as low as 90 K.8 For the latter one, gold can also be reactive on non-reducible supports if catalysts are properly prepared and pretreated. Li and He et al.9 prepared the single gold atoms supported on aluminum oxide clusters, showing high performance for CO oxidation. However, at present, most gold catalysts still suffer from the easy sintering of gold particles at elevated temperature and poor long-term stability, which is the major obstacle to the practical application of gold catalysts.1–4 Clearly, it remains a formidable challenge to design the catalyst with both high catalytic activity and good stability in high temperature environment.
Recently, some metal salts such as metal carbonates,10 orthovanadates (LaVO4)11 and bismuth phosphates (BiPO4)12 have been selected as the third type of gold support. It was found that Au/HAP exhibited good reactive stability for CO oxidation because of the abundant PO43− and OH−.2,4,5 Phonthammachai et al.13 decorated HAP ceramic foam scaffolds with well-dispersed gold nanoparticles, and the Au–HAP composite showed high stability for CO oxidation. Cao et al.5 studied the effect of calcination atmosphere (H2, He and O2) on the activity of Au/HAP catalyst in CO oxidation and found Au/HAP showed the best stability and highest steady state activity when it was calcined in O2 environment. Liu et al.14 later suggested gold nanoparticles performed higher initial activity using higher crystallinity HAP as carrier materials. Nevertheless, HAP supported gold nanocatalysts had relatively lower catalytic activity in CO oxidation than metal oxide supported gold catalysts. While some studies in catalysis have demonstrated that the presence of transition metal oxides could improve the activity and stability of the gold catalyst.15–20 Zhang et al.21 successfully confined Au–Cu Alloy nanoparticles with sizes of about 3 nm in the confined space of SBA-15, and achieved higher activity than monometallic particles for preferential CO oxidation in rich H2 stream. Thereafter, they found that the Au–CuOx was the most active form among Au–Cu alloy nanoparticles that they have studied for the CO oxidation reaction.22 Lin and Zhong et al.23 prepared the Au–Cu/SBA-15 catalyst and found that the CuO particles could reduce the gold particle size, stabilize the gold particles, and facilitate the activation of molecular oxygen for CO oxidation. Dai and co-workers24 have developed silica-supported Au–CuOx hybrid catalysts for the selective oxidation of ethanol into acetaldehyde and confirmed that the active catalyst was the Au core with a thin CuOx shell. Moreover, gold catalysts containing copper or copper oxide have been studied for a wide range of other reactions including water gas shift (WGS),25 total oxidation of methane, ethane and propane18 and epoxidation of propene.19
Au/HAP exhibited good stability, while transition metal oxides could enhance the activity of the gold catalysts. If HAP was modified by transition metal oxides, its supported gold catalyst might possess enhanced activity and stability simultaneously. Zhang et al.4b reported that FeOx played a significant role for the excellent activity and sintering-resistance of Au/HAP in CO oxidation. But thus far there have been no examples on copper or copper oxide-modified HAP supported gold catalysts for CO oxidation catalysis.
Herein, Au–Cu/HAP catalyst was studied. Poriferous HAP nanorods were synthesized via a deposition–precipitation approach using degreasing cotton as template. Au–Cu/HAP catalysts were obtained using a two-step impregnation method. The samples were characterized by XRD, TEM, SEM, UV-Vis, ICP, XPS, H2-TPR and O2-TPD. Their catalytic performance was tested for CO oxidation reaction. The results showed that Au–Cu/HAP catalyst exhibited the better activity and stability compared with Au/HAP and Cu/HAP catalysts for CO oxidation. The possible reason for the superior property of Au–Cu/HAP might be the synergistic interaction between the gold and CuOx phase in Au–Cu/HAP catalysts. To the best of our knowledge, this is the first report that highly efficient and stable Au–Cu/HAP catalysts were obtained through a facile synthesis route and that gold and copper could synergistically catalyze the CO oxidation.
2. Experimental
All the regents were of analytic grade and were used without further treatment.
2.1 HAP support preparation
The poriferous HAP nanorods (ratio: Ca/P = 1.67) were prepared through a deposition–precipitation method using degreasing cotton (Caoxian Country Hualu Hygienic Material Co., Ltd.) as template (0.1 g). The (NH4)2HPO4 solution (0.03 mol L−1, 100 mL, Tianjin Fengchuan Chemical Reagent Technology Co., Let.) was added drop-wise to Ca(NO3)2·4H2O solution (0.05 mol L−1, 100 mL, Tianjin Guangfu Fine Chemical Research Institute), maintaining pH about 10.5 with NH3·H2O (Tianjin Fengchuan Chemical Reagent Technology Co., Let.) through all preparation process. The system was processed at 85 °C for 2 h. After aging for 12 h at room temperature, filtered, washed, dried at 80 °C and then calcined at 300 °C for 5 h in air.
2.2 Catalysts preparation
Au–Cu/HAP catalysts with a nominal total gold and copper loading of 2.0 wt% were prepared by a two-step impregnation method. Firstly, a certain amount of Cu(NO3)2·3H2O (Aladdin Industrial Corporation) was added to the solution of HAP (0.4 g in 100 mL distilled water) under continuous stirring for 1 h, and adjusted pH to 10.5 with Na2CO3 solution (0.05 mol L−1, Tianjin Chemical Reagent Factory), followed by ultrasound for 1 h. Then the precipitate was centrifuged, washed, dried at 80 °C and finally calcined in air at 300 °C for 2 h. Secondly, 100 mL of distilled water was mixed with 0.4 g of the product above and a certain amount of HAuCl4 (0.01 mol L−1, Tianjin Guangfu Fine Chemical Research Institute). The resulting mixture was stirred for 12 h, and then refluxed at 100 °C for 4 h. The suspension was centrifuged, washed several times with distilled water to remove Cl1− and dried at 80 °C, then the product was calcined in air at the same temperature as the first step (300 °C) for 2 h. The amount of HAuCl4 and Cu(NO3)2·3H2O was varied to get the desired Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu atomic ratios, and Au–Cu/HAP catalysts were identified as Au–Cu(x–y)/HAP in which x–y was the nominal Au:Cu atomic ratio.
:Cu atomic ratios, and Au–Cu/HAP catalysts were identified as Au–Cu(x–y)/HAP in which x–y was the nominal Au:Cu atomic ratio.
Au/HAP and Cu/HAP catalysts were prepared in the same way with Au–Cu/HAP.
2.3 Characterization
The X-ray diffraction (XRD) patterns were carried out using a Rigaku D/Max-2500 X-ray diffractometer (Cu Kα λ = 0.154 nm) to identify the crystal phase. Transmission electron microscopy (TEM) images were taken using JEM-2100 TEM and Tecnai G2F20 TEM working at 200 kV. EDX spectra were collected on Mo TEM grids. Scanning electron microscopy (SEM) was performed on a JSM-7500F SEM.
Ultraviolet-Visible spectroscopy (UV-Vis) spectroscopy was recorded by UV-Vis NIR spectrophotometer (SHIMADZU UV-3600). The actual gold and copper loadings were determined by inductively coupled plasma (ICP) on Varian 725 ES ICP instrument. X-ray photoelectron spectra (XPS) were recorded to investigate the chemical states of Au and Cu on a PHI5000VersaProbe. The H2 temperature-programmed reduction (H2-TPR) profiles were measured on a ChemiSorb 2720. Sample (100 mg) was pretreated in Ar at 300 °C for 1 h, cooled down and then reduced with H2 from 50 to 800 °C at a flow rate of 50 mL min−1 with a heating ramp of 10 °C min−1. Temperature-programmed desorption of oxygen (O2-TPD) was carried out using a quantachrome chemBET TPD. 50 mg of sample was pretreated in He at 300 °C for 1 h, cooled downed to room temperature, and then exposed to O2 gas for adsorption until saturated. O2 desorption was performed in 15 mL min−1 He with a heating ramp of 10 °C min−1 to 800 °C.
