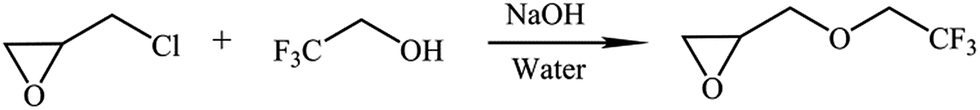DOI:
10.1039/C7RA08929E
(Paper)
RSC Adv., 2017,
7, 47271-47278
Fluorinated glycidyl azide polymers as potential energetic binders†
Received
12th August 2017
, Accepted 2nd October 2017
First published on 6th October 2017
Abstract
To improve the mechanical properties of glycidyl azide polymer (GAP)-based polyurethane network binders, a novel fluorinated glycidyl azide polymer, (2,2,2-trifluoro-ethoxymethyl epoxy-r-glycidyl azide) copolymer (poly(TFEE-r-GA)) was synthesized through an initial cationic copolymerization of epichlorohydrin and 2,2,2-trifluoro-ethoxymethyl epoxy, followed by azidation. The structure of poly(TFEE-r-GA) was characterized by FTIR, 1H NMR, 13C NMR and GPC. DSC and TGA were used to investigate the thermal behavior of poly(TFEE-r-GA), the glass transition temperature and decomposition temperature of poly(TFEE-r-GA) were found to be −49.5 and 250 °C, respectively. The copolyurethane networks were further synthesized by cross-linking poly(TFEE-r-GA) using trimethylolpropane as a chain extender agent, using isophorone diisocyanate as a cross-linking agent. In comparison with GAP, the poly(TFEE-r-GA) based copolyurethane networks exhibited relatively better mechanical properties, which had a tensile strength of 5.52 MPa, and an elongation at break of 162.8%. All the results indicated that the fluorine-containing GAP might serve as a potential energetic binder for future propellant formulations.
1. Introduction
In recent years, the replacement of conventional binders (hydroxyl terminated polybutadiene (HTPB),1 hydroxyl terminated polyether (HTPE),2 etc.) by energetic polymeric binders (polymer backbones containing energetic functional groups such as azido, nitro (C-nitro, O-nitro and N-nitro) and difluoramine groups)3,4 to develop high performance explosives and advanced rocket propellants is a new trend in the field of energetic material formulations. It is required of these energetic polymeric binders to not only improve the internal energy of the formulations, but also improve the overall oxygen balance of the propellant.5,6 Among the energetic polymeric binders, glycidyl azide polymers (GAP) and their related energetic polymer binders are considered as promising candidates for propellant binders.7,8 It is well known that GAP has a high positive heat of formation (+957 J g−1), low detonation tendency, low glass transition temperature (Tg = −45 °C), and also has good compatibility with high-energy oxidizers. However, GAP which has high polarity azide groups grafted on the polymer backbone, possesses poor flexibility of backbone resulting in lack of intermolecular interactions, exhibits inferior mechanical properties in propellants formulations.9
To overcome these problems, GAP-based copolymer binders which were prepared via the incorporation of flexible-structural polymer (HTPB, polyethylene glycol (PEG) or polycaprolactone (PCL)) into GAP are widely developed to improve their mechanical behavior.10,11 Byoung Sun M. prepared plasticized GAP-based copolymer binders through introduction of PEG and PCL to improve the flexibility of polymer backbone, the results indicated that the mechanical properties of the GAP-based propellant were considerably improved.12 Recently, GAP based prepolymers which were prepared through the cationic copolymerization with monomers such as tetrahydrofuran, ethylene oxide, or other vinyl monomers giving flexibility to the polymer backbone have attracted growing interest.13,14 The copolymerization of additional monomer will improve the chain flexibility and intermolecular interactions of GAP polymer backbone. Accordingly, the mechanical properties of GAP based binders can be enhanced.15
In the past decades, fluorinated polymers have garnered increasing attention as high explosive binders in the energetic material community, owing to their long-term chemical stability, low coefficients of friction, high densities, compatibilities with the energetic materials, and broad operating temperature ranges.16,17 Moreover, fluoropolymer–metal compositions can give specific high reaction energy.18 For example, magnesium, Teflon, and Viton system (MTV) was reported to give an especially large specific reaction energy of 9.4 kJ g−1, in comparison with TNT and RDX yield just 3.72 kJ g−1 and 6.569 kJ g−1.19 Therefore, synthesis of fluorinated GAP via copolymerization of fluorinated monomers may be a promising method to improve its application property.
In this work, (2,2,2-trifluoro-ethoxymethyl epoxy-r-glycidyl azide) copolymer (poly(TFEE-r-GA)) was synthesized by a combination of cationic copolymerization of 1,1,1-trifluoro-2,3-epoxypropane (TFEE) and epichlorohydrin (ECH) using butane diol (BDO) as the initiator and boron trifluoride etherate (BF3·OEt2) as the catalyst, and followed by azidation with sodium azide and N,N-dimethylformamide (DMF) as solvents. The chemical structure, molecular weight and thermal properties were characterized by fourier transform infrared (FTIR), NMR, gel permeation chromatography (GPC), differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA), respectively. The reaction energy of poly(TFEE-r-GA)/Al mixture was investigated by cook-off test. The copolyurethane networks was prepared using poly(TFEE-r-GA) as prepolymer, trimethylolpropane (TMP) as chain extender agent, and isophorone diisocyanate (IPDI) as cross-linking agent. The mechanical properties of the copolyurethane networks were described by tensile test and scanning electron microscopy (SEM).
2. Experimental
2.1 Materials
2,2,2-Trifluoroethanol, TMP, IPDI were purchased from J&K scientific Ltd. (Shanghai). ECH, sodium azide, DMF, dichloromethane (DCM), 1,2-dichloroethane, BDO, BF3·OEt2 and dibutyltin dilaurate (DBTDL) were supplied by Chengdu Kelong Chemical Reagents Company. BDO and BF3·OEt2 were distilled under reduced pressure prior to use. All solvents for the reactions were analytical grade and were dried before use.
