
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical insights into the reaction of Cp*(Cl)Hf(diene) with isonitriles†
Ming-Ran Du‡
a,
Xiang-Biao Zhang *a,
Sheng-Meng Si‡a and
Lei Wang*b
*a,
Sheng-Meng Si‡a and
Lei Wang*b
aSchool of Chemical Engineering, Anhui University of Science and Technology, Huainan, Anhui 232001, People's Republic of China. E-mail: xbzhang_theochem@yahoo.com
bDepartment of Chemistry, Huaibei Normal University, Huaibei, Anhui 235000, People's Republic of China. E-mail: leiwang@chnu.edu.cn
First published on 20th September 2017
Abstract
The migratory insertion of isonitriles into a metal–C bond is a potentially important method for C–C bond construction in organic and pharmaceutical syntheses. In this context, the reaction mechanism of Cp*(Cl)Hf(diene) (Cp* = pentamethylcyclopentadienyl) with isonitriles was studied using density functional theory calculations. Hf-imido complexes and α-methylene cyclopentenimines are the confirmed products for N-tert-butyl- or N-1-adamantyl-substituted isonitriles. They are also the thermodynamically favored products of N-2,6-dimethylphenyl (Ar)-substituted isonitriles. The β-H elimination reaction pathway is responsible for the formation of α-methylene cyclopentenimines. Its elementary reactions include the isomerization of Cp*(Cl)Hf(diene), migratory insertion of the first isonitrile into the Hf–C bond, C–C reductive elimination, β-H elimination, migratory insertion of the second isonitrile into the Hf–H bond, C–C reductive coupling, addition of a Hf–H bond, and fragmentation of the six-membered hafnacycle. For methyl-, ethyl-, and Ar-substituted isonitriles, the kinetically favored products are diazahafnacyclopentanes (“σ complexes”). These are formed via an isomerization reaction pathway that comprises isomerization of the Hf complex, C–C reductive coupling, and insertion of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. The effects of different substituents on the isonitrile nitrogen on the main elementary reactions are discussed.
N bond. The effects of different substituents on the isonitrile nitrogen on the main elementary reactions are discussed.
Introduction
Hf complexes have attracted considerable attention from chemists owing to their unique reactivities in alkene/olefin polymerization and copolymerization,1–4 Friedel–Crafts reactions,5 activation of small molecules such as N2 and CO2,6,7 ring-opening polymerization of cyclic esters,8 epoxidation,9 and other important organic reactions.10–14 Chirik et al. prepared a base-free dihafnocene μ-nitrido complex via CO-induced dinitrogen cleavage. They proved that this complex could serve as a versatile platform to construct N–C bonds via reactions with activated alkynes, organonitriles, CO2, isocyanates, Me3SiI, and alkyl triflates.6 On the other hand, [1,4]-addition of CO to enynes and dienes is very useful in synthesis.15 As potential replacement of CO, isonitriles could react with radicals, electrophiles, and nucleophiles.16–22 There are several advantages for using isonitriles over CO. For example, isonitrile reactions do not require high pressures, and the reactivities of isonitriles can be easily modulated by introducing different-sized electron-donating/withdrawing substituents to the isonitrile nitrogen.Currently, the migratory insertion of isonitriles into metal–C bonds is becoming a potentially important method for the construction of C–C bonds.23–26 The resulting iminoacyl and metallaaziridine complexes have also been proven to be versatile intermediates in numerous transition metal-promoted stoichiometric and catalytic transformations. Xie et al. have reported the migratory insertion of isonitriles into Ta–C bonds. They suggested that alkyl and aryl isonitriles exhibit different reactivity patterns, and that the reaction products depend on the type and stoichiometry of these isonitriles.24a,b Norton et al. described the migratory insertion of isonitriles into Zr–C bonds. They revealed the reversibility of migratory insertion of tert-butyl (tBu) isonitrile.24d The migratory insertion of isonitrile into titanacyclobutane complexes bearing two Cp* (Cp* = pentamethylcyclopentadienyl) moieties provides a pathway for the stereocontrolled synthesis of valuable organic cyclobutanimines.26f Recently, Norton et al. reported the reaction of Cp*(Cl)M(2,3-dimethylbutadiene) (M = Ti, M1a; M = Hf, 1a) with isonitriles.27 The reaction of M1a with N-tBu- and N-1-adamantyl (1-Ad)-substituted isonitriles afforded the bis-inserted titanaaziridines at room temperature. Under an elevated temperature and in the presence of pyridine, the titanaaziridine fragments formed the complex cyclic α-methylene cyclopent-3-enimines and Ti imido complexes, while the reaction of 1a with N-2,6-dimethylphenyl-substituted isonitrile (ArNC) generated a diazahafnacyclopentane with Ar groups (“σ complex”). They pointed out that group 4 metals do mediate the cycloaddition of isonitrile to 2,3-dimethylbutadiene, and the product structure depends upon the nature of the metal. However, the reaction of 1a with isonitriles in the presence of pyridine has not been investigated. Ballmann et al. proved that for the reaction of Hf complexes with isonitriles, the steric effects of different ligands influence the product formation.28
In order to expand the application of Hf complexes to C–C bond formation, a comprehensive and unambiguous understanding of the reaction pattern of 1a with isonitriles is required. In this work, density functional theory (DFT) calculations were performed for the reactions of 1a with isonitriles bearing tBu, methyl (Me), ethyl (Et), Ar, and 1-Ad groups to answer the following questions: (1) whether α-methylene cyclopentenimines and Hf imido complexes can be produced via the reaction of 1a with isonitriles in the presence of pyridine; (2) if yes, what is the detailed mechanism for their formation; (3) what is the detailed mechanism for the formation of diazahafnacyclopentanes; and (4) the effects of substituent groups on the nitrogen of isonitrile in terms of the reaction mechanisms, and how these substituent groups influence the competition between the afforded products (diazahafnacyclopentanes and α-methylene cyclopentenimines).
Computational details
All calculations were performed using the Gaussian 09 program package.29 Molecular structures were optimized by the B3LYP/BS1 method.30,31 Benzene was experimentally adopted as the reaction medium.27 Thus, benzene solvent effects were taken into account by invoking the polarizable continuum model (PCM) solvation method.32 In BS1, the Hf and Cl atoms were described by the LANL2DZ basis sets and improved with a set of f- or d-polarization functions (α = 0.784 for Hf; α = 0.640 for Cl) with effective core potentials (ECP).33–37 Other atoms were represented by the 6-311+G(d,p) basis sets, except for the atoms on the tBu, Me, Et, Ar, and 1-Ad substituent groups that were described by 6-31G basis sets.38–40 This theoretical model has proven to be reliable, affording results that were in good agreement with the experimental data [ESI†]. Additionally, frequency calculations were performed to confirm the calculated species as minima (no imaginary frequency) or transition states (only one imaginary frequency), and to provide thermodynamic corrections at 101![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 325 Pa and 298.15 K. Moreover, intrinsic reaction coordinate (IRC) calculations were carried out to ensure that the afforded transition states were correctly connected to the intended intermediates.41 Finally, more accurate energies were calculated using the 6-311G(d,p) basis sets instead of the 6-31G basis sets. Corrected Gibbs free energies were used to describe the free-energy profiles of the reaction.