2.4 Catalytic activity
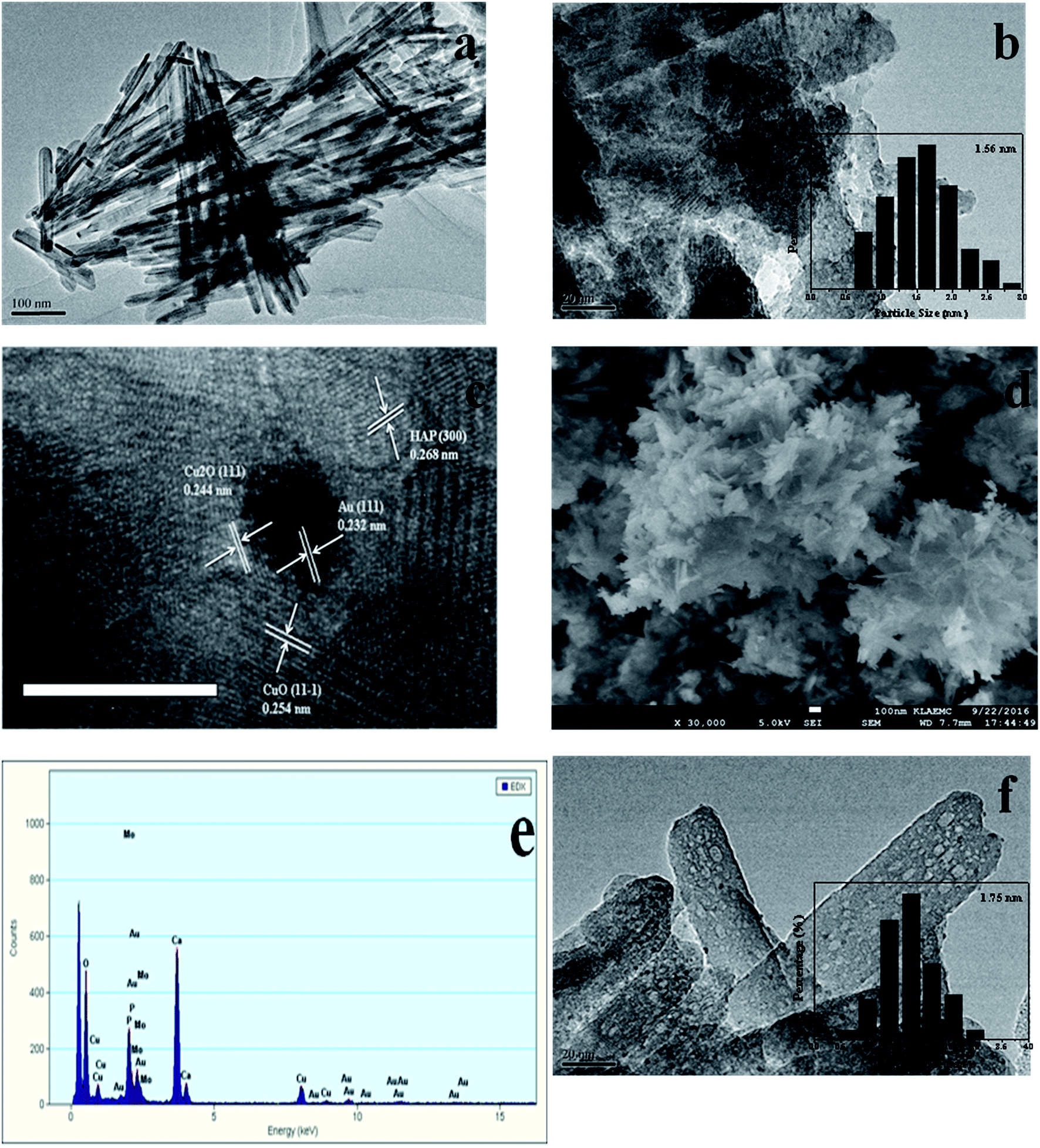
Catalytic reaction tests were carried out in a similar manner as reported in a previous work.26 The catalytic activity was evaluated in a fixed-bed flow millireactor under atmospheric pressure. Sample (200 mg) was diluted by chemically inert quartz sand and then was loaded into a stainless steel tube with an inner diameter of 8 mm. The feed gas containing of 10% CO and balanced air was flowed through reactor at a total flow rate of 36.3 mL min−1. The sample was heated with a ramping rate of 5 °C min−1 and held at the reaction temperature for 17 min. Then the effluent gases were analyzed on-line by GC-508A gas chromatography using H2 as carrier gas. The degree of conversion of CO was calculated to test the activity, which was outlined in the following equation:
3. Results and discussion
3.1 XRD
Fig. 1 presents the XRD patterns of HAP support and various catalysts (nominal total gold and copper loading: 2.0 wt%). As shown in Fig. 1, the prepared HAP support was well-defined crystalline phase-hexagonal structure of pure apatite (JCPD no. 09-0432). The XRD patterns of catalysts were similar to that of HAP, which indicated that the crystal structure of the HAP support was not changed after the deposition of gold and copper. Au/HAP gave metallic gold diffraction peak (111) at 2θ = 38.2°, which might be due to the relatively high loading of gold or large particle size. For Cu/HAP and Au–Cu/HAP, there were no apparent changes in the overall patterns of the samples after deposition of gold and copper compared with HAP. Since the typical peaks 2θ = 44.5°, 35.5° and 42.6° assigned to diffractions of Au (200), CuO (002) and Cu2O (200) could be overlaid by HAP (400), (301) and (302), respectively, it was difficult to deduce the existence form of gold and copper. TEM and XPS studies were subsequently adapted to investigate the oxidation state of gold and copper species in catalysts.
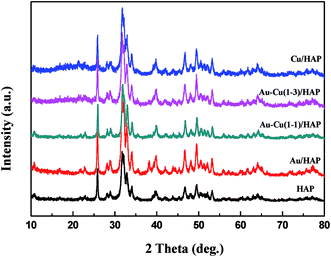 |
| | Fig. 1 XRD patterns of HAP support and various catalysts (nominal total gold and copper loading: 2.0 wt%). | |
3.2 TEM and SEM
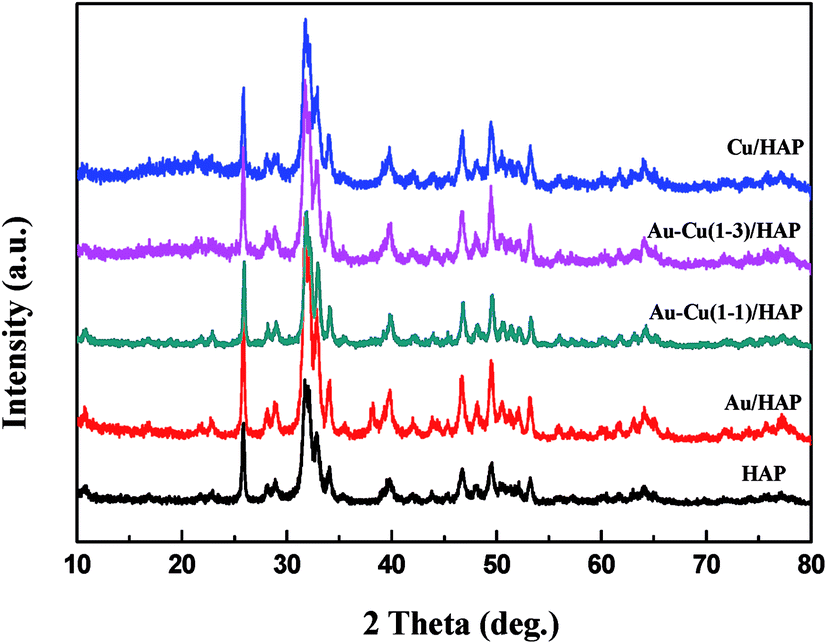
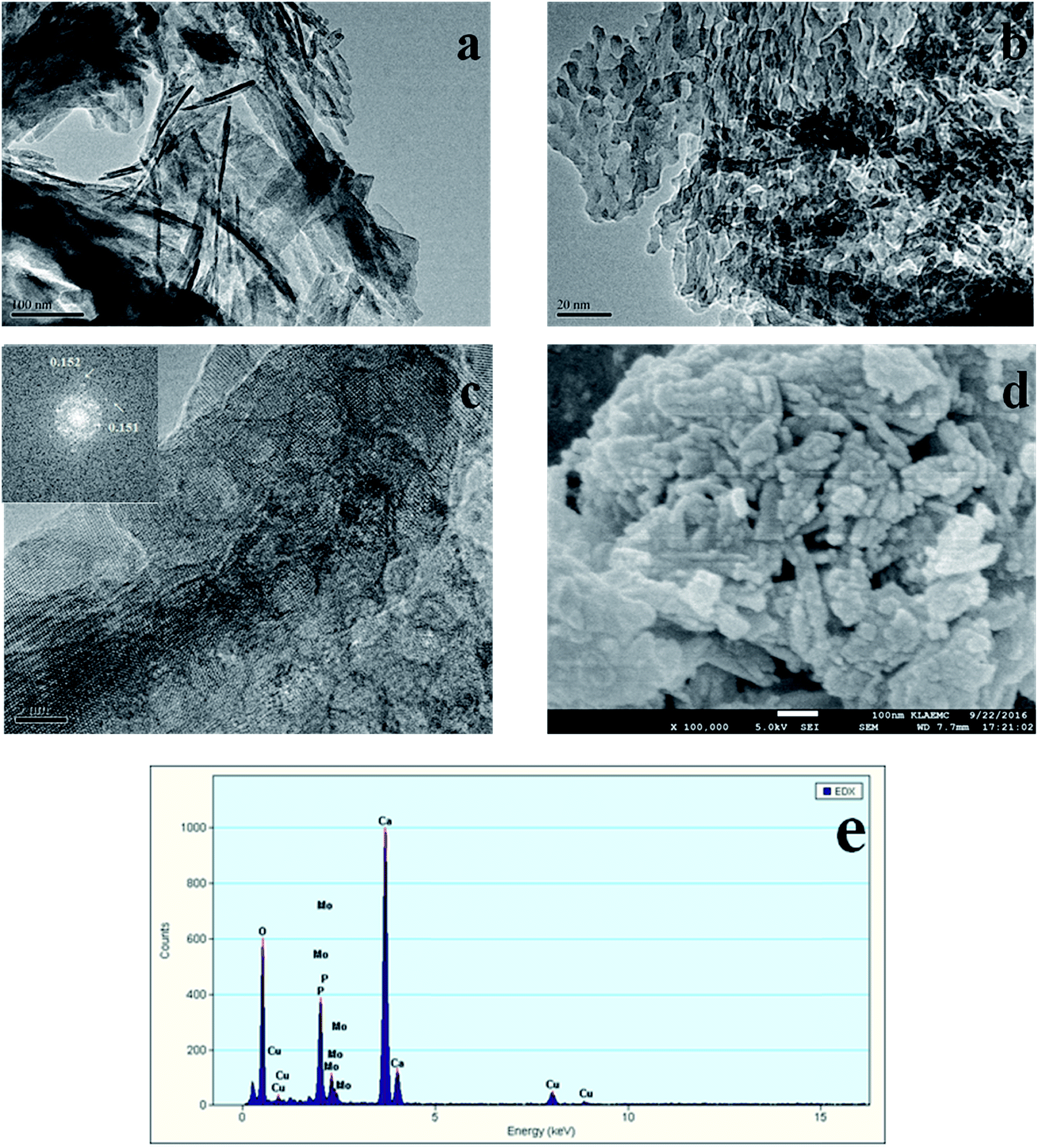
The TEM and SEM images of Au/HAP (Au: 2.0 wt%) are given in Fig. 2. HAP support was clearly observed with nearly uniform nanorod structure with a diameter of 30–40 nm and a length of more than hundreds of nanometers, and many pores were presented on the surface of HAP nanorods. As seen from Fig. 2a and b, gold nanoparticles with the mean size of 3.04 nm were distributed homogeneously on the surface of HAP nanorods. Fig. 2c is a high-resolution (HR)-TEM image of Au/HAP catalyst, showing the observed nanoparticles were gold nanoparticles because of the lattice fringe of 0.232 nm.21,27 Fig. 3 performs the typical TEM and SEM images of Cu/HAP (Cu: 2.0 wt%) catalysts. The lattice fringes measured from HR-TEM image (Fig. 3c) corresponded to that of crystalline CuO (JCPDS 48-1548), which had spacing of 0.150 nm for {113} plane.21 This confirmed that CuO nanoparticles were formed in the Cu/HAP catalyst. However, no obvious CuO nanoparticles were observed on the surface of HAP support, probably because of the low contrast between CuO particles and HAP support (Fig. 3a and b). The similar result was reported for the catalyst of CuO supported on SBA-15.23 It should be mentioned that HAP nanorods broke after the deposition of copper. This might be contributed to the interaction between CuO and HAP. The EDX image (Fig. 3e) illustrated the uniform distribution of copper element on the HAP support almost everywhere without any other impurities. The TEM images of Au–Cu(1–3)/HAP catalyst (Fig. 4a and b) show that nanosized metal particles were well dispersed on the HAP support with the mean size of 1.56 nm, which was smaller than that on Au/HAP catalyst. As seen from Fig. 4c, Au, CuO and Cu2O with the lattice distances of 0.232, 0.254 and 0.244 nm which corresponded to the {111} plane of Au, {11−1} plane of CuO and {111} plane of Cu2O were observed on the surface of the HAP,21,27 indicating that CuO and Cu2O species coexisted in Au–Cu/HAP catalyst. It could be speculated from the EDX image of Au–Cu(1–3)/HAP (Fig. 4e) that gold and copper elements were homogeneously distributed throughout the nanocrystal. In addition, gold and copper compositions were detected with an atomic ratio of 1:4, which agreed well with the actual Au:Cu atomic ratio of 1:4.57 (Table 1).