2.2 Characterization
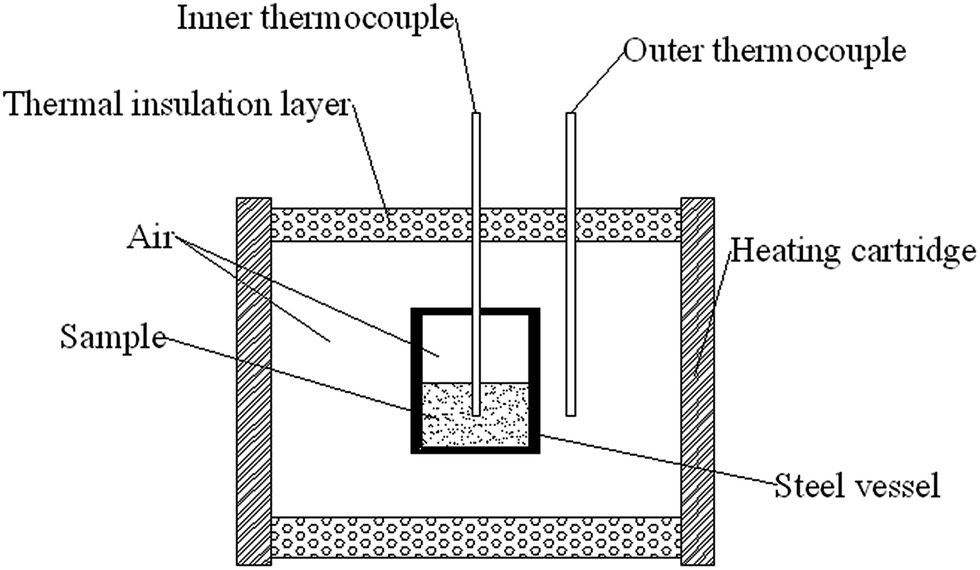
FTIR spectra were collected using a bruker tensor 27 FTIR spectrometer (KBr pellet) in the spectral range of 4000–400 cm−1 with 64 scans at a spectral resolution of 4 cm−1. 1H NMR and 13C-NMR spectra were recorded in a Bruker 500 MHz instrument in deuterated chloroform. GPC was conducted on a Waters GPC, tetrahydrofuran was used as the mobile phase and the calibration was carried out with polytetrahydrofuran standards. The viscosity of polymers was measured using a Brookfield viscometer (Cap 2000+) at 25 °C. DSC equipped with a TA instruments DSC Q1000 and TGA equipped with a SDT Q600 TGA instrument (TA Instruments) were used to thermally characterize the samples in two experiments: 25–550 °C and −100–50 °C at a heating rates of 10 °C min−1 under nitrogen atmosphere at 20 mL min−1. The sample weight was 8–10 mg. The glass transition temperature (Tg) was reported as the midpoint temperature of the baseline shift measured during the transition. A method of drop hammer impact sensitivity test for energetic polymer was developed. The weight of the hammer was 3 kg and the drop height was between 0–1.29 m. A schematic geometry of equipment for slow cook-off test used in the research was shown in Scheme 1. In the experiment, 10 g poly(TFEE-r-GA)/10 g Al mixture or 10 g GAP/10 g Al mixture were charged into steel vessel, and the system was heated by cartridge wall. The heating rate of the electric heating cord was set at 1 °C min−1, and the temperature range from the room temperature to 240 °C. Temperature changes at the center of heating cartridge wall and inner of equal heating sample components were recorded by sheathed thermocouples. Their temperature vs. time curves were obtained to investigate the reaction energy of polymer/Al mixtures. Mechanical properties of all the GAP films were measured on a Shimadzu AG-X Plus universal testing machine (Shimadzu, Japan) with a tensile rate of 50 mm min−1. The copolyurethane networks were cut into dumbbell-shaped specimens (dimensions: 35 mm × 6 mm × 2 mm) and kept at 0% humidity for 7 days before measurement. A mean value of five replicates from each film was taken. SEM observation was carried out on a VEGA 3 LMU scanning electron microscope (TESCAN, Czech Republic). All the copolyurethane networks films were frozen in liquid nitrogen and then snapped immediately. The fracture surfaces of the fractured films were sputtered with gold and then photographed.
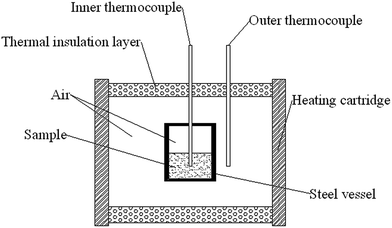 |
| | Scheme 1 Schematic geometry of cook-off test. | |
2.3 Synthesis of 2,2,2-trifluoro-ethoxymethyl epoxy (TFEE)
TFEE was first synthesized via the reaction of ECH, 2,2,2-trifluoroethanol, sodium hydroxide and a catalytic amount of water as described in Scheme 2. 2,2,2-Trifluoroethanol (30 g, 0.3 mol), ECH (138.4 g, 1.5 mol), sodium hydroxide (12 g, 0.3 mol) and a catalytic amount of water (0.4 mL) were charged into a dried 250 mL three-necked round-bottom flask equipped with a mechanical stirrer and condenser. The reaction mixture was stirred at 70 °C for 7 h. After the reaction, the impure product was distilled under reduced pressure and the fraction at the boiling point of 117 °C was redistilled using a Fischer–Spaltrohr concentric tube distillation apparatus. The TFEE was obtained by distillation and the fraction of 123–124 °C was collected and the product was shown to be a single component by GC analysis (37 g, yield, 80%).