325 Pa and 298.15 K. Moreover, intrinsic reaction coordinate (IRC) calculations were carried out to ensure that the afforded transition states were correctly connected to the intended intermediates.41 Finally, more accurate energies were calculated using the 6-311G(d,p) basis sets instead of the 6-31G basis sets. Corrected Gibbs free energies were used to describe the free-energy profiles of the reaction.
Results and discussion
Mechanism of the reaction of 1a with tBuNC
The proposed mechanisms for the reaction of 1a with tBuNC to access α-methylene cyclopentenimine and diazahafnacyclopentane are illustrated in Scheme 1. The reaction pathways in Scheme 1(a) were calculated first. With a free-energy barrier of 58.6 kJ mol−1 [Fig. 1(a)], 1a isomerizes to 1b via TS1a-1b; this reaction is endergonic by 25.1 kJ mol−1. While the 2,3-dimethylbutadiene ligand in 1a coordinates in a “supine” π-fashion (Fig. 2), the same ligand in 1b coordinates in a “prone” π-fashion.42 The two Hf–C(terminal) diene distances (1a, Hf–C1 = 2.25 Å, Hf–C4 = 2.25 Å; 1b, Hf–C1 = 2.24 Å, Hf–C4 = 2.24 Å) differ from the two Hf–C(internal) diene distances (1a, Hf–C2 = 2.50 Å, Hf–C3 = 2.50 Å; 1b, Hf–C2 = 2.54 Å, Hf–C3 = 2.54 Å). tBuNC coordinates to 1b with the end-on C atom to afford 2a. This makes the electronic energy of the system decrease by 21.6 kJ mol−1, and the Gibbs free energy of the system increases by 38.1 kJ mol−1 due to the loss of entropy (−171.2 J mol−1 K−1). The Hf–C(terminal) diene distances (Hf–C1 = 2.37 Å, Hf–C4 = 2.45 Å) in 2a are longer than those in 1b. The structure of 2a maintains the coordinated CC π-bond, and the two Hf–C(internal) diene distances (Hf–C2 = 2.45 Å, Hf–C3 = 2.50 Å) exhibit a stronger interaction between the Hf center and the CC π-bond than that observed in 1b. The CC double bond distance of the butadiene fragment (C3–C2 = 1.41 Å) in 2a is longer than those in 1a and 1b. Subsequent migratory insertion of tBuNC into the neighboring Hf–C bond requires 35.2 kJ mol−1 to furnish 3a with an uncoordinated N atom, and this is slightly endergonic by 2.1 kJ mol−1. As the C–C σ-bond (C5–C4 = 1.92 Å) begins to form in TS2a-3a, one Hf–C(terminal) diene distance (Hf–C4 = 2.60 Å) becomes longer. Moreover, the end-on C atom of tBuNC begins to transition from an sp hybridization to an sp2 state, accompanied by bending of the tBuNC moiety. In the “supine” oriented 3a, the CC π double bond moiety is repelled out of