 |
| | Fig. 2 TEM and SEM images of Au/HAP (Au: 2.0 wt%). TEM images (a, b), HR-TEM image (c), SEM image (d) and TEM image after reaction (e). | |
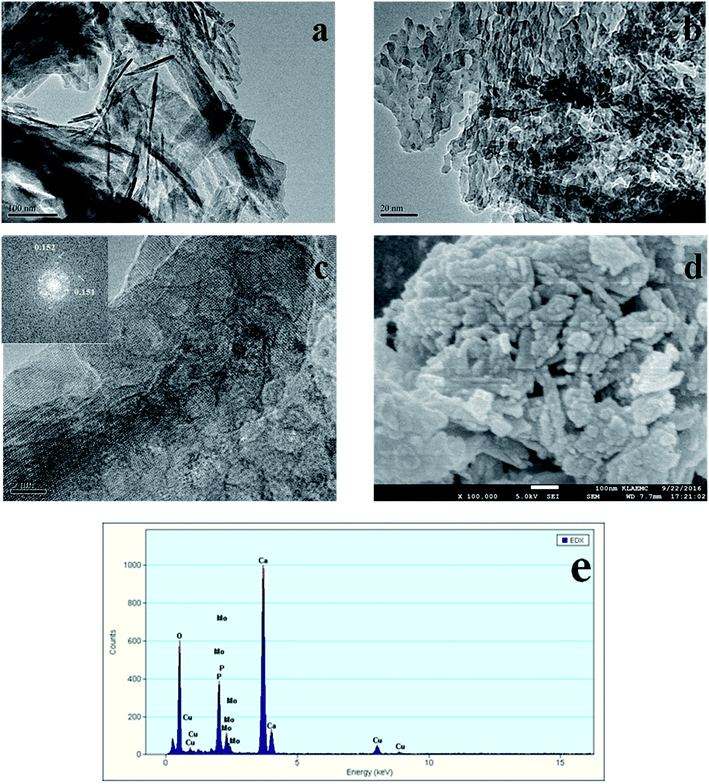 |
| | Fig. 3 TEM and SEM images of Cu/HAP (Cu: 2.0 wt%). TEM images (a, b), HR-TEM image with the corresponding FFT image shown in the inset (c), SEM image (d) and EDX spectrum (e). | |
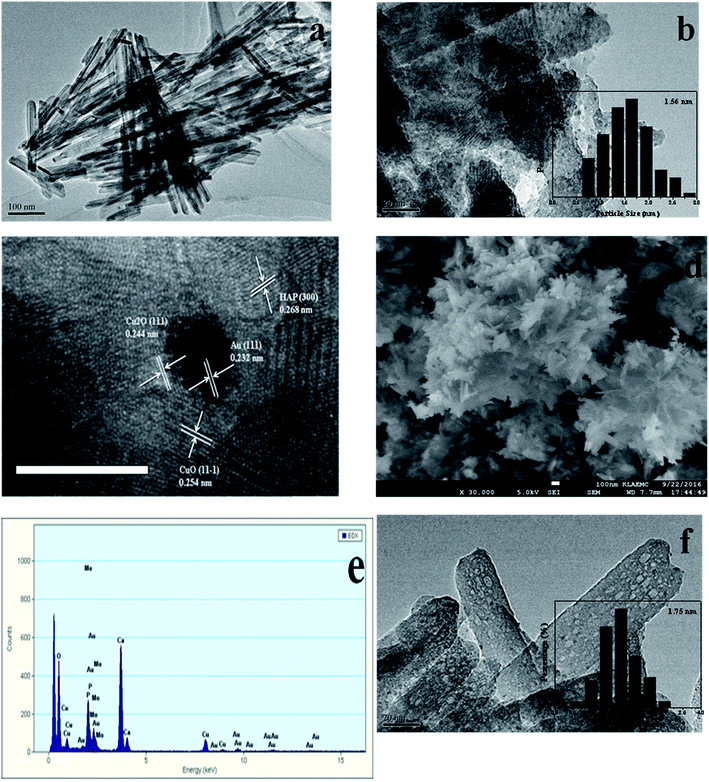 |
| | Fig. 4 TEM and SEM images of Au–Cu(1–3)/HAP (nominal total gold and copper loading: 2.0 wt%). TEM images (a, b), HR-TEM image (c), SEM image (d), EDX spectrum (e) and TEM image after reaction (f). | |
Table 1 The nominal loading and the actual loading of gold and copper
| Sample |
Nominal gold loading (wt%) |
Nominal total gold and copper loading (wt%) |
Actual total gold and copper loading (wt%) |
Nominal Au:Cu molar ratio |
Actual Au:Cu molar ratio |
| Au/HAP |
2.0 |
2.0 |
1.49 |
1:0 |
1:0 |
| Cu/HAP |
0 |
2.0 |
1.83 |
0:1 |
0:1 |
| Au–Cu/HAP |
1.5 |
2.0 |
1.52 |
1:1 |
1:1.73 |
| Au–Cu/HAP |
1.0 |
2.0 |
1.74 |
1:3 |
1:4.57 |
| Au–Cu/HAP |
1.0 |
3.0 |
2.83 |
1:6 |
1:9.19 |
| Au–Cu/HAP |
1.0 |
6.0 |
6.15 |
1:15 |
1:18.93 |
| Au–Cu/HAP |
1.0 |
11.0 |
11.11 |
1:30 |
1:34.50 |
| Au–Cu/HAP |
1.0 |
16.0 |
15.58 |
1:45 |
1:49.61 |
| Au/HAP |
1.0 |
1.0 |
0.61 |
1:0 |
1:0 |
The SEM micrograph of Au/HAP (Fig. 2d) indicates that Au/HAP was basically rod shape with a diameter of about 30–40 nm and a length of 200–300 nm. Fig. 3d performs the SEM image of Cu/HAP. It could be seen the Cu/HAP had the morphology of shorter nanorods compared with Au/HAP. As can be seen from Fig. 4d, the Au–Cu/HAP had a flower-like morphology consisting of nanorods. It was likely that CuOx phase in Au–Cu/HAP had a stronger interaction with gold nanoparticles than HAP, and CuOx anchored gold nanoparticles to the support, stabilizing gold particles in an optimal size range to catalyze CO oxidation reaction. Fig. 2e and 4f are the TEM images of Au/HAP and Au–Cu(1–3)/HAP after CO oxidation reaction, respectively. Compared with their TEM images before reaction (Fig. 2b and 4b), it was strikingly found that no obvious changes in particle size after catalytic reaction were observed for Au–Cu(1–3)/HAP catalyst. The mean diameter was 1.56 and 1.75 nm for the sample before and after reaction, respectively. There was a significant increase of gold particle from 3.04 to 7.01 nm for Au/HAP, which suggested the presence of CuOx in Au–Cu/HAP preserved the gold particle size during CO oxidation reaction.