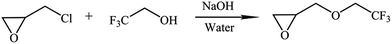 |
| | Scheme 2 Synthesis route of TFEE. | |
1H NMR (500 MHz, CDCl3, δ): 2.63 and 2.82 (s, 2H, CH2 in ring), 3.18 (s, 1H, CH), 3.53 and 3.92 (s, 2H, CH2), 3.96 (s, 2H, CH2–CF3); 13C NMR (500 MHz, CDCl3, δ): 43.66 (CH2 in ring), 50.36 (CH), 68.38 (CH2), 72.72 (CH2–CF3), 120.5–127.2 (CF3); 19F NMR(500 MHz, CDCl3, δ): −74.4 (CF3).
2.4 Copolymerization of poly(TFEE-r-GA)
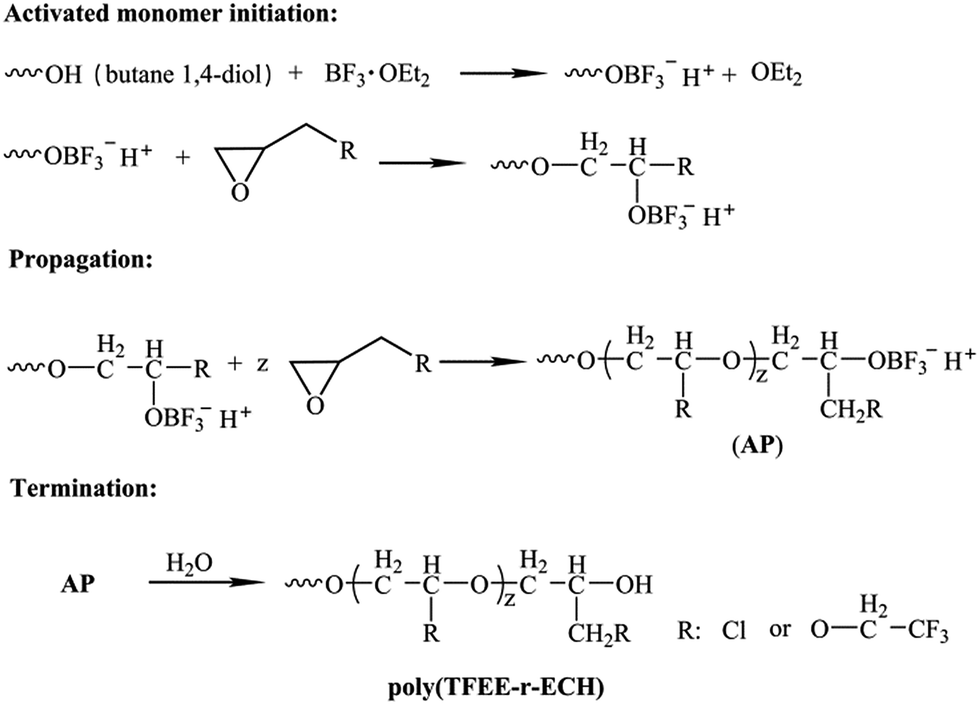
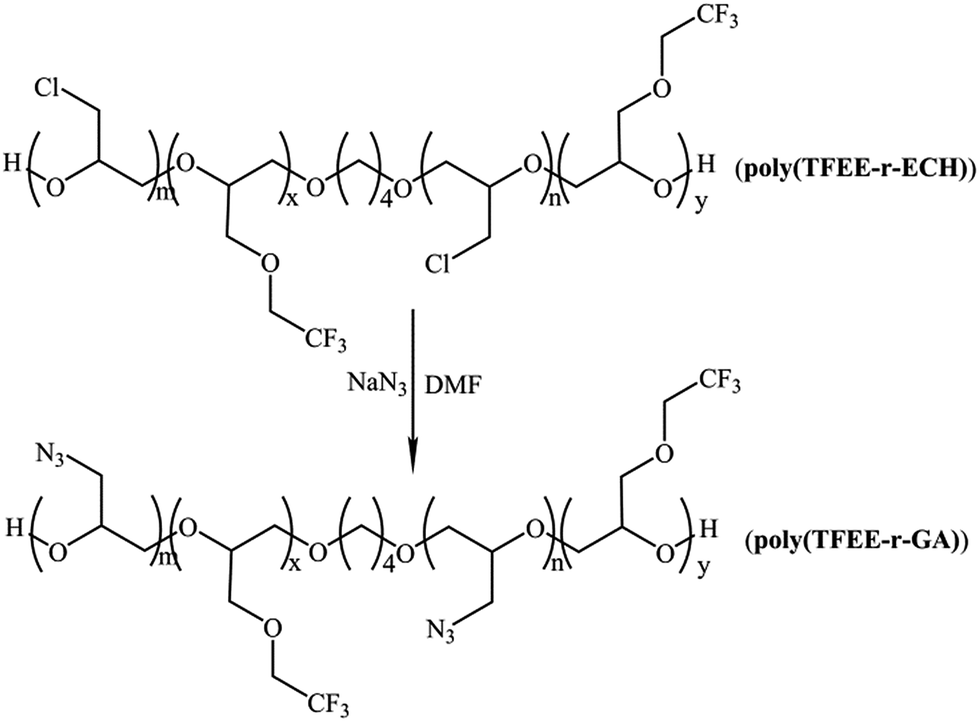
The poly(TFEE-r-GA) copolymer was synthesized in two steps as described in Schemes 3 and 4. First step was the synthesis of (2,2,2-trifluoro-ethoxymethyl epoxy-r-epichlorohydrin) copolymer (poly(TFEE-r-ECH)) through cationic ring-opening copolymerization. Second step was the conversion of poly(TFEE-r-ECH) to poly(TFEE-r-GA) copolymer by azidation. The steps involved in polymer synthesis are described below.
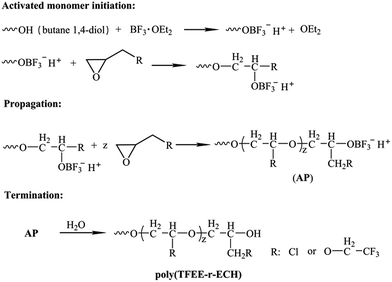 |
| | Scheme 3 Synthesis route of poly(TFEE-r-ECH) via cationic polymerization. | |
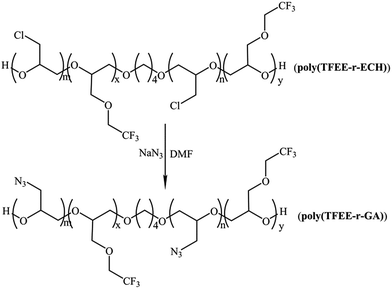 |
| | Scheme 4 Synthesis of poly(TFEE-r-GA) by azidation. | |
Step 1. BDO (1.80 g, 0.02 mmol) was dissolved in freshly distilled DCM (50 mL) in a three necked flask fitted with a thermometer, BF3·OEt2 (0.852 g, 0.006 mol) was injected into the reaction mixture and stirred at room temperature for 30 min. After cooling the reaction vessel to 0 °C using ice-salt mixture, a solution of ECH (44.4 g, 0.48 mol) and TFEE (18.72 g, 0.12 mol) was added drop by drop (at the rate of 0.1 mL min−1) over a period of 8 h. After addition of the monomers, the reaction was allowed to come to room temperature and left to react for another 24 h. And then, the reaction was halted by the addition of 1% of sodium bicarbonate solution (50 mL). The organic phase was separated and washed with distilled water, and then was removed by vacuum evaporation. 60.2 g of yellowish poly(TFEE-r-ECH) was obtained after dried under vacuum at 30 °C (91.7% yield).
Step 2. Poly(TFEE-r-ECH) (40 g) was reacted with molar excess of sodium azide (25 g) in 80 mL of DMF in a three necked flask. The reaction was then heated to 95 °C and left under stirring for 36 h. Thereafter, the reaction was stopped by the addition of distilled water under stirring. The organic phase was extracted into DCM and washed with distilled water for three times. The organic layer was separated, dried with sodium sulfate, and concentrated by vacuum evaporation to acquire the poly(TFEE-r-GA) (91.7% yield).
2.5 Preparation of copolyurethane networks
Poly(TFEE-r-GA)-based polyurethane networks were prepared via mixing poly(TFEE-r-GA) copolymer, TMP and IPDI at a NCO/OH ratio of 1.0, the networks prepared from GAP at same conditions were used as control. A typical synthesis procedure was as follows: a given amount poly(TFEE-r-GA) copolymer was first taken in a three necked flask fitted with a thermometer and nitrogen inlet. After addition of TMP, IPDI and catalyst in an amount calculated to have the desired NCO/OH ratio, the solution was allowed to mix for another 20 min in a vacuum for degassing. Thereafter, the mixture was transferred to a Teflon-coated mold, and left to react 7 days at 60 °C. The obtained copolyurethane networks were cut into dumbbell-shaped specimens to measure the mechanical properties using the universal testing machine and SEM.