the Hf center. This is suggested by the Hf–C2 (2.71 Å) and Hf–C3 (3.06 Å) distances. Since tBuNC directly coordinates to 1a [Fig. 1(a)] and is subsequently inserted into the Hf–C bond to generate 3d, the system requires 112.6 kJ mol−1 of free energy. This is 14.2 kJ mol−1 larger than that required for the favored reaction pathway mentioned above. These results indicate that the isomerization of 1a to 1b is indispensable to decrease the free-energy barrier for the insertion to occur. The calculated free-energy barrier for the insertion of tBuNC into the Hf–C bond in 2a is much lower than the 64.5 kJ mol−1 required for the migratory insertion of tBuNC into the Pd(III)–C bond reported by Yan et al.23c In turn, the latter is much lower than the 102.1 kJ mol−1 required in 2b. Subsequently, the N atom coordinates to the Hf center to afford 3b, and this reaction is highly exergonic (58.6 kJ mol−1). The large exergonicity stems from the higher stability of 3b (having a 14-valence-electron configuration) when compared to that of 3a (12-valence-electron configuration). The coordination process of the N atom easily takes place and only requires a free-energy barrier <3.8 kJ mol−1. Up to this point, the migratory insertion of the first tBuNC is complete and the process is reversible. This is consistent with the reversibility observed for the insertion of tBuNC into the Zr–C bond.24d
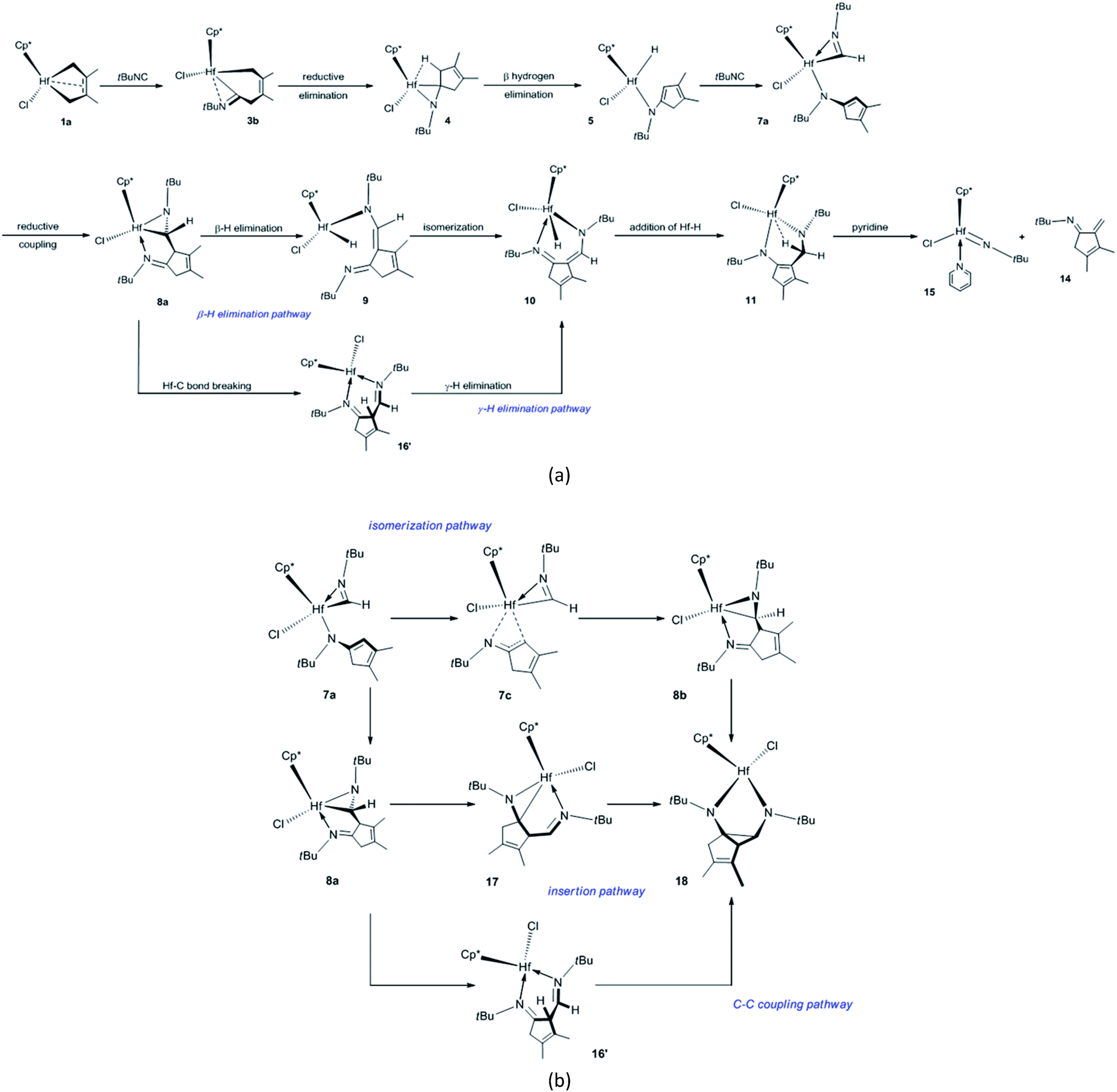 | ||
| Scheme 1 Proposed mechanisms for (a) the formation of N-tert-butyl (tBu)-substituted α-methylene cyclopentenimine and (b) formation of diazahafnacyclopentane 18. | ||
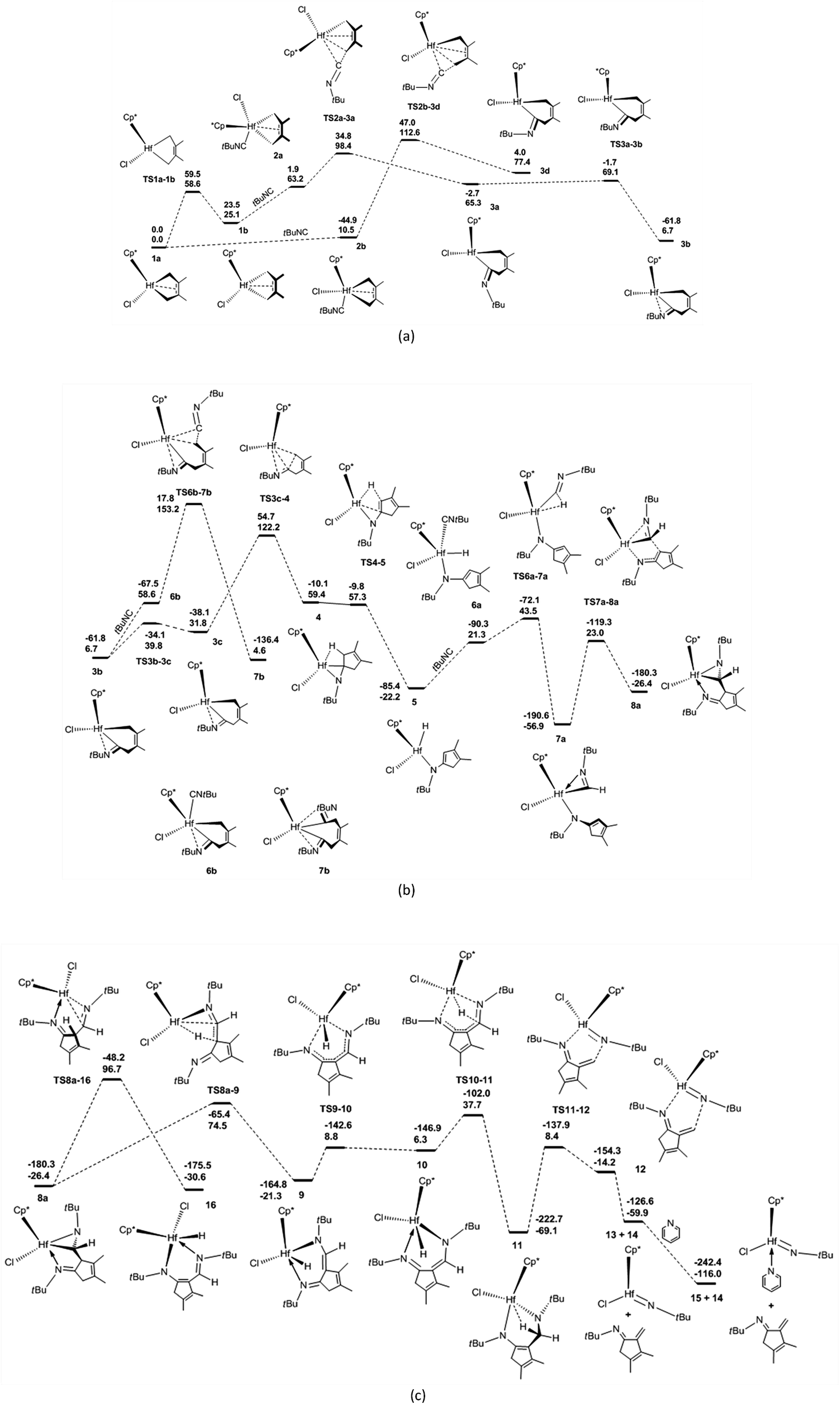 | ||
| Fig. 1 Free-energy profiles for (a) the insertion of the first tert-butyl-substituted isonitrile (tBuNC) into Hf–C bond, (b) the insertion of the second tBuNC into Hf–H bond as well as into Hf–C bond in 6b, and (c) the fragmentation of hafnaaziridine 8a. The electronic energies (above) and Gibbs free energies (below) are given in kJ mol−1. | ||
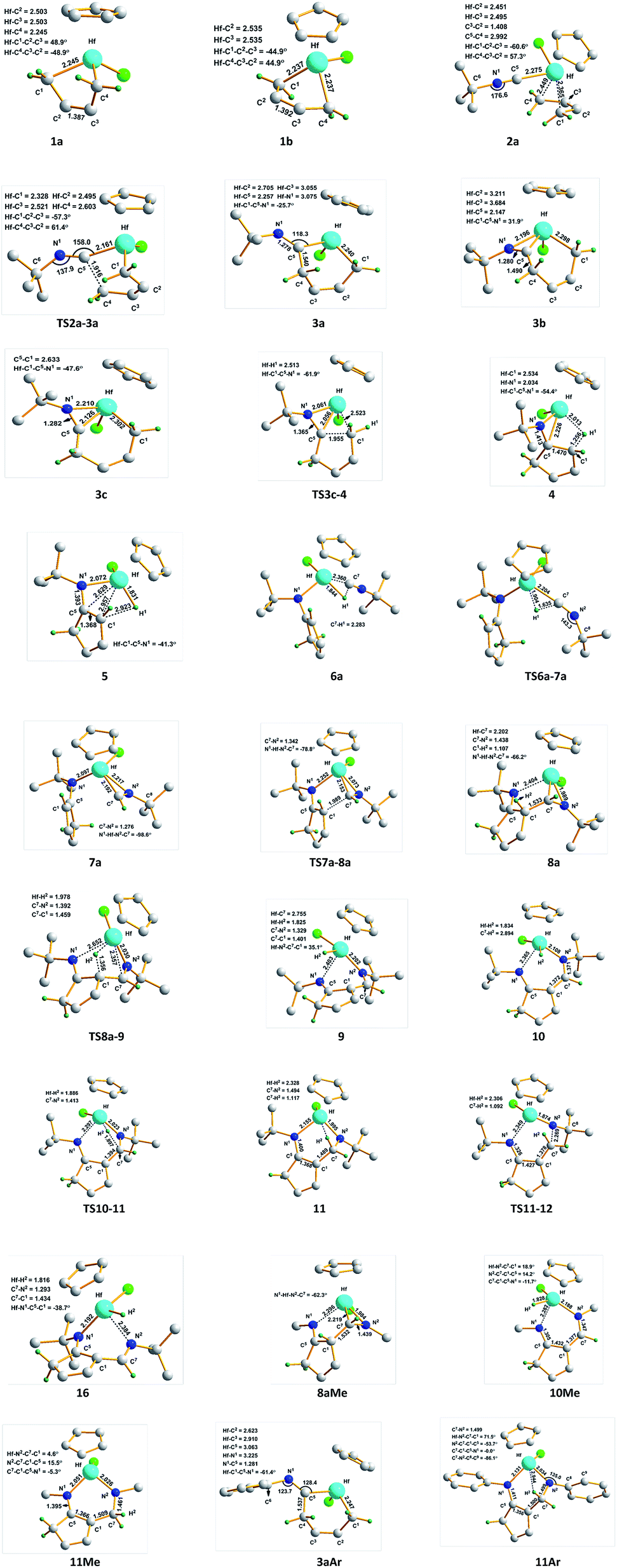 | ||
| Fig. 2 Optimized structures of selected Hf complexes. The methyl (Me) groups and the H atoms, except for those involved in the reactions, are omitted for clarity. Distances and angles are given in Å and degree, respectively. | ||