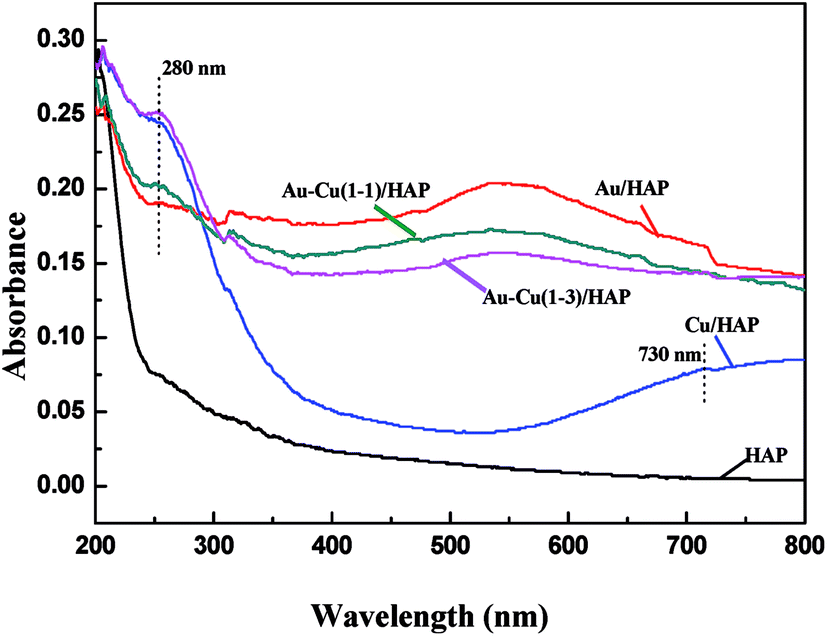
3.3 UV-Vis
Fig. 5 performs the UV-Vis diffuse reflectance spectra (DRS) of HAP support and various catalysts (nominal total gold and copper loading: 2.0 wt%). It is apparently seen that the absorption spectra of Au/HAP and Au–Cu/HAP exhibited an absorption band with a maximum at 500–600 nm, which was characteristic for the plasmon resonance of metallic gold particles.28–30 It is well known that the Plasmon band of metallic nanoparticles was effected by the particle size, loading, shape, and surrounding environment.27,31 Au–Cu/HAP showed less sharp absorbance peak at 500–600 nm than Au/HAP, indicating the addition of copper to Au/HAP caused a much smaller gold particle size and the higher particle dispersion in Au–Cu/HAP.32 The low energy peaks (280 nm) assigned to an band from copper element were all observed in Cu/HAP and Au–Cu/HAP catalysts,33 while the weaker absorption presented in Au–Cu(1–1)/HAP compared with Cu/HAP and Au–Cu(1–3)/HAP catalysts might be due to the small particle size and relatively low concentration of copper. Moreover, a small new peak at about 730 nm appeared in Cu/HAP. This could be ascribed to the additional formation of large CuO particles, which had a negative effect on the catalytic performance of catalyst.21 Therefore, it could be understandably deduced that CuOx phase could depress the movement and growth of gold nanoparticles in Au–Cu/HAP on account of the stronger interaction with gold nanoparticle than HAP.
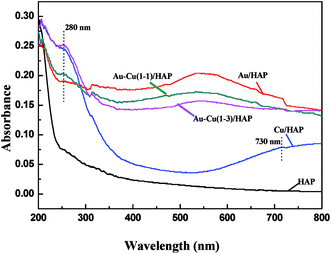 |
| | Fig. 5 UV-Vis DRS of HAP support and various catalysts (nominal total gold and copper loading: 2.0 wt%). | |
3.4 ICP
ICP was used to determine the actual gold and copper loadings of catalysts, and the chemical compositions were listed in Table 1. The results showed that actual Au:Cu molar ratios were slightly higher than nominal ones. It might be because copper was more easily deposited on the surface of HAP in comparison with gold, which was related to the low isoelectric point of HAP and to the base and acid sites of metal precursors.4a,34
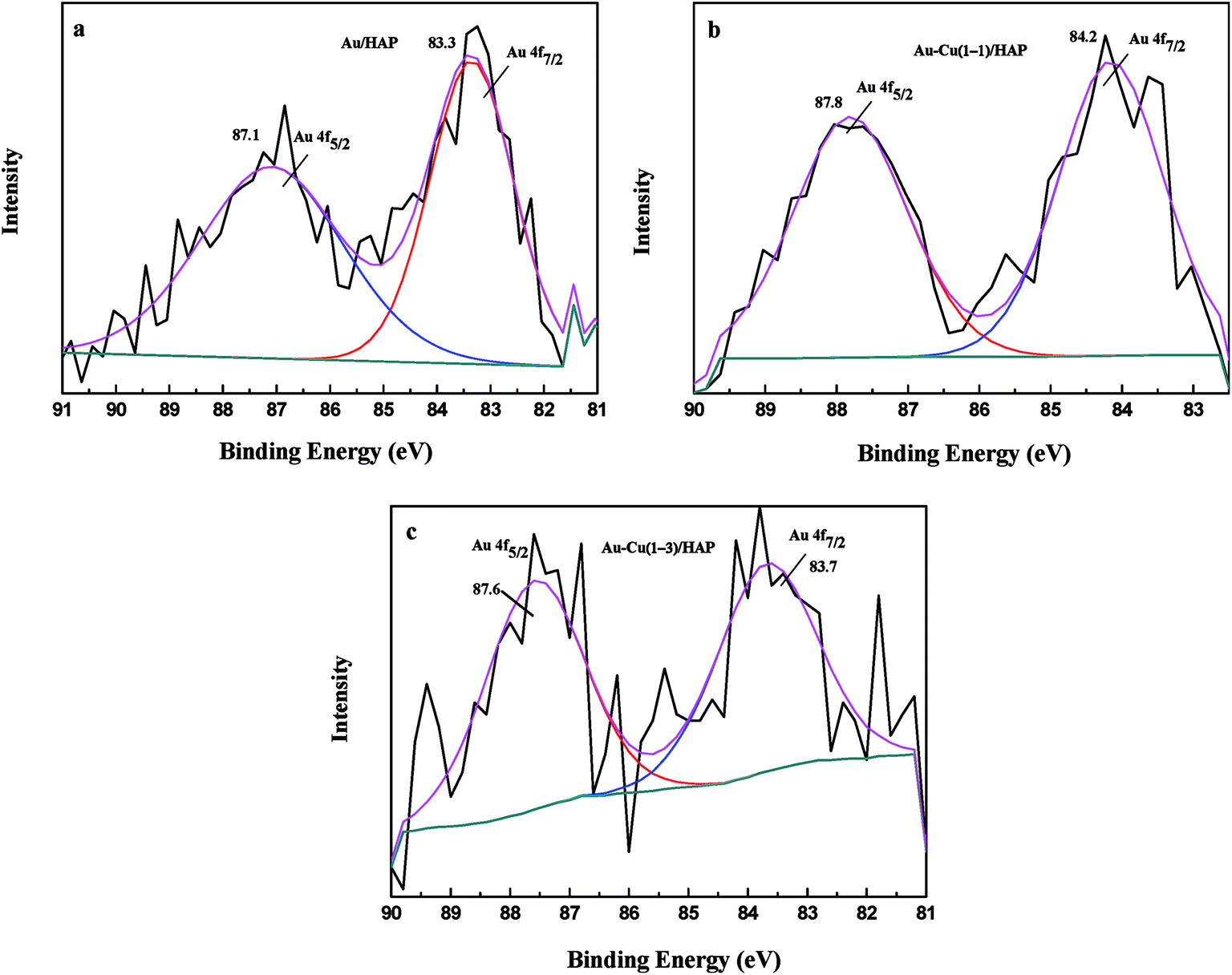
3.5 XPS
XPS analysis was performed to investigate information about chemical states of Au and Cu. Fig. 6 presents Au 4f XPS spectra of catalysts (nominal total gold and copper loading: 2.0 wt%). The Au 4f7/2 peaks for Au/HAP, Au–Cu(1–1)/HAP and Au–Cu(1–3)/HAP were located at binding energy of 83.3, 84.2 and 83.7 eV, respectively, which were characteristic for zero valent gold and closed to the ones observed for metallic gold.4a,31 Peaks assigned to oxidized gold species, which would be located around 85.5 and 86.3 eV, have been not detected.4a,14
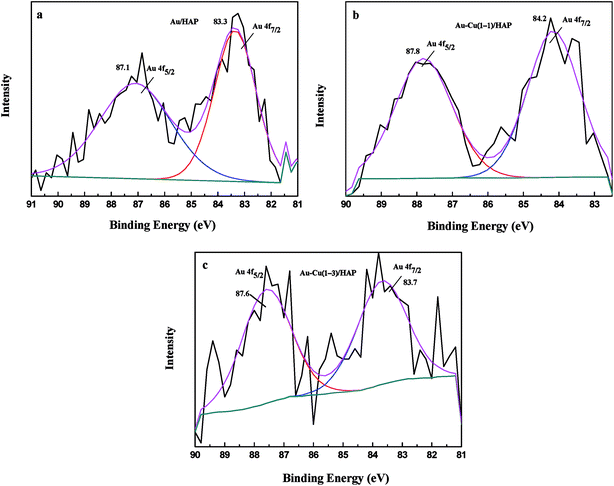 |