3. Results and discussion
3.1 Preparation and structure of poly(TFEE-r-GA)
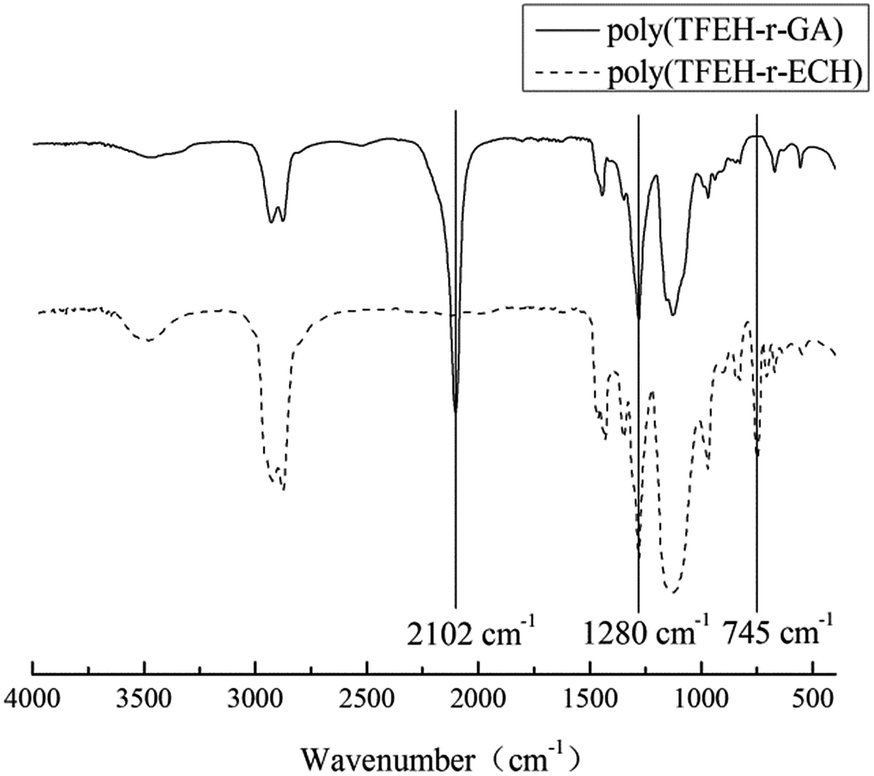
As stated above, the synthesis of the poly(TFEE-r-GA) was done by cationic polymerization of ECH and TFEE, using BDO as an initiator and BF3·OEt2 as a catalyst, followed by azidation with sodium azide in DMF medium. The reaction steps were outlined in Schemes 2–4. The structures of poly(TFEE-r-GA) were unambiguously assigned based on their FTIR, 1H NMR and 13C NMR spectra. The FTIR spectra (Fig. 1) showed changes in the chemical structures of poly(TFEE-r-ECH) and poly(TFEE-r-GA). As shown in Fig. 1A, the characteristic adsorption peak at 3480 cm−1 explained the O–H stretching vibration of poly(TFEE-r-ECH), and the bands around 2923 and 2873 cm−1 accounted for the C–H symmetric and antisymmetric stretching vibration of poly(TFEE-r-ECH). A strong peak at 1127 cm−1 informed about the C–O group present in poly(TFEE-r-ECH), the strong peak at 1280 cm−1 were attribute to the CF3,20,21 and the bands at 745 cm−1 in Fig. 1A were attributed to the C–Cl symmetric stretching vibrations.22,23 When compared with the poly(TFEE-r-ECH) (Fig. 1A), a disappearance of the C–Cl stretching band in FTIR spectra at 745 cm−1 and appearance of a new stretching band for C–N3 at 2102 cm−1 was shown in Fig. 1B, this resulted from the reaction of azidation.24
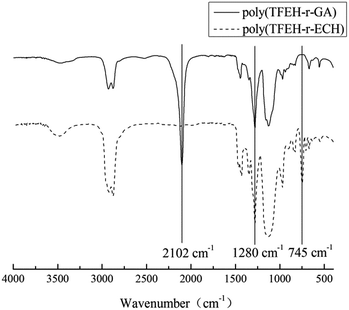 |
| | Fig. 1 FTIR spectra of (A) poly(TFEE-r-ECH) and (B) poly(TFEE-r-GA). | |
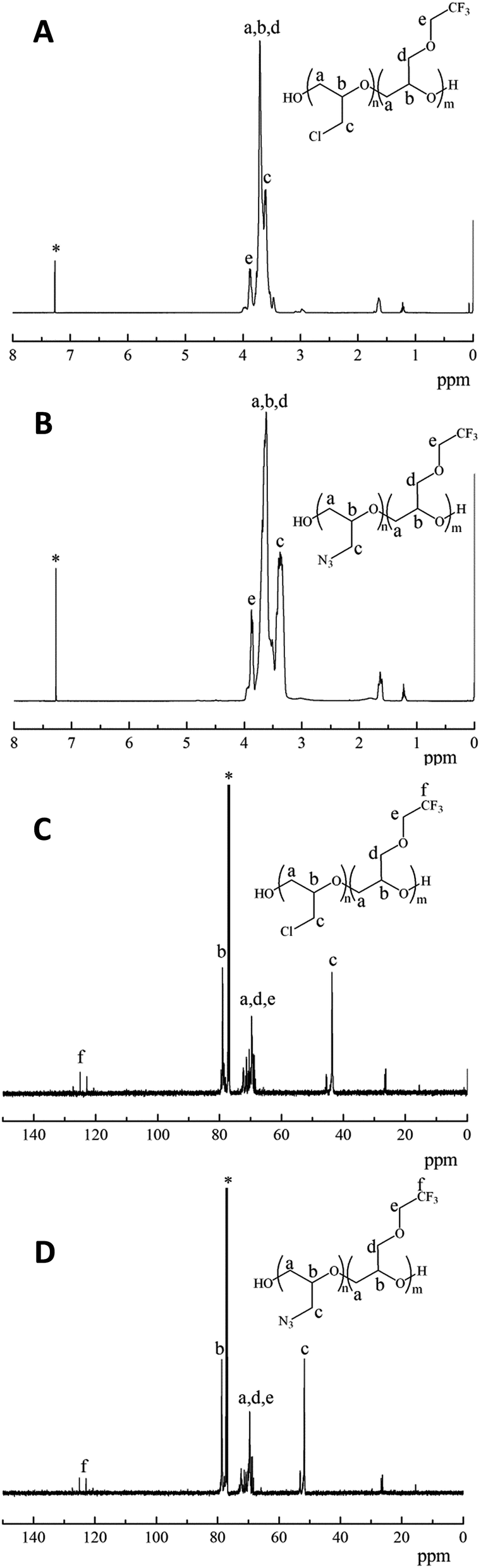
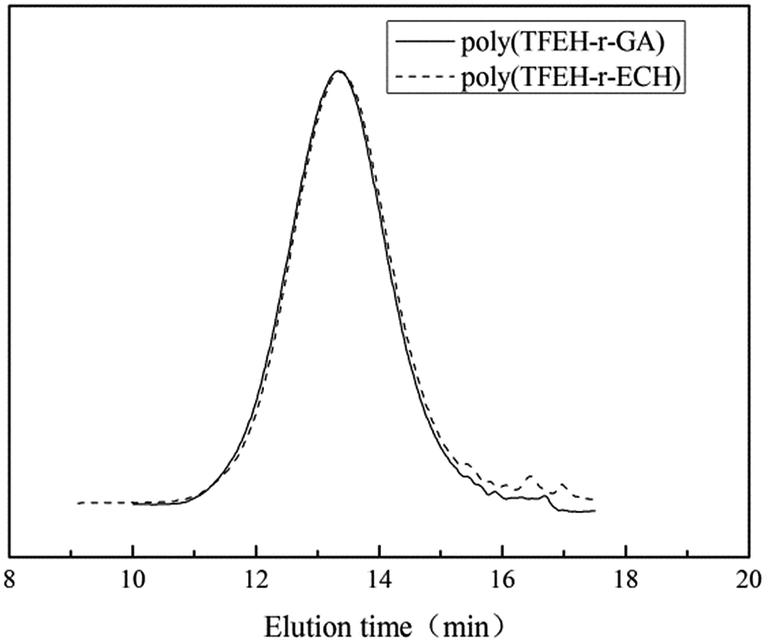
The structures of poly(TFEE-r-ECH) and poly(TFEE-r-GA) were also characterized by 1H-NMR and 13C NMR spectra as presented in Fig. 2. As shown in Fig. 2A, the signals observed at 3.61 ppm were ascribed to the methylene protons (denoted c) of the side chain. The signals at 3.7–3.9 ppm were due to the methine (denoted b) and methylene protons (denoted a, d, e) of the main chain and other side chain. After azidation, the peak of the methylene protons of chloromethyl groups at 3.61 ppm disappeared and a new signal observed at 3.37 ppm belonged to the resonance of the methylene protons (denoted c) of azidomethyl groups, as shown in Fig. 2B. The azidation reaction of poly(TFEE-r-ECH) and sodium azide forming poly(TFEE-r-GA) was also confirmed by their 13C NMR spectra. As shown in Fig. 2C, the resonances of the methylene carbons (denoted a, d, e) and methine carbons (denoted b) in poly(TFEE-r-ECH) main chain were presented at 68–72 ppm and 79 ppm respectively. The signals for the carbons in the chloromethyl group appeared at 43.5 ppm, and the signals for the carbons in the CF3 appeared at 120–127 ppm.25,26 After azidation, the resonance of azidomethyl carbon (Fig. 2D, denoted c) appeared in further downfield (51.6 ppm) than the corresponding signals for the chloromethylic carbon of CH2Cl (Fig. 2C, denoted c).27 Therefore, the 1H NMR and 13C NMR spectra of poly(TFEE-r-ECH) and poly(TFEE-r-GA) strictly corroborated the FTIR analyses that the reaction of poly(TFEE-r-ECH) with sodium azide had occurred by forming azide and all of the chlorine atoms were substituted by azide groups in the structure of poly(TFEE-r-GA). Moreover, as showed in Fig. 3, it could be observed that the average molecular weight of poly(TFEE-r-ECH) increased from 2732 g mol−1 (PD = 1.45) to 2876 g mol−1 (PD = 1.47) after azidation. The GPC result was lower than the theoretical molecular weight of poly(TFEE-r-ECH) (around 3000 g mol−1), it may attribute to the unexpected transfer and termination processes involved in the copolymerization.28 These results confirmed the successful synthesis of the poly(TFEE-r-GA).