With a free-energy barrier of 33.1 kJ mol−1 [Fig. 1(b)], 3b isomerizes to 3c via TS3b-3c. An increase in tension leads to an endergonicity of 25.1 kJ mol−1. C–C reductive elimination from 3c generates 4 through TS3c-4. This process involves Hf–C bond breaking and C–C bond formation, and it needs to overcome a free-energy barrier of 90.4 kJ mol−1 and is endergonic by 27.6 kJ mol−1. In 4, there is a strong agostic interaction between the Hf center and C–H bond. This is indicated by the lengths of the Hf–H1 (2.01 Å) and C1–H1 (1.26 Å) bonds. Subsequent β-H elimination rapidly occurs to furnish the Hf hydride 5. In 5, the newly formed CC π double bond conjugates with another of its kind, and the Hf–C bond is completely broken. The conjugation and the release of tension in the three-membered ring make 5 relatively stable, causing the system to be highly exergonic (81.6 kJ mol−1). In 5, the distance between the Hf center and newly formed π double bond moiety (Hf–C5 = 2.63 Å, Hf–C1 = 2.96 Å) indicates that there is almost no interaction between them. The coordination of 5 with the second tBuNC generates 6a with an endergonicity of 43.5 kJ mol−1. The coordination makes the electronic energy and entropy of the system decrease by 4.9 kJ mol−1 and 146.9 J mol−1 K−1, respectively. Compound 6a satisfies the 14-valence-electron configuration. Subsequently, tBuNC is inserted into the Hf–H bond to afford 7a. The formation of 7a requires the free-energy barrier of 22.2 kJ mol−1 and is exergonic by 78.2 kJ mol−1. TS6a-7a belongs to an early transition state where the length of its Hf–H bond is increased by only 0.06 Å. In 7a, the N atom is coordinated to the Hf center. Compound 7a is converted to 8a via TS7a-8a. This C–C reductive coupling reaction requires a free-energy barrier of 79.9 kJ mol−1 and is endergonic by 30.5 kJ mol−1. TS7a-8a belongs to a late transition state with a forming C–C bond (1.99 Å) in length.
Clearly, 7a is the most stable species in the overall tBuNC bis-insertion process, its formation requires the free-energy barrier of 122.2 kJ mol−1 and is exergonic by 56.9 kJ mol−1, and C–C reductive elimination is rate-determining. The fused metallabicyclic hafnaaziridine 8a is an important intermediate. Unfortunately, it cannot be experimentally observed because its formation is thermodynamically unfavorable.
The coordination of tBuNC to 3b [Fig. 1(b)] and the subsequent migratory insertion into the Hf–C bond to generate bis(iminoacyl) hafnacycle 7b need to overcome a free-energy barrier of 146.5 kJ mol−1; this value is so high that the reaction does not proceed.