| | Fig. 6 Au 4f XPS spectra of Au/HAP (a), Au–Cu(1–1)/HAP (b) and Au–Cu(1–3)/HAP (c) (nominal total gold and copper loading: 2.0 wt%). | |
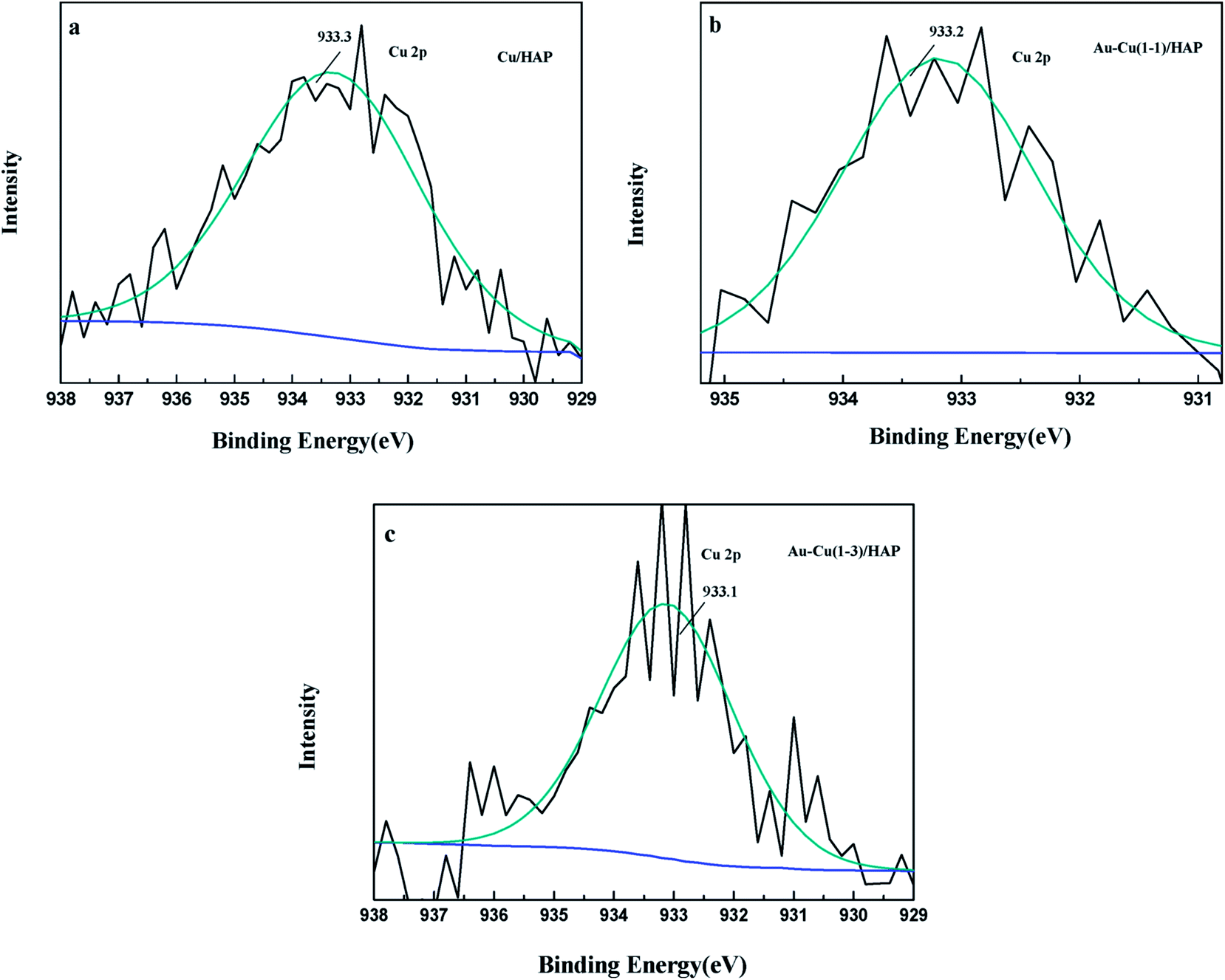
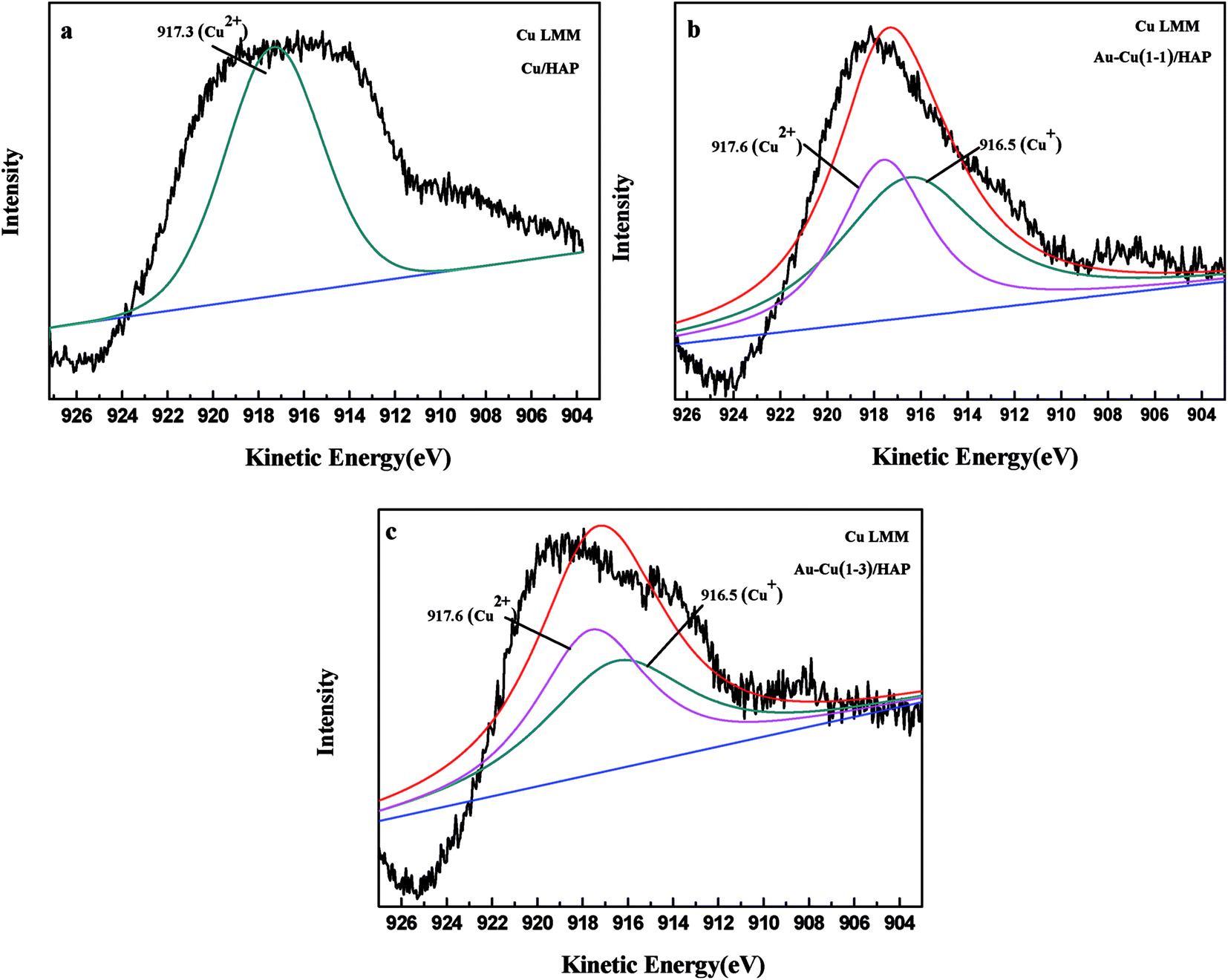
The Cu 2p3/2 XPS spectra of catalysts are presented in Fig. 7. Cu 2p3/2 characteristic peaks of Cu/HAP, Au–Cu(1–1)/HAP and Au–Cu(1–3)/HAP were observed at binding energies of 933.3, 933.2 and 933.1 eV, respectively, indicating the presence of Cu2+/CuO, which was in accord with the reported literature values.35 As is well known, the Cu 2P3/2 binding energy and peak shape could not be work to distinguish between Cu+ and Cu0, because they are essentially identical. Therefore, X-ray Cu L3M45M45 (LMM) Auger lines were carried out to identify the Cu value (Cu2+ 917.6 eV; Cu+ 916.5 eV; Cu0 918.5 eV).36 As shown in Fig. 8, copper species existed as Cu2+ in Cu/HAP catalyst, while copper coexisted as Cu2+ and Cu+ in the two Au–Cu/HAP catalysts. It was reasonable to speculate that the strong interaction between CuO and gold particles induced the reduction of Cu2+ to Cu+ that was regarded as a more active adsorption site in CO oxidation, which might change the electronic property of gold.12,37
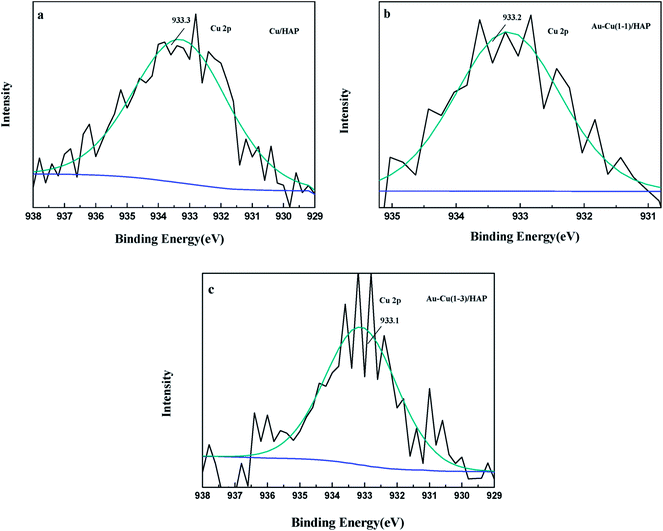 |
| | Fig. 7 Cu 2p3/2 XPS spectra of Cu/HAP (a), Au–Cu(1–1)/HAP (b) and Au–Cu(1–3)/HAP (c) (nominal total gold and copper loading: 2.0 wt%). | |
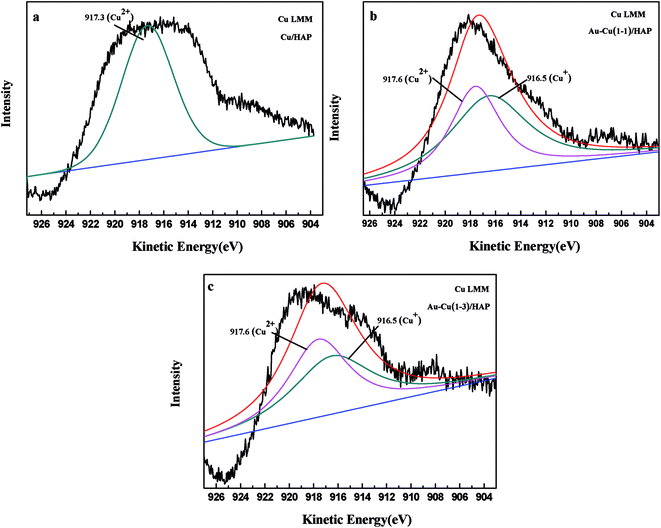 |
| | Fig. 8 Kinetic energy spectra of the Cu Auger LMM for Cu/HAP (a), Au–Cu(1–1)/HAP (b) and Au–Cu(1–3)/HAP (c) (nominal total gold and copper loading: 2.0 wt%). | |
3.6 H2-TPR
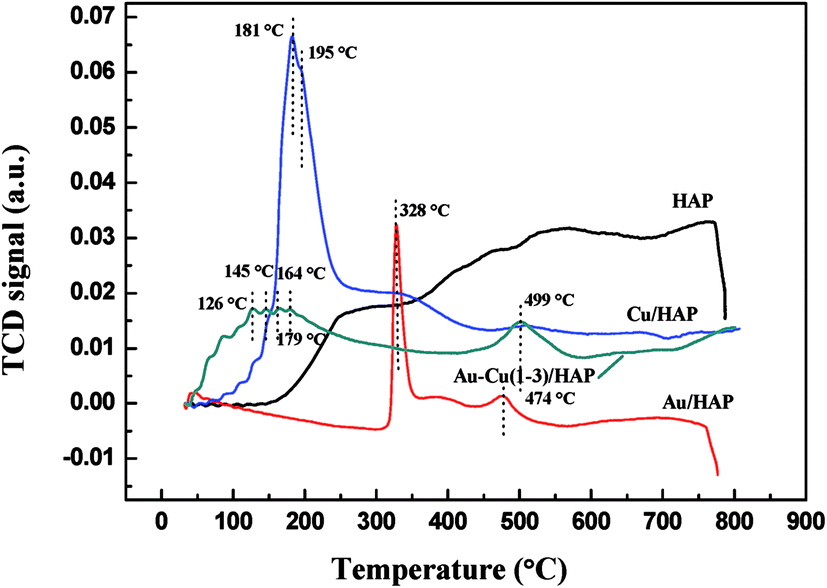
Fig. 9 presents the TPR profiles of HAP support and various catalysts (nominal total gold and copper loading: 2.0 wt%). The TPR profile of HAP showed no obvious reduction peaks from 30 to 800 °C, while Au/HAP exhibited two reduction peak centered at 328 and 474 °C, respectively. The former one was attributed to the reduction of surface oxygen and the later one was representative of bulk oxygen species.38 That was to say, the deposition of gold significantly increased the reducibility of support. In H2-TPR profile of Cu/HAP catalyst, the reduction peak at 181 °C with a shoulder at 195 °C could be assigned to the reduction of small CuO particles and large ones, respectively.21,39 It is well established that the smaller CuO particles were, the more easily they could be reduced.40 For Au–Cu(1–3)/HAP catalyst, three reduction peaks at 126, 145 and 164 °C were ascribed to a multistep reduction process of CuO (CuO → Cu2O → Cu). These reduction temperatures were lower than those that reported in the literature.21 One possible reason was that gold could disperse CuOx particles, thereby