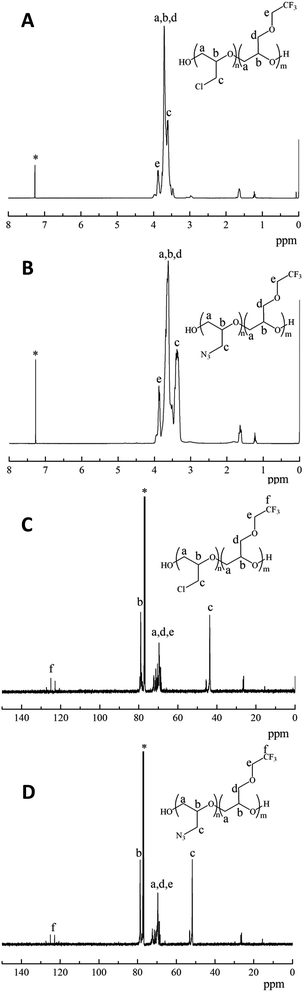 |
| | Fig. 2 1H NMR spectra of (A) poly(TFEE-r-ECH), (B) poly(TFEE-r-GA), 13C NMR spectra of (C) poly(TFEE-r-ECH), (D) poly(TFEE-r-GA). | |
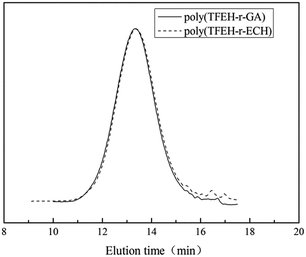 |
| | Fig. 3 GPC elution curves of poly(TFEE-r-ECH) and poly(TFEE-r-GA). | |
3.2 Density, viscosity and sensitivity
The density of poly(TFEE-r-GA) was determined and compared with GAP. The density of poly(TFEE-r-GA) is 1.36 cm−3, which was higher than pure GAP (1.28 g cm−3). It was reported that the introduction of CF3 groups increases the density of polymer, which was also seen in our current work.29 The viscosity of poly(TFEE-r-GA) at 25 °C is 2.66 Pa s in comparison to the GAP based system which had 11.9 Pa s. It attributed to the fluoropolymers possess low coefficients of friction,30,31 and the copolymerization of TFEE would be effective to decrease the viscosity of the GAP which was advantageous for machinability. For safe handling of the copolymers, the sensitivity of the copolymers was also measured by the drop weight test. In comparison to pure GAP (H50 = 42.2 cm), poly(TFEE-r-GA) showed significantly less sensitive (H50 > 129 cm). It may attribute to poly(TFEE-r-GA) containing lower azide group density than GAP, and it can be handled with the common safety precautions.
3.3 Glass transition temperature
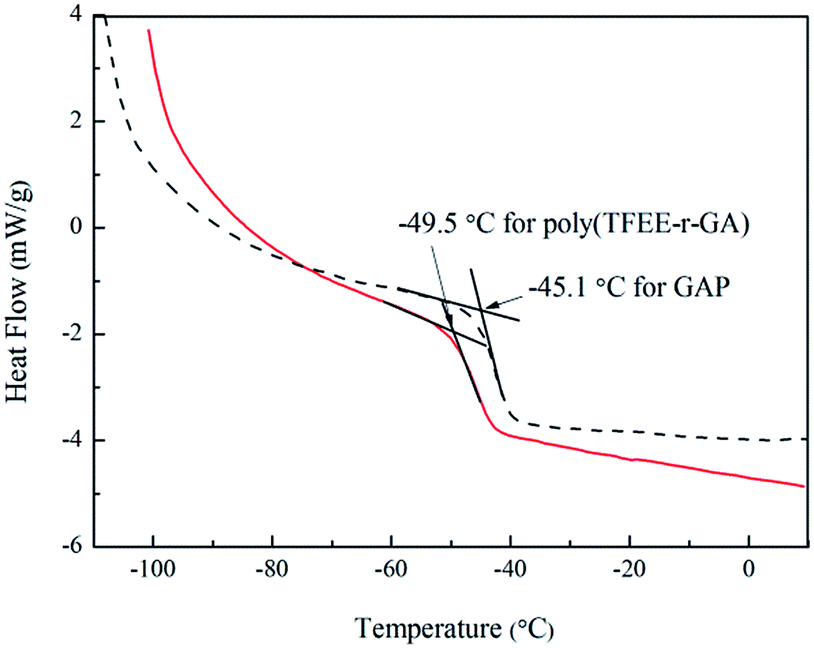
The glass transition temperature (Tg) is an important property of polymers because it determines the processing and working temperature range.32 The Tg of poly(TFEE-r-GA) was measured by second heating DSC of the poly(TFEE-r-GA), as shown in Fig. 4. In comparison of pure GAP (Tg = −45.1 °C), the DSC scan of poly(TFEE-r-GA) showed a lower Tg of −49.5 °C, which is useful and benefits for low temperature properties. It may attribute to the internal plasticisation among polymer caused by the –CH2OCH2CF3 group, and the improvement of chain flexibility of GAP via the copolymerization.33
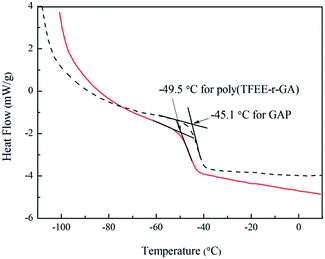 |
| | Fig. 4 DSC curves of poly(TFEE-r-GA) and GAP. | |
3.4 Thermal decomposition of poly(TFEE-r-GA)
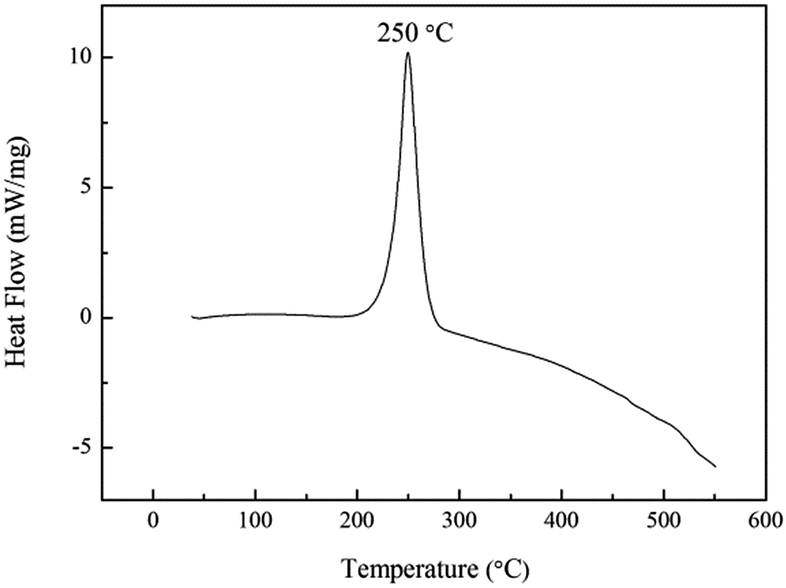