Compound 8a undergoes β-H elimination to afford Hf hydride 9 via TS8a-9 [Fig. 1(c)], the system requires 100.9 kJ mol−1 and is endergonic by 5.1 kJ mol−1. In TS8a-9, the Hf–H bond is almost formed, and the length of the breaking C–H bond is 0.25 Å longer than that of 8a (Fig. 2); moreover, the Hf–C bond begins to break. Hf hydride 9 comprises a 14-valence-electron configuration. The ketimine N atoms retain η1 bonding with the Hf center, while Hf–C is completely broken. Compound 9 overcomes a free-energy barrier (30.1 kJ mol−1) and isomerizes into 10, the system is endergonic by 27.6 kJ mol−1. The two Hf–N bond lengths in 10 differ by 0.26 Å from each other. With a free-energy barrier of 31.4 kJ mol−1, 10 undergoes the addition of a Hf–H bond across its 1,5-diazapentadiene double bond and is converted to the six-membered hafnacycle 11; the reaction is exergonic by 75.4 kJ mol−1. TS10-11 is an early transition state with a breaking Hf–H bond slightly longer than that of 10. Additionally, the length of the C–H bond being formed is 1.90 Å, which is much longer than the usual C–H bond. The length of Hf–H (2.33 Å) in 11 indicates that there is a strong agostic interaction between the Hf center and the newly formed C–H σ-bond. Through TS11-12, 11 fragments into Hf imido complex 13 and α-methylene cyclopentenimine 14. The system requires a free-energy barrier of 77.5 kJ mol−1 and is endergonic by 9.2 kJ mol−1. In TS11-12, the Hf–N and C–N bonds begin to break, and their lengths are increased by 0.19 Å and 0.78 Å relative to 11, respectively. The length of Hf–H in TS11-12 is shortened by 0.02 Å, indicative of a stronger agostic interaction than that observed in 11. Compound 13 is trapped by pyridine to generate 15 with an exergonicity of 56.1 kJ mol−1.
According to the γ-H elimination reaction pathway [Scheme 1(a)], the calculations show that while the Hf–C bond in 8a is broken, the H atom spontaneously transfers to the Hf center and 8a is directly converted to Hf hydride 16 [Fig. 1(c) and 2]; and the hypothesized non-hydride 16′ [Scheme 1(a)] is not obtained. The Hf–C bond requires 123.1 kJ mol−1 to break, a value that is larger than the 100.9 kJ mol−1 required for β-H elimination from 8a. Thus, the γ-H elimination reaction pathway is ruled out.
Fig. 1 illustrates that 7a is a crucial intermediate. The formation of N-tBu-substituted α-methylene cyclopentenimine 13 and Hf imido complex 15 requires the free-energy barrier of 131.4 kJ mol−1 and is exergonic by 116.0 kJ mol−1, and β-H elimination from 8a is rate-determining.
There are three proposed reaction mechanisms for the formation of diazahafnacyclopentane 18 [Scheme 1(b)]. (1) In the insertion reaction pathway, 8a is converted to 17 through the formation and breaking of the Hf–C bonds. Subsequently, a CN moiety is inserted into the Hf–C bond in 17 to generate 18. (2) In the isomerization reaction pathway, 7a first isomerizes into 7c that in turn undergoes C–C reductive coupling to afford 8b. This compound is converted to the target compound 18 via the insertion of the CN moiety into the Hf–C bond. (3) The C–C coupling reaction pathway involves the non-hydride 16′. However, the above discussion indicates that Hf hydride 16 instead of 16′ is produced after the breaking of the Hf–C bond in 8a. Thus, this reaction pathway is unfeasible. The calculated free-energy profiles of the insertion and isomerization reaction pathways are shown in Fig. 3. The results show that the isomerization reaction pathway is kinetically more favorable than the insertion one (Fig. 3) but requires high free-energy barrier (146.1 kJ mol−1), and the reaction is endergonic by 39.3 kJ mol−1 [with reference to 7a; Fig. 3(b)]. This analysis demonstrates that for tBuNC, the