presenting smaller CuOx particles. The other reason was that gold weakened the bond strength of Cu–O and enhanced the reduction of CuO.39 Besides, the reduction peak at 328 °C of Au/HAP disappeared after the deposition of copper and a new peak at 179 °C was observed in TPR profile of Au–Cu(1–3)/HAP catalyst. It could be attributed to the surface oxygen species becoming the shared oxygen on Au–Cu(1–3)/HAP because of the synergistic effect between gold and CuOx phase.41
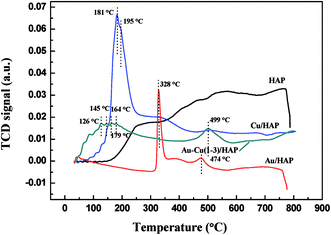 |
| | Fig. 9 H2-TPR profiles of HAP support and various catalysts (nominal total gold and copper loading: 2.0 wt%). | |
3.7 O2-TPD
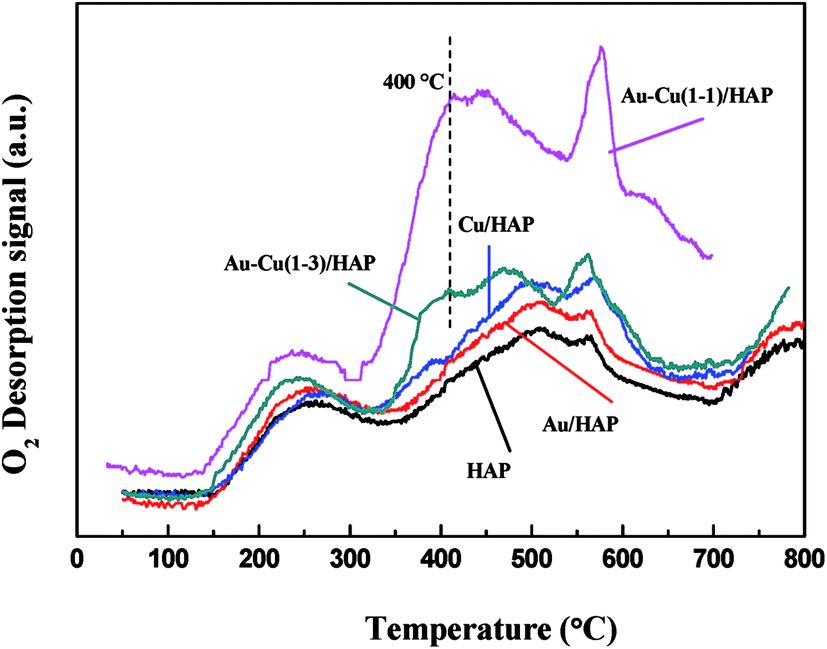
The oxygen adsorption and desorption behaviors of samples were investigated using O2-TPD techniques, and the results were showed in Fig. 10. The peak below 300 °C could be ascribed to the desorption of O2 physically absorbed on the support.42 The two peaks in the 500–600 °C range were due to the desorption of chemisorbed O2 molecules in the pores of samples (in the case of HAP, Au/HAP and Cu/HAP),31 while Au–Cu(1–1)/HAP and Au–Cu(1–3)/HAP exhibited much lower desorption temperature and larger amount of desorbed O2, which suggested they had bigger O2 adsorption capacity. As we know, catalytic activity depends on the adsorption of reactants (CO and O2), and more adsorption contributes to more reaction.42 This is possibly because more adsorption of CO and O2 generates more activated CO and O2 molecules, and then the activated molecules interact with each other to form product (CO2), promoting the catalytic oxidation of CO. In addition, a small new desorption peak at 400 °C was observed in Au–Cu(1–1)/HAP and Au–Cu(1–3)/HAP, respectively. This might be attributed to the adsorption of O2 at metal support interfacial sites.42 Thus, TPD results revealed that the addition of copper into Au/HAP gave rise to new O2 adsorption sites at Au/HAP interfaces and created the bigger O2 adsorption capacity.
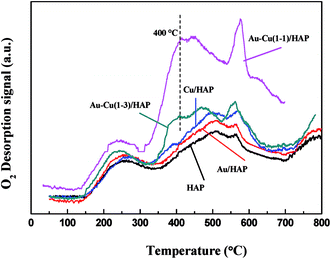 |
| | Fig. 10 O2-TPD profiles of HAP support and various catalysts (nominal total gold and copper loading: 2.0 wt%). | |
3.8 Catalytic activity
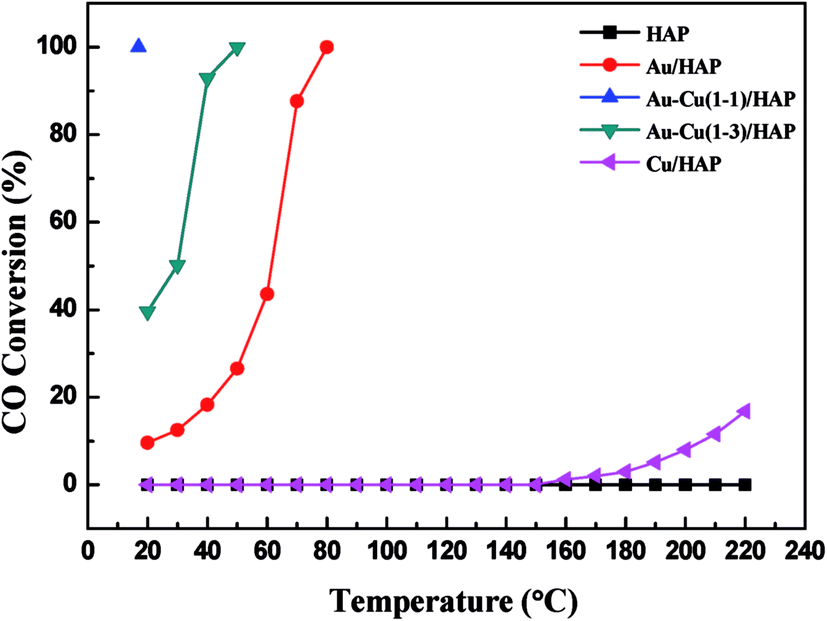
Fig. 11 shows the curves of CO conversion over HAP support and various catalysts (nominal total gold and copper loading: 2.0 wt%). It was showed that HAP had no activity and Cu/HAP had little activity from 20–220 °C. The T100 (T100: the temperature at which 100% CO conversion was achieved) of Au/HAP, Au–Cu(1–1)/HAP and Au–Cu(1–3)/HAP was 80, 17 and 50 °C, respectively. Au–Cu(1–1)/HAP and Au–Cu(1–3)/HAP catalysts showed the higher activity than Au/HAP and Cu/HAP catalysts, which was consistent with O2-TPD results (Fig. 10).