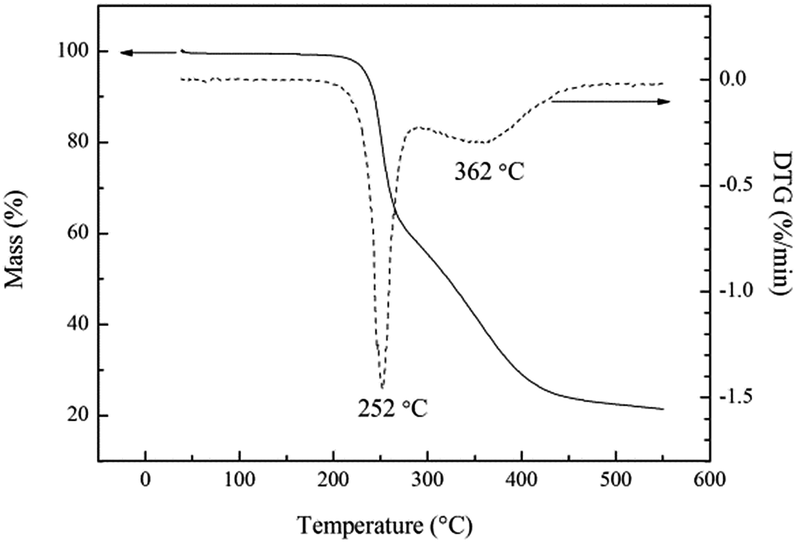
The thermal stability of energetic materials plays an important role in their preparation, processing, storage, and application. Hence, the thermal decomposition behaviors of poly(TFEE-r-GA) were studied using DSC and TGA methods. As shown in Fig. 5, the DSC curve of poly(TFEE-r-GA) presented a main single exothermic event with maximum at 250 °C. This phenomenon may account for the decomposition of the azide groups to give nitrogen molecules.34 TGA is one of the commonly used techniques for rapid evaluation of the thermal stability of polymer materials. It was carried out to investigate the thermal stability, decomposition temperature of poly(TFEE-r-GA). The TGA and derivative thermogravimetry (DTG) curves of poly(TFEE-r-GA) are shown in Fig. 6. The TGA thermogram of the poly(TFEE-r-GA) displays two distinct regions of weight loss. The first weight loss temperature occurred at 252 °C with a sharp weight loss of around 40–45% with respect to the total, corresponded to the decomposition of the azide groups of the side chain for nitrogen releasing.
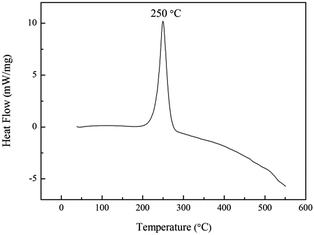 |
| | Fig. 5 DSC curve of poly(TFEE-r-GA). | |
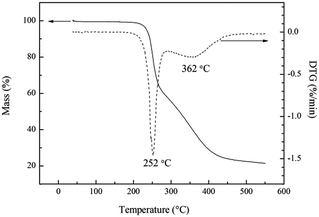 |
| | Fig. 6 TGA/DTG curves of poly(TFEE-r-GA). | |
The second main weight loss occurred at 362 °C with a gradual weight loss, observed in Fig. 6, was due to the main-chain thermal decomposition of poly(TFEE-r-GA).35,36 Therefore, both DSC and TGA results gave strong evidence that poly(TFEE-r-GA) had satisfactory thermal stability.
3.5 Cook-off test of poly(TFEE-r-GA)/Al
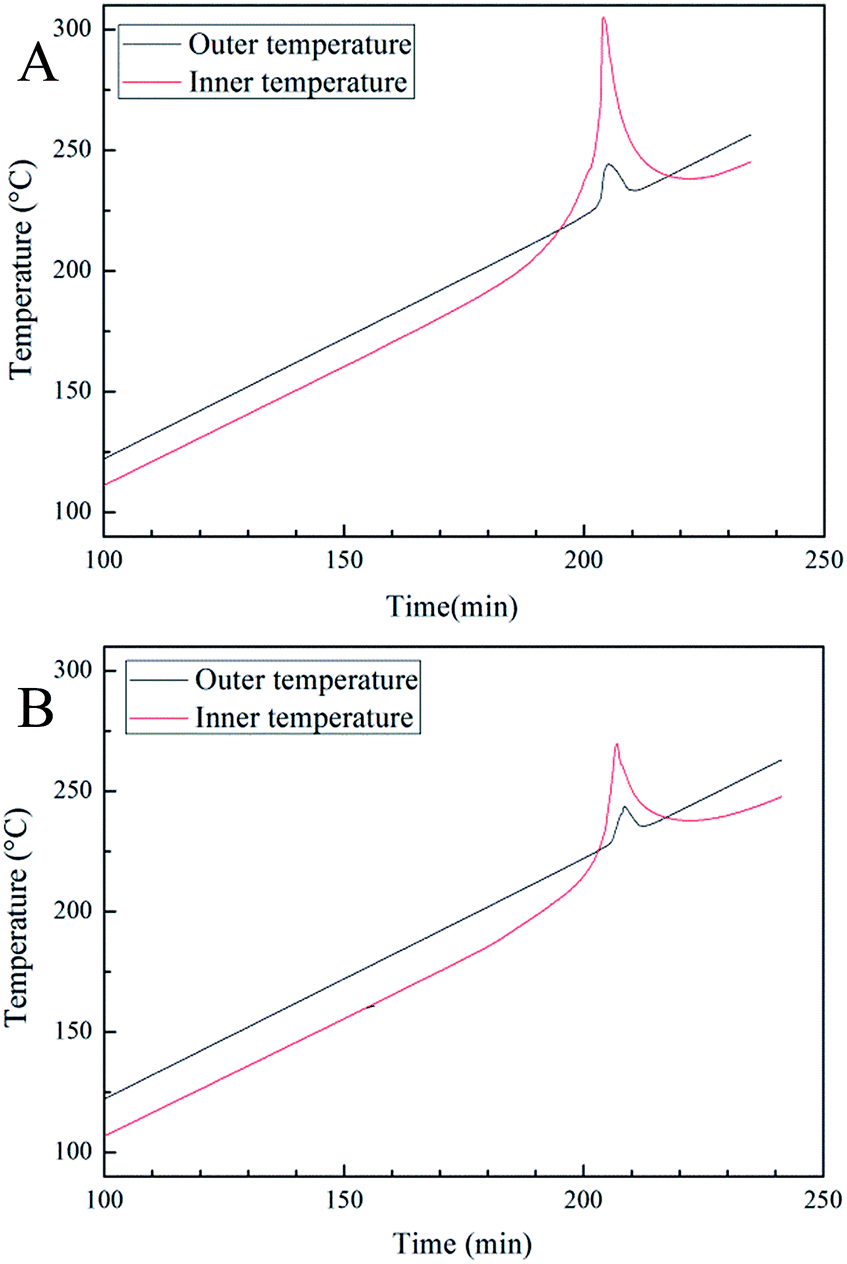
The cook-off experiments were used to investigate the reactively of poly(TFEE-r-GA) and Al. Generally, the integral of inner and outer temperature curves reflect the reaction heat of sample (Qr = Cs·ΔT, Qr: reaction heat, Cs: heat capacity of the system, ΔT: temperature variation of the system). As shown in Fig. 7, the integral of poly(TFEE-r-GA)/Al mixture was approximately 1.5 times higher than the integral of GAP/Al mixture. The results implied that the poly(TFEE-r-GA)/Al mixture may give a larger reaction energy than GAP/Al mixture.