diazahafnacyclopentane (18) formation is kinetically and thermodynamically unfavorable compared to the α-methylene cyclopentenimine (15) formation. However, in the experiment of Norton et al., the diazahafnacyclopentane 18 was characterized by NMR spectrum.27 The inconsistency between the calculation result and experiment could be due to influences of ligands (such as PMe3), and these influences were not taken into account in our study. In addition, the β-H elimination from 8b is also unfavorable because the required free energy is too high [146.5 kJ mol−1; Fig. 3(b)].
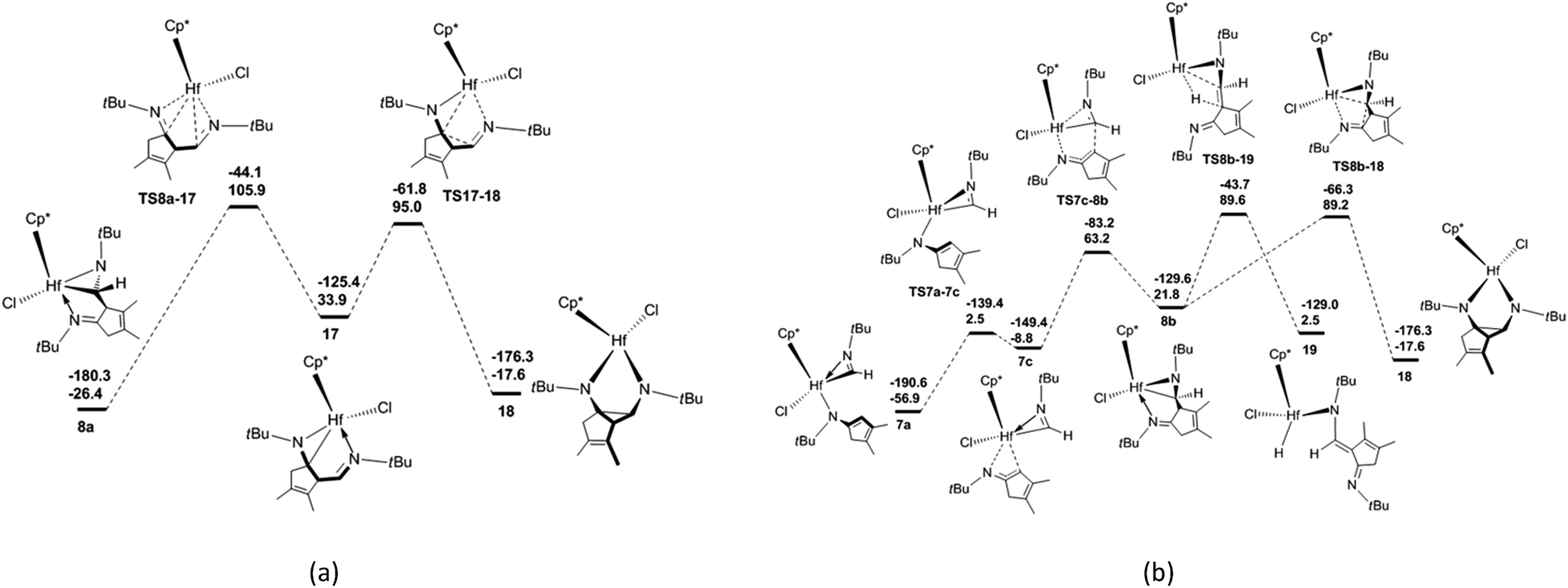 | ||
| Fig. 3 Free-energy profiles for the formation of diazahafnacyclopentane 18 via (a) the insertion reaction pathway and (b) the isomerization reaction pathway and the free-energy profile for β-H elimination from Hf complex 8b. The electronic energies (above) and Gibbs free energies (below) are given in kJ mol−1. | ||
Effects of substituent groups on the mechanisms and products
In order to reveal the effects of isonitrile N-substituent groups on the reaction mechanisms and products, the energetic profiles of the reactions using MeNC, EtNC, ArNC, and 1-AdNC instead of tBuNC were calculated (Fig. S1–S5†). The main calculated results are summarized in Scheme 2.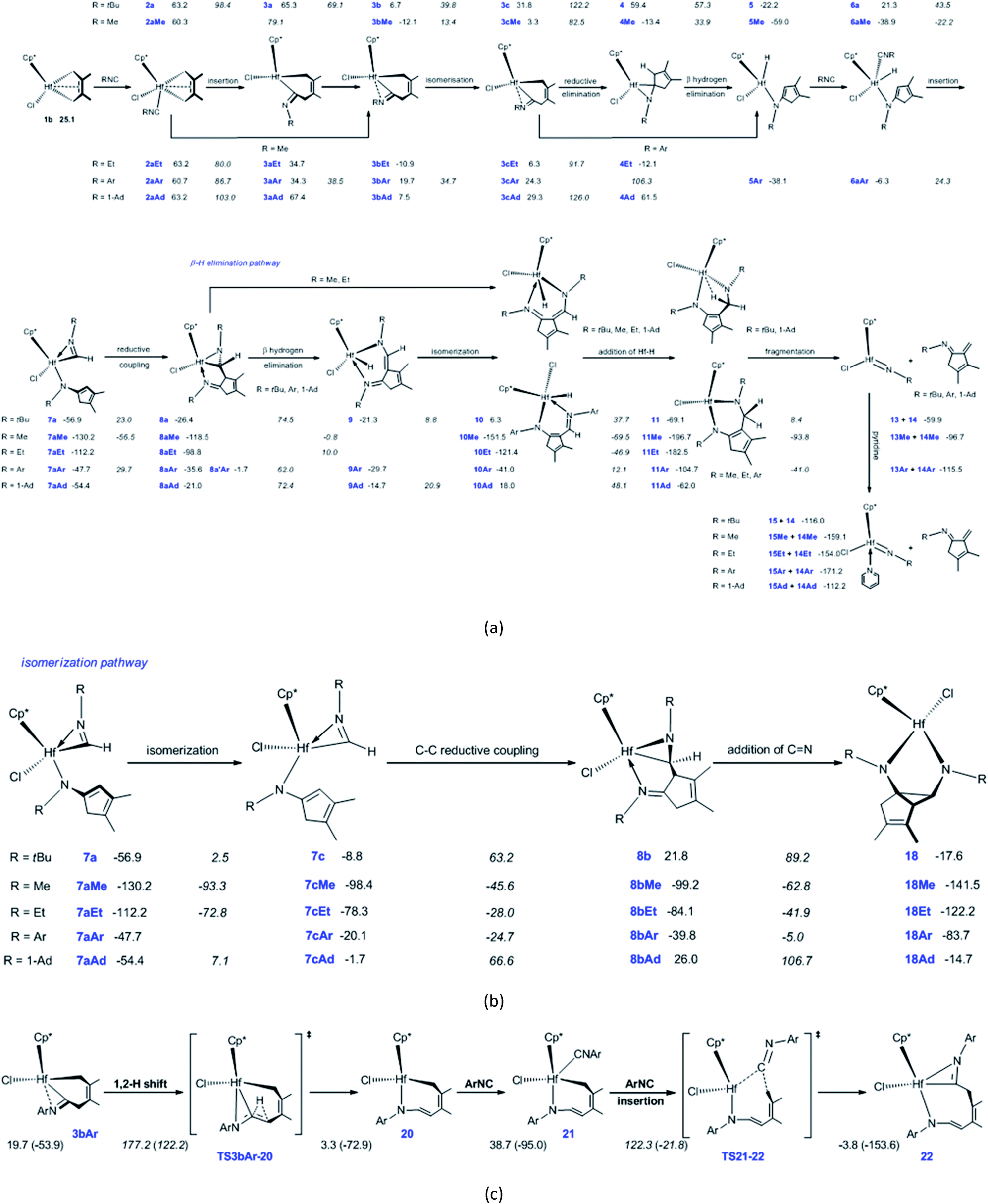 | ||
| Scheme 2 Summary of the calculated reaction pathways and results for (a and b) Cp*(Cl)Hf(2,3-dimethylbutadiene) (1a) with N-R-substituted isonitriles (values denote Gibbs free energies in kJ mol−1 and values in italics are the activation free-energy barriers for elementary reactions), (c) for 1,2-H shift in 3bAr and ArNC insertion into the Hf–C bond in 21 [the electronic energies (in parentheses) and Gibbs free energies (outside parentheses) are given in kJ mol−1]. | ||
The bis-insertion reaction of MeNC [Scheme 2(a)] reveals a number of differences. (1) The coordination of MeNC to 1b and hydride 5Me is endergonic by 35.2 and 20.1 kJ mol−1, respectively, and these values are less than that observed in tBuNC. (2) The migratory insertion of MeNC into the Hf–C bond in 2aMe directly affords the N-coordinated six-membered hafnacycle 3bMe, overcoming a free-energy barrier of 18.8 kJ mol−1; the reaction is exergonic by 72.4 kJ mol−1. (3) Compared to tBuNC, for MeNC the isomerization of the Hf complex 3bMe, C–C reductive elimination, migratory insertion into the Hf–H bond in 6aMe, and C–C reductive coupling require less energies (25.5 vs. 33.1, 79.2 vs. 90.4, 16.7 vs. 22.2, and 73.7 vs. 79.9 kJ mol−1, respectively). Moreover, the isomerization and reductive coupling reaction steps are less endergonic (15.4 vs. 25.1 and 11.7 vs. 30.5 kJ mol−1, respectively) while MeNC insertion into the Hf–H bond in 6aMe is more exergonic (91.3 vs. 78.2 kJ mol−1). Additionally, while C–C reductive elimination for tBuNC is endergonic, this reaction is exergonic by 16.7 kJ mol−1 in MeNC. These differences are all attributable to the smaller steric effects exhibited by the Me group than that by the tBu group. (4) Finally, β-H elimination from hydride 4Me requires 47.3 kJ mol−1 to proceed; this is much larger than the value observed for tBuNC. This step is exergonic by 45.6 kJ mol−1. Unlike the hydride 4, there is no strong agostic interaction between the Hf center and the C–H bond in 4Me, which is responsible for the higher free-energy barrier for the β-H elimination. With a free-energy barrier of 117.7 kJ mol−1 and an exergonicity of 33.0 kJ mol−1, 8aMe undergoes β-H elimination and is converted to hydride 10Me. The two Hf–N bond lengths (Hf–N1 = 2.30 Å and Hf–N2 = 1.98 Å) in 8aMe are shorter than those in 8a. This indicates that the electron density of the Hf center in the former is higher. Furthermore, the Hf–C bond length (2.22 Å) in 8aMe is longer than that of 8a (2.20 Å). These factors led us to determine that the required free-energy barrier for β-H elimination from 8aMe is larger than that from 8a. Compared to β-H elimination, the breaking of the Hf–C bond in 8aMe is kinetically unfavorable because it requires 125.6 kJ mol−1 to overcome the free-energy barrier. Subsequent addition of a Hf–H bond in 10Me overcomes the free-energy barrier of 82.0 kJ mol−1 to afford the six-membered hafnacycle 11Me, and this step is exergonic by 45.2 kJ mol−1. Compared to 10, the configuration of 10Me does not favor the addition of a Hf–H bond. In 11Me, there is no agostic interaction between the Hf center and the C–H bond. This is ascribed to the smaller steric effect of the Me group on the nitrogen atom that renders the structure of six-membered hafnacycle 11Me more planar. With pyridine, the fragmentation of 11Me to Hf imido complex 15Me and N-Me-substituted α-methylene cyclopentenimine 14Me is thermodynamically unfavorable, because the system is endergonic by 37.6 kJ mol−1. The six-membered hafnacycle 11Me requires 129.4 kJ mol−1 to form and is exergonic by 196.7 kJ mol−1, thus it is a possible product.
In the formation of diazahafnacyclopentane 18Me, the isomerization reaction pathway is kinetically more favorable than the insertion reaction pathway (Fig. S2 in ESI†). The isomerization of the Hf complex 7aMe, C–C reductive coupling, and insertion of CN into the Hf–C bond in 8bMe require less energies [36.9 vs. 59.4, 52.8 vs. 72.0, and 36.4 vs. 67.4 kJ mol−1, respectively; Scheme 2(b)] than the corresponding reaction steps for tBuNC. Moreover, the C–C reductive coupling is slightly exergonic (0.8 kJ mol−1). This differs from the endergonicity (30.6 kJ mol−1) of the corresponding reaction for tBuNC. The insertion of a CN bond is more exergonic (42.3 kJ mol−1) relative to that of tBuNC (39.4 kJ mol−1). For MeNC, the formation of diazahafnacyclopentane 18Me is kinetically favorable, overcoming a free-energy barrier of 94.6 kJ mol−1 and is exergonic by 141.5 kJ mol−1.
Conclusions similar to those for MeNC were reached for the EtNC substrate. The electronic effects of the substituent on the isonitrile nitrogen on some main elementary reactions were examined. Thus, compared to MeNC, (1) the required free-energy barrier for the migratory insertion of EtNC into the Hf–C bond slightly decreases [16.8 vs. 18.8 kJ mol−1; Scheme 2(a)]. Moreover, the formed intermediate 3aEt features an uncoordinated N atom that is related to the relative larger size of the Et moiety. The required free-energy barrier for the C–C reductive elimination increases (85.4 vs. 79.2 kJ mol−1). (2) In the β-H elimination reaction pathway, the required energies for β-H elimination and the addition of a Hf–H bond decrease (108.8 vs. 117.7 and 74.5 vs. 82.0 kJ mol−1, respectively). (3) In the isomerization reaction pathway, isomerization of the Hf complex 7aEt and insertion of the CN bond in 8bEt both overcome higher free-energy barriers [39.4 vs. 36.9 and 42.2 vs. 36.4 kJ mol−1, respectively; Scheme 2(b)] while C–C reductive coupling in 7cEt requires a slightly reduced free energy (50.3 vs. 52.8 kJ mol−1). These differences are ascribed to the ethyl moiety that renders the Hf center more electron-rich, thus influencing the reactivities of the Hf complexes. According to the shape of the MeNC free-energy profiles (Fig. S2 in ESI†), it can be inferred from Fig. S3 in ESI† that in the case of EtNC, the formation of six-membered hafnacycle 11Et requires 122.2 kJ mol−1 and exhibits an exergonicity of 182.5 kJ mol−1. Furthermore, the formation of diazahafnacyclopentane 18Et overcomes a lower free-energy barrier (102.6 kJ mol−1) and is less exergonic (122.2 kJ mol−1).