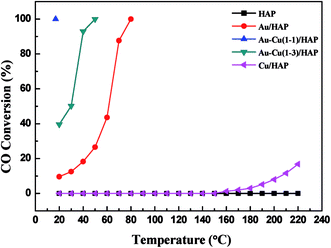 |
| | Fig. 11 Catalytic activity of HAP support and various catalysts (nominal total gold and copper loading: 2.0 wt%). | |
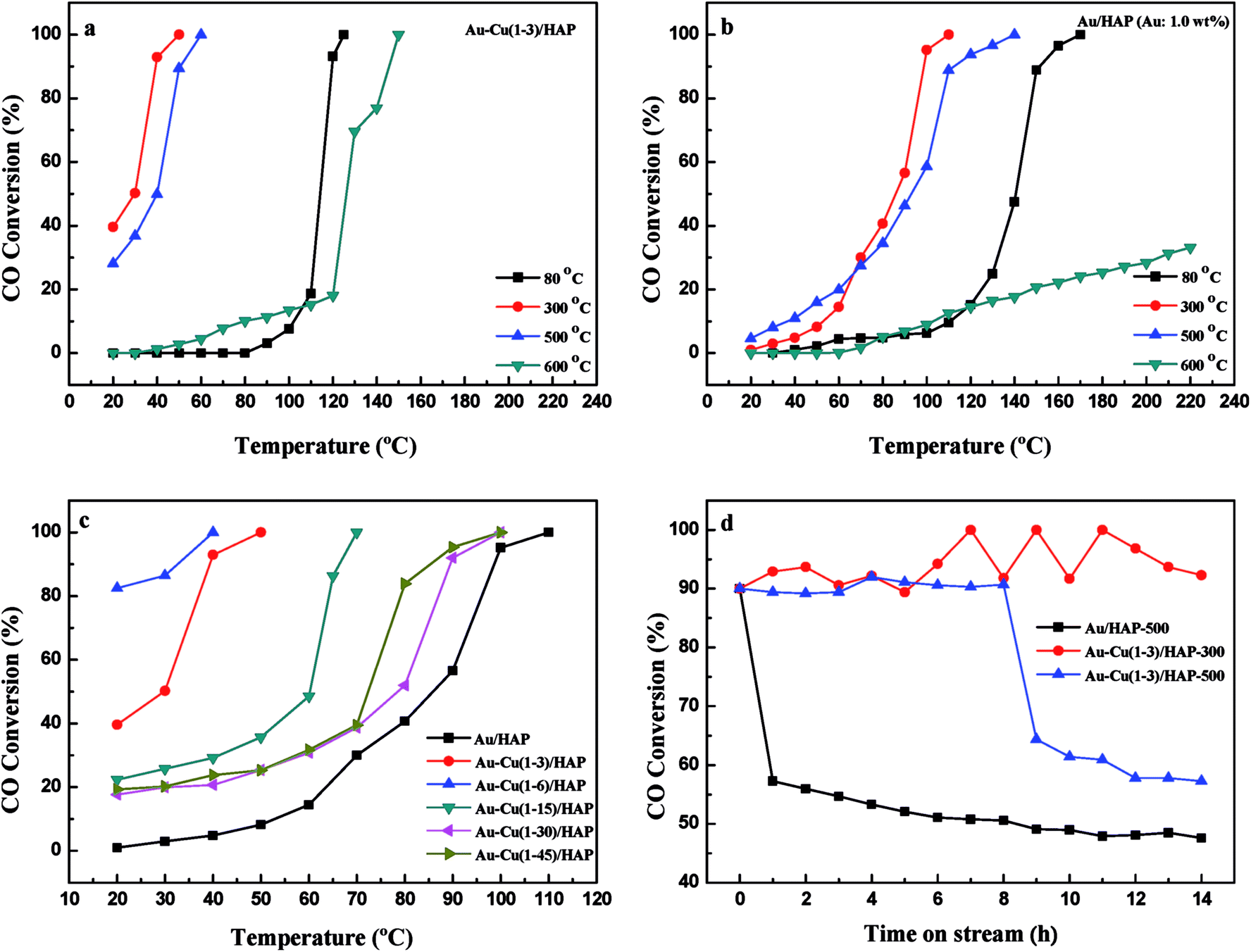
It is well known that gold particles tend to sinter at high temperatures, which leads to a dramatic decrease in activity.23,31,42 To investigate the catalytic activity and sintering-resistance of Au–Cu(1–3)/HAP (Au: 1.0 wt%) catalyst compared with Au/HAP (Au: 1.0 wt%), the catalysts at various calcination temperatures were prepared and their activities were monitored in Fig. 12a and b. It was noted that the CO oxidation activity of the catalysts whether Au–Cu(1–3)/HAP or Au/HAP followed the order of 300 °C > 500 °C > 80 °C > 600 °C of the calcination temperature, and the Au–Cu(1–3)/HAP was significantly more active compared with the Au/HAP at any one of the same calcination temperatures, indicating sufficiently that Au–Cu(1–3)/HAP catalyst had the better efficiency and sintering-resistance at high temperature than Au/HAP catalyst. Surprisingly, Au–Cu(1–3)/HAP calcined 500 °C still retained very high activity with a T100 of 60 °C when compared with that calcined at 300 °C with a T100 of 50 °C, which further confirmed that few gold nanoparticles sintered in Au–Cu(1–3)/HAP at elevated temperature. As determined from the curves, the specific rate over Au–Cu(1–3)/HAP calcined at 300 and 500 °C reached 2.20 and 1.61 molCO gAu−1 h−1 at 30 °C, which was 4.6 and 3.4 times higher than 0.48 molCO gAu−1 h−1 of Au/TiO2 catalyst (World Gold Council) at the same temperature, respectively.23 To our knowledge, the higher reduction temperature was, the less content of reduced copper existed on the catalyst surface. Cu+ species was the main adsorption and active center of CO oxidation, and Cu2+–CO species was unstable and could not exist at room temperature. Therefore, Cu+ was more active site than Cu2+ for CO oxidation.43 It could be concluded that the presence of Cu+ not only preserved the gold particle size but also provided the active component in Au–Cu/HAP catalyst during calcination process.39
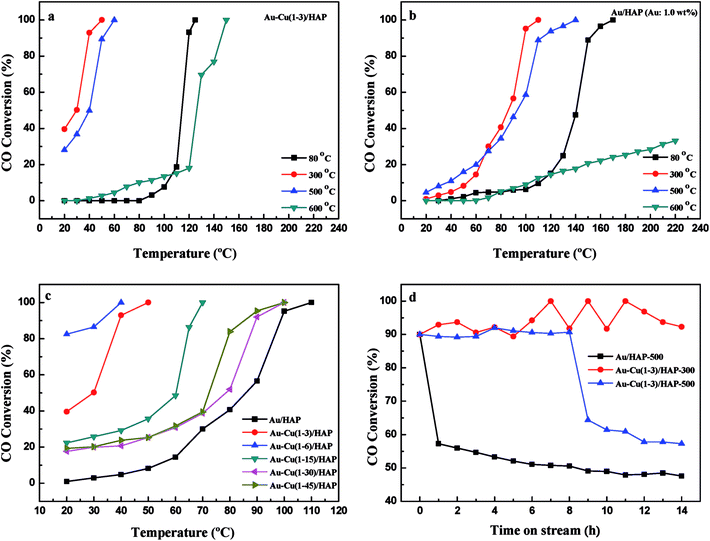 |
| | Fig. 12 Catalytic activity of Au–Cu(1–3)/HAP (Au: 1.0 wt%) (a) and Au/HAP (Au: 1.0 wt%) (b) calcined at different temperatures; Au–Cu/HAP catalysts with different Au:Cu atomic ratios (Au: 1.0 wt%) for CO oxidation (c); the stability of Au–Cu(1–3)/HAP (Au: 1.0 wt%) and Au/HAP (Au: 1.0 wt%) catalysts (d). | |
To show the effect of CuOx on the catalysis of CO oxidation, Au–Cu/HAP catalysts with various Au:Cu atomic ratios (Au: 1.0 wt%) were prepared and calcined at 300 °C. The chemical compositions of these catalysts were listed in Table 1 and the catalytic activities were showed in Fig. 12c. The Au–Cu(1–6)/HAP behaved higher activity with a T100 of 40 °C than Au–Cu(1–3)/HAP and then the activity decreased with the increasing of copper amount. Additionally, all the Au–Cu/HAP catalysts were more active that Au/HAP (Au: 1.0 wt%) with a T100 of 110 °C. These results showed that CuOx played a significant role in improving the activity of Au/HAP catalyst for CO oxidation and excessive copper could hinder access to the active sites on gold.22
Fig. 12d demonstrates the stability of Au–Cu(1–3)/HAP (Au: 1.0 wt%) calcined at 300 and 500 °C and Au/HAP (Au: 1.0 wt%) calcined at 500 °C under the reaction condition. At a conversion level of 90%, Au–Cu(1–3)/HAP calcined at 300 °C was found to be the best stable among the catalysts, and it maintained the CO conversion at about 90% over 14 h reaction time. Au–Cu(1–3)/HAP catalyst calcined at 500 °C had the conversion of about 90% in the first 8 h, and then the conversion decreased to about 60% over next six hours. However, the Au/HAP catalyst calcined at 500 °C was significantly deactivated, especially in the first 1 h, and then declined to about 50% during the same 14 h reaction time. The possible reason for the drop of the catalytic activity of Au/HAP in CO conversion might be the particle sintering. It was likely that the synergistic interaction between gold and CuOx phase in Au–Cu(1–3)/HAP catalyst was responsible for the enhanced stability for CO oxidation.