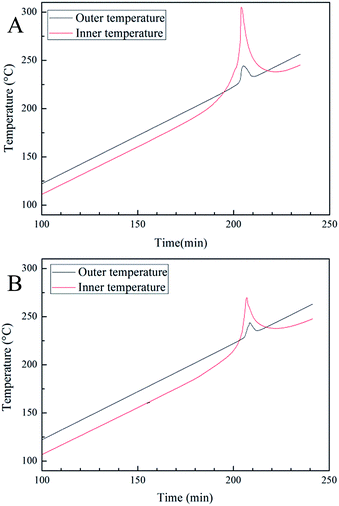 |
| | Fig. 7 Cook-off curves of (A) poly(TFEE-r-GA)/Al mixture and GAP/Al mixture (B). | |
3.6 Mechanical properties of copolyurethane networks
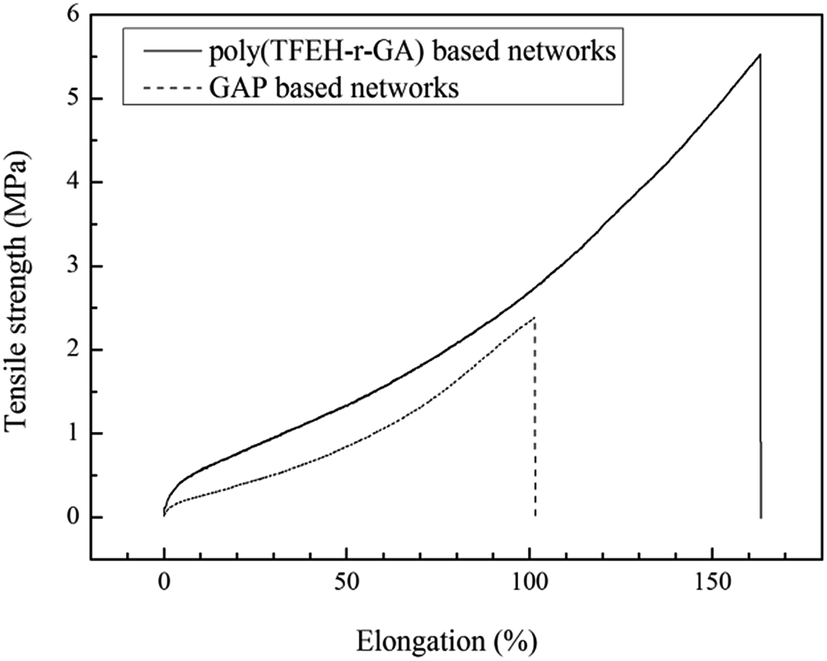
The mechanical properties of poly(TFEE-r-GA) based copolyurethane networks at 25 °C were investigated by static tensile tests and the stress–strain curves were shown in Fig. 8, networks prepared from GAP were used as control. It was worth of noting that, comparing with pure GAP based networks (2.4 MPa in tensile strength, 101% in elongation at break), the poly(TFEE-r-GA) based copolyurethane networks exhibited superior mechanical properties of similar formulation, which both had a higher tensile strength of 5.52 MPa and an elongation at break of 162.8%. It is known that GAP based polymeric binder possesses inferior mechanical properties owing to its plenty of azide groups grafted on the polymer backbone resulting in bad polymer backbone flexibility and poor intermolecular interaction.37,38 In this study, compared with pure GAP, poly(TFEE-r-GA) which prepared from copolymerization had better flexibility of main chains resulting in better intermolecular interaction of polymer. Thus, the poly(TFEE-r-GA) based copolyurethane networks exhibited the improvement of mechanical behaviors. This clearly revealed that the synthesis a flexible structural GAP based copolymer to overcome the poor mechanical properties may be a promising solution.
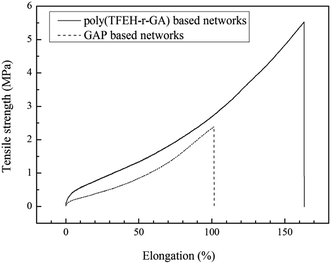 |
| | Fig. 8 Tensile testing of polyurethane networks prepared from poly(TFEE-r-GA) and GAP. | |
3.7 Fracture morphologies of the poly(TFEE-r-GA) based polyurethane networks
To investigate the difference of the mechanical properties between the gels prepared from GAP and poly(TFEE-r-GA), the fracture morphologies of gels were studied by SEM. Fig. 8 showed the SEM images of the gels prepared from GAP (A, B) and poly(TFEE-r-GA) (C, D). As shown in Fig. 9, in comparison with GAP based gel, the fractured stripes of the gel prepared from poly(TFEE-r-GA) showed few and scattered while the fractured surface gradually became smooth. Furthermore, micrographs have also revealed no obvious two-phase structure observed in the poly(TFEE-r-GA) composites. It indicated that GAP and TFEE have no great-scale phase separation, have a good miscibility and compatibility.39
 |
| | Fig. 9 SEM images for the fracture surface of the films prepared from (A, B) poly(TFEE-r-GA) and (C, D) GAP. | |
4. Conclusions
A novel random copolymer, poly(TFEE-r-GA), was synthesized to produce a material for potential application as energetic binder for solid rocket propellant. The synthetic route started with the synthesis of the halogenated polymeric precursor and then by azidation. The structures of these compounds were confirmed by FTIR, 1H NMR, 13C NMR and GPC, and their thermal stability was estimated by DSC and TGA. The DSC and TGA-DTG curves indicated that poly(TFEE-r-GA) had excellent resistance to thermal decomposition up to 200 °C and began to decompose gradually at about 230 °C. Moreover, sensitivity test results showed that poly(TFEE-r-GA) was an insensitive material. Cook-off test results indicated that the poly(TFEE-r-GA)/Al mixture may give a larger reaction energy than GAP/Al mixture. The copolyurethane networks were synthesized by cross-linking poly(TFEE-r-GA) using TMP as chain extender agent and IPDI as cross-linking agent. The copolyurethane networks exhibited the highest tensile strength of 5.52 MPa, and the corresponding elongation at break of 162.8%. These results indicated that poly(TFEE-r-GA) exhibited a good stability and might serve as a potential energetic binder in propellant formulations.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support from the China Postdoctoral Science Foundation (2016M592851).
Notes and references
- J. K. Chen and T. B. Brill, Combust. Flame, 1991, 87, 217–232 CrossRef CAS.
- R. I. Caro and J. M. Bellerby, Int. J. Energ. Mater. Chem. Propul., 2008, 7, 171–185 Search PubMed.
- D. M. Badgujar, M. B. Talawar, S. N. Asthana and P. P. Mahulikar, J. Hazard. Mater., 2008, 151, 289–305 CrossRef CAS PubMed.
- B. Gaur, B. Lochab, V. Choudhary and I. K. Varma, J. Macromol. Sci., Part C: Polym. Rev., 2003, 43, 505–545 CrossRef.
- T. M. Klapoetke and S. M. Sproll, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 122–127 CrossRef CAS.
- H. F. R. Schoyer, A. J. Schnorhk, P. Korting, P. J. Vanlit, J. M. Mul, M. Gadiot and J. J. Meulenbrugge, J. Propul. Power, 1995, 11, 856–869 CrossRef CAS.
- K. Selim, S. Ozkar and L. Yilmaz, J. Appl. Polym. Sci., 2000, 77, 538–546 CrossRef CAS.
- T. H. Hagen, T. L. Jensen, E. Unneberg, Y. H. Stenstrom and T. E. Kristensen, Propellants, Explos., Pyrotech., 2015, 40, 275–284 CrossRef CAS.
- Y. Ding, C. Hu, X. Guo, Y. Che and J. Huang, J. Appl. Polym. Sci., 2014, 131, 40007–40015 Search PubMed.
- S. Mathew, S. K. Manu and T. L. Varghese, Propellants, Explos., Pyrotech., 2008, 33, 146–152 CrossRef CAS.
- B. S. Min, G. Baek and S. W. Ko, J. Ind. Eng. Chem., 2007, 13, 373–379 CAS.
- B. S. Min, Propellants, Explos., Pyrotech., 2008, 33, 131–138 CrossRef CAS.
- R. R. Soman, N. T. Agawane, A. Hazarika, K. J. Sharma, T. Mukundan and S. N. Asthana, J. Polym. Mater., 2003, 20, 423–433 CAS.
- Y. M. Mohan, Y. Mani and K. M. Raju, Des. Monomers Polym., 2006, 9, 201–236 CrossRef CAS.
- J. Deng, G. Li, M. Xia, Y. Lan and Y. Luo, J. Appl. Polym. Sci., 2016, 133, 43840–43847 CrossRef.
- Y. Long, Y. G. Liu, F. D. Nie and J. Chen, Colloid Polym. Sci., 2012, 290, 1855–1866 CAS.
- D. M. Dattelbaum, S. A. Sheffield, D. Stahl, M. Weinberg, C. Neel and N. Thadhani, J. Appl. Phys., 2008, 104, 113525–113537 CrossRef.
- C. D. Yarrington, S. F. Son and T. J. Foley, J. Propul. Power, 2010, 26, 734–743 CrossRef CAS.
- K. B. Rider, B. K. Little, S. B. Emery and C. M. Lindsay, Propellants, Explos., Pyrotech., 2013, 38, 433–440 CrossRef CAS.
- A. Cardone, C. Martinelli, V. Pinto, F. Babudri, M. Losurdo, G. Bruno, P. Cosma, F. Naso and G. M. Farinola, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 285–291 CrossRef CAS.
- A. P. Kharitonov, G. V. Simbirtseva, V. M. Bouznik, M. G. Chepezubov, M. Dubois, K. Guerin, A. Hamwi, H. Kharbache and F. Masin, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3559–3573 CrossRef CAS.
- A. M. Kawamoto, J. A. S. Holanda, U. Barbieri, G. Polacco, T. Keicher, H. Krause and M. Kaiser, Propellants, Explos., Pyrotech., 2008, 33, 365–372 CrossRef CAS.
- T. Cai, W. J. Yang, K. G. Neoh and E. T. Kang, Ind. Eng. Chem. Res., 2012, 51, 15962–15973 CrossRef CAS.
- S. Pisharath and H. G. Ang, Polym. Degrad. Stab., 2007, 92, 1365–1377 CrossRef CAS.
- X. Wang, J. Hu, Y. Li, J. Zhang and Y. Ding, J. Fluorine Chem., 2015, 176, 14–19 CrossRef CAS.
- D. V. Vorobyeva, A. S. Peregudov, G. V. Roeschenthaler and S. N. Osipov, J. Fluorine Chem., 2015, 175, 60–67 CrossRef CAS.
- V. Vasudevan and G. Sundararajan, Propellants, Explos., Pyrotech., 1999, 24, 295–300 CrossRef CAS.
- D. Hemant, PhD thesis, Aston University, 1995.
- R. Sarangapani, S. T. Reddy and A. K. Sikder, J. Mol. Graphics Modell., 2015, 57, 114–121 CrossRef CAS PubMed.
- N. K. Bourne and G. T. Gray, J. Appl. Phys., 2003, 93, 8966–8969 CrossRef CAS.
- Y. Y. Durmaz, E. L. Sahkulubey, Y. Yagci, E. Martinelli and G. Galli, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4911–4919 CrossRef CAS.
- X. Wang, B. Jin, R. Peng, Q. Zhang, W. Gong, H. Huang, S. Chu and H. Dong, J. Polym. Res., 2015, 22, 1–11 CrossRef CAS.
- A. Singh, P. K. Soni, T. Shekharam and A. Srivastava, J. Appl. Polym. Sci., 2013, 127, 1751–1757 CrossRef CAS.
- J. K. Nair, R. R. Soman, N. T. Agawane, R. S. Satpute, T. Mukundan, S. D. Kakade, M. Gupta and S. N. Asthana, J. Polym. Mater., 2005, 22, 87–95 CAS.
- Y. M. Mohan and K. M. Raju, Des. Monomers Polym., 2005, 8, 159–175 CrossRef CAS.
- K. Ghosh, J. Athar, S. Pawar, B. G. Polke and A. K. Sikder, J. Energ. Mater., 2012, 30, 107–123 CrossRef CAS.
- C. Hu, X. Guo, Y. Jing, J. Chen, C. Zhang and J. Huang, J. Appl. Polym. Sci., 2014, 131, 40636–40643 CrossRef.
- J. D. Yeager, A. M. Dattelbaum, E. B. Orler, D. F. Bahr and D. M. Dattelbaum, J. Colloid Interface Sci., 2010, 352, 535–541 CrossRef CAS PubMed.
- K. Malkappa and T. Jana, Ind. Eng. Chem. Res., 2013, 52, 12887–12896 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08929e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Zhongxue Geb,
Xianming Lub,
Hongchang Mob,
Yueping Jib and
Huaiming Huc
*ab,
Zhongxue Geb,
Xianming Lub,
Hongchang Mob,
Yueping Jib and
Huaiming Huc