In the bis-insertion reaction of ArNC, the electron-withdrawing characteristics of the Ar moiety lead to an increase in the free-energy barrier for the insertion of ArNC into the Hf–C bond in 2aAr, compared to that of EtNC [26.0 vs. 16.8 kJ mol−1; Scheme 2(a)]. The formed 3aAr is a N-uncoordinated six-membered hafnacycle. The isomerization of N-coordinated hafnacycle 3bAr requires less free-energy (15.0 kJ mol−1) and is less endergonic (4.6 kJ mol−1) compared to the values observed for tBuNC and MeNC. These differences are related to the expanded conjugation of the π electrons for ArNC. C–C reductive elimination from 3cAr overcomes a free-energy barrier of 82.0 kJ mol−1 and directly forms Hf hydride 5Ar, and this step is exergonic by 62.4 kJ mol−1. The required free energy ranges between 90.4 kJ mol−1 for tBuNC and 79.2 kJ mol−1 for MeNC. The insertion of ArNC into the Hf–H bond in 6aAr overcomes a higher free-energy barrier (30.6 kJ mol−1) and is much less exergonic (41.4 kJ mol−1) compared to that observed for tBuNC and MeNC. This is attributed to the more electron-deficient Hf center in the case of ArNC. The required free-energy barrier for C–C reductive coupling in 7aAr is 3.7 kJ mol−1 higher than that of MeNC, and 2.5 kJ mol−1 lower than that of tBuNC. The shape of the free-energy profile for the pathway leading to α-methylene cyclopentenimine 14Ar [Fig. S4(b) in ESI†] resembles that of tBuNC. However, the electron-withdrawing characteristics of the Ar group lower the free-energy [63.7 kJ mol−1; Scheme 2(a)] required for β-H elimination. Moreover, addition of the Hf–H bond in 10Ar requires more free energy (53.1 kJ mol−1) for ArNC than for tBuNC. Similar to MeNC and EtNC, there is no agostic structure present in the Hf–H bond addition product 11Ar. Because π-electron delocalization stabilizes the transition state during the fragmentation of 11Ar, this step requires less free energy (63.7 kJ mol−1) for ArNC than for tBuNC and MeNC. In addition, when Ar replaced the tBu group in TS8a-16, the afforded TS(9′-18)Ar was calculated [Fig. S4(b) in ESI†]. This was proved to be connected to the Hf hydride 9′Ar and diazahafnacyclopentane 18Ar; however, the required free energy (156.6 kJ mol−1) is too high. The free-energy profile for the isomerization reaction pathway leading to diazahafnacyclopentane 18Ar exhibits similar traits to those of MeNC and EtNC. The C–C reductive coupling in 7cAr and the insertion of CN in 8bAr require lower energies [44.8 and 34.8 kJ mol−1, respectively; Scheme 2(b)]. The Ar moiety is an electron-withdrawing but bulkier group. However, its orientation in the Hf complexes can efficiently avoid strong repulsions with other groups. This leads to ArNC reactivities that are similar to those of tBuNC as well as MeNC and EtNC. For ArNC, the formation of α-methylene cyclopentenimine 14Ar and Hf imido complex 15Ar overcomes a free-energy barrier of 109.7 kJ mol−1 and is exergonic by 171.2 kJ mol−1 while the formation of diazahafnacyclopentane 18Ar requires 106.3 kJ mol−1 and is exergonic by 83.7 kJ mol−1. Diazahafnacyclopentane 18Ar has been experimentally produced and characterized by X-ray crystallography.27 Based on the NMR spectrum, Teuben et al. proposed that 18′Ar (see Chart 1) was afforded in the reaction of 1a with 2ArNC through a 1,2-H shift in 3bAr followed by the second ArNC insertion into the Hf–C bond.43 The calculated results show that 1,2-H shift in 3bAr requires the free-energy barrier of 157.5 kJ mol−1 [Scheme 2(c)], and that the apparent activation free energy for the pathway to 18′Ar as proposed by Teuben et al. is not less than 177.2 kJ mol−1. Thus, compared to 18Ar, the formation of 18′Ar is kinetically unfavorable.
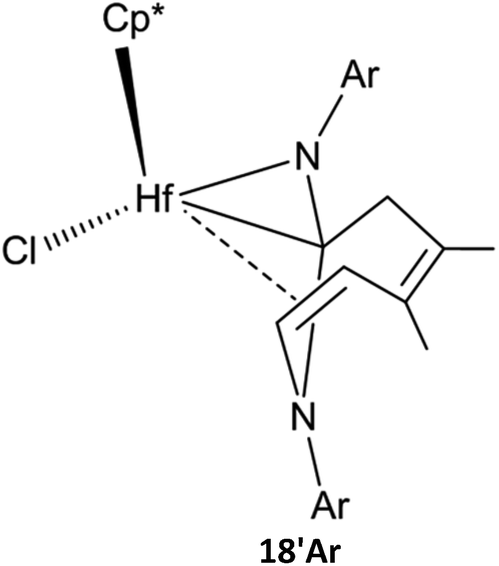 | ||
| Chart 1 | ||
For 1-AdNC, the calculated reaction free-energy profiles are similar to those of tBuNC (Fig. S5 in ESI†). Fig. S5(a)–(c) in ESI† suggest that N-1-Ad-substituted α-methylene cyclopentenimine 14Ad and Hf imido complex 15Ad can be experimentally afforded: their formation requires 126.8 kJ mol−1 and is exergonic by 112.2 kJ mol−1.
Conclusions
The migratory insertion of an isonitrile into a metal–C bond is becoming an increasingly important method for the construction of C–C bonds in organic and pharmaceutical syntheses. In this work, we theoretically studied the reaction of Cp*(Cl)Hf(2,3-dimethylbutadiene) with isonitriles using DFT calculations. Reaction mechanisms for tBuNC, MeNC, EtNC, ArNC, and 1-AdNC were proposed and discussed. Unlike the reaction of Cp*(Cl)Ti(2,3-dimethylbutadiene) with isonitriles, the fused three-five metallabicyclic bis-insertion product (hafnaaziridines) predicted in our study could not be experimentally observed, because their formation is thermodynamically unfavorable. For the bulky group (tBu and 1-Ad)-substituted isonitriles, the formation of α-methylene cyclopentenimines and Hf imido complexes was both kinetically and thermodynamically favorable. These reactions exhibited a free-energy barrier of >126 kJ mol−1 and were exergonic by ∼112 kJ mol−1. For the smaller MeNC and EtNC, the formation of six-membered hafnacycles was thermodynamically more favorable (free-energy barrier of ∼122 kJ mol−1; exergonicity >182 kJ mol−1) compared to the formation of the corresponding α-methylene cyclopentenimines and Hf imido complexes. On the other hand, the diazahafnacyclopentanes were the kinetically favored products, with a free-energy barrier of formation <105 kJ mol−1 and exergonicity of <142 kJ mol−1. The Hf imido complex and the α-methylene cyclopentenimine were the thermodynamically favored products for the electron-withdrawing Ar-substituted isonitrile, and the reaction required 109.7 kJ mol−1 and was exergonic by 171.2 kJ mol−1. On the other hand, diazahafnacyclopentane was the kinetically favored product with a lower formation free-energy barrier of 106.3 kJ mol−1 and an exergonicity of 83.7 kJ mol−1. Moreover, in the formation of α-methylene cyclopentenimines and hafnacycles, the β-H elimination reaction pathway was found to be dominant. This reaction comprises several elementary reaction steps including: the isomerization of Cp*(Cl)Hf(2,3-dimethylbutadiene), migratory insertion of the isonitrile into a Hf–C bond, C–C reductive coupling, migratory insertion of isonitrile into Hf–H bond, β-H elimination, addition of a Hf–H bond, and fragmentation. Finally, an isomerization reaction pathway was found to be responsible for the formation of diazahafnacyclopentanes. This pathway comprises the isomerization of the Hf complex, C–C reductive coupling, and CN bond insertion as elementary reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Natural Sciences Foundation of Anhui Province (1608085MB42), Scientific Research Foundation of Anhui University of Science and Technology (ZY538), Beijing National Laboratory for Molecular Sciences (1G129), and the Undergraduate Innovation and Entrepreneurship Training