3.9 Mechanism
On the basis of above results, the possible reaction mechanism was proposed to explain the enhanced activity and stability of Au–Cu/HAP catalyst compared with Au/HAP and Cu/HAP catalysts in CO oxidation. Firstly, CO molecules were absorbed on the metal support interfacial sites. Subsequently, on the one hand, absorbed CO molecules were oxidized by the active O donated by CuOx. On the other hand, the presence of copper gave rise to more oxygen vacancies which could activate molecular oxygen by forming superoxide species. Finally, the adsorbed CO molecules reacted with active O to form CO2. Moreover, the reducibility of a reducible oxide support (TiO2, CeO2, Fe2O3, CuO etc.) at the interface enhanced when gold was supported on it, which increased the mobility of lattice oxygen to react with absorbed CO in the reaction.22 These results indicated that in Au–Cu/HAP, Cu2+ shifted to a more reduced Cu+, and CuOx phase worked in synergy efficiently with gold, creating small metal particles and preserving the active component in supported gold catalysts. Thus, the role of CuOx in CO oxidation was extremely significant in Au–Cu/HAP catalyst.
4. Conclusion
In conclusion, the Au–Cu/HAP catalyst has been successfully prepared for the first time. It has been indicated that Au–Cu/HAP catalyst had the superior activity and stability compared with Au/HAP and Cu/HAP. The enhanced performance could be ascribed to the synergistic effect between gold and CuOx phase in Au–Cu/HAP. The addition of copper into Au/HAP created more new O2 adsorbed sites and bigger O2 adsorption capacity, and excessive copper could hinder access to the active sites on gold. Studies of this type of supports might shed new light on reaction mechanism for CO oxidation. Of more importance, we believe the synergistic effect between gold and CuOx phase is expected to provide further value for the wide application in other catalytic oxidation reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21271110, 21373120 and 21271107) and MOE Innovation Team of China (IRT13022). This work was supported by Tianjin Key Laboratory for photoelectric Materials and Devices, Tianjin University of Technology, Tianjin 300384, China.
References
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405–408 CrossRef.
- H. L. Tang, J. K. Wei, F. Liu, B. T. Qiao, X. L. Pan, L. Li, J. Y. Liu, J. H. Wang and T. Zhang, J. Am. Chem. Soc., 2016, 138, 56–59 CrossRef CAS PubMed.
- E. del Río, A. B. Hungría, M. Tinoco, R. Manzorro, M. A. Cauqui, J. J. Calvino and J. A. Pérez-Omil, Appl. Catal., B, 2016, 197, 86–94 CrossRef.
-
(a) K. F. Zhao, B. T. Qiao, J. H. Wang, Y. J. Zhang and T. Zhang, Chem. Commun., 2011, 47, 1779–1781 RSC;
(b) K. F. Zhao, B. T. Qiao, Y. J. Zhang and J. H. Wang, Chin. J. Catal., 2013, 34, 1386–1394 CrossRef CAS.
- J. Huang, L. C. Wang, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Appl. Catal., B, 2011, 101, 560–569 CrossRef CAS.
- S. H. Overbury, L. Ortiz-Soto, H. G. Zhu, B. Lee, M. D. Amiridis and S. Dai, Catal. Lett., 2004, 95, 99–106 CrossRef CAS.
- M. M. Schubert, S. Hackenberg, A. C. van Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113–122 CrossRef CAS.
- Y. F. Yang, P. Sangeetha and Y. W. Chen, Ind. Eng. Chem. Res., 2009, 48, 10402–10407 CrossRef CAS.
- Z. Y. Li, Z. Yuan, X. N. Li, Y. X. Zhao and S. G. He, J. Am. Chem. Soc., 2014, 136, 14307–14313 CrossRef CAS PubMed.
- H. L. Lian, M. J. Jia, W. C. Pan, Y. Li, W. X. Zhang and D. Z. Jiang, Catal. Commun., 2005, 6, 47–51 CrossRef CAS.
- J. Liu, W. Chen, X. Liu, K. Zhou and Y. Li, Nano Res., 2008, 1, 46–55 CrossRef CAS.
- P. Zhang, H. H. Yu, J. J. Li, H. Zhao, B. L. Zhu, W. P. Huang and S. M. Zhang, RSC Adv., 2016, 6, 15304–15312 RSC.
- N. Phonthammachai, Z. Y. Zhong, J. Guo, Y. F. Han and T. J. White, Gold Bull., 2008, 41, 42–50 CrossRef CAS.
- J. D. Wang, J. K. Liu, Y. Lu, D. J. Hong and X. H. Yang, Mater. Res. Bull., 2014, 55, 190–195 CrossRef CAS.
- E. Smolentseva, N. Bogdanchikova, A. Simakov, A. Pestryakov, I. Tusovskaya, M. Avalos, M. H. Farías, J. A. Díaz and V. Gurin, Surf. Sci., 2006, 600, 4256–4259 CrossRef CAS.
- S. D. Lin, A. C. Gluhoi and B. E. Nieuwenhuys, Catal. Today, 2004, 90, 3–14 CrossRef CAS.
- A. Venugopal, J. Aluha and M. S. Scurrel, Catal. Lett., 2003, 90, 1–6 CrossRef CAS.
- B. E. Solsona, T. Garcia, C. Jones, S. H. Taylor, A. F. Carley and G. J. Hutchings, Appl. Catal., A, 2006, 312, 67–76 CrossRef CAS.
- R. J. Chimento, F. Medina, J. L. G. Fierro, J. Llorca, J. E. Sueiras, Y. Cesteros and P. Salagre, J. Mol. Catal. A: Chem., 2007, 274, 159–168 CrossRef.
- P. Haider and A. Baiker, J. Catal., 2007, 248, 175–187 CrossRef CAS.
- X. Y. Liu, A. Q. Wang, X. D. Wang, C. Y. Mou and T. Zhang, Chem. Commun., 2008, 3187–3189 RSC.
- X. Y. Liu, A. Q. Wang, L. Li, T. Zhang, C. Y. Mou and J. F. Lee, J. Catal., 2011, 278, 288–296 CrossRef CAS.
- X. Li, S. S. S. Fang, J. Teo, Y. L. Foo, A. Borgna, M. Lin and Z. Y. Zhong, ACS Catal., 2012, 2, 360–369 CrossRef CAS.
- J. C. Bauer, G. M. Veith, L. F. Allard, Y. Oyola, S. H. Overbury and S. Dai, ACS Catal., 2012, 2, 2537–2546 CrossRef CAS.
- A. Venugopal, J. Aluha and M. S. Scurrel, Catal. Lett., 2003, 90, 1–6 CrossRef CAS.
- H. Zhao, P. Zhang, Y. D. Wang, W. P. Huang and S. M. Zhang, J. Sol-Gel Sci. Technol., 2014, 71, 406–412 CrossRef CAS.
- T. Okuno, G. Kawamura, H. Muto and A. Matsuda, J. Sol-Gel Sci. Technol., 2015, 74, 748–755 CrossRef CAS.
- R. Zanella, S. Giorgio, C. H. Shin, C. R. Henry and C. Louis, J. Catal., 2004, 222, 357–367 CrossRef CAS.
- G. Carrot, J. C. Valmalette, C. J. G. Plummer, S. M. Scholz, J. Dutta, H. Hofmann and J. G. Hilborn, Colloid Polym. Sci., 1998, 276, 853–859 CAS.
- S. L. Westcott, S. J. Oldenburg, T. R. Lee and N. J. Halas, Langmuir, 1998, 14, 5396–5401 CrossRef CAS.
- K. M. Parida, N. Sahua, P. Mohapatra and M. S. Scurrell, J. Mol. Catal. A: Chem., 2010, 319, 92–97 CrossRef CAS.
- Z. Y. Zhong, S. Patskovskyy, P. Bouvrette, J. H. T. Luong and A. Gedanken, J. Phys. Chem. B, 2004, 108, 4046–4052 CrossRef CAS.
- H. F. Lang, S. Maldonado, K. J. Stevenson and B. D. Chandler, J. Am. Chem. Soc., 2004, 126, 12949–12956 CrossRef CAS PubMed.
- G. J. H Crisel, P. J. Kooyman and B. E. Nieuwenhuys, J. Catal., 2000, 191, 430–437 CrossRef.
- C. Z. Sun, J. Zhu, Y. Y. Lv, L. Qi, B. Liu, F. Gao, K. Q. Sun, L. Dong and Y. Chen, Appl. Catal., B, 2011, 103, 206–222 CrossRef CAS.
- S. M. Zhang, W. P. Huang, X. H. Qiu, B. Q. Li, X. C. Zheng and S. H. Wu, Catal. Lett., 2002, 80, 1–2 CrossRef.
- S. S. Sun, D. S. Mao, J. Yu, Z. Q. Yang, G. Z. Lu and Z. Ma, Catal. Sci. Technol., 2015, 5, 3166–3181 CAS.
- Y. W. Chen, H. J. Chen and D. S. Lee, J. Mol. Catal. A: Chem., 2012, 363–364, 470–480 CrossRef CAS.
- T. C. Ou, F. W. Chang and L. S. Roselin, J. Mol. Catal. A: Chem., 2008, 293, 8–16 CrossRef CAS.
- C. S. Chen, T. C. Chen, C. C. Chen, Y. T. Lai, J. H. You, T. M. Chou, C. H. Chen and J. F. Lee, Langmuir, 2012, 28, 9996–10006 CrossRef CAS PubMed.
- T. J. Huang and Y. C. Kung, Catal. Lett., 2003, 85, 49–55 CrossRef CAS.
- N. Sahua, K. M. Paridaa, A. K. Tripathib and V. S. Kambleb, J. Mol. Catal. A: Chem., 2011, 399, 110–116 Search PubMed.
- X. L. Guo and R. X. Zhou, Catal. Sci. Technol., 2016, 6, 3862–3872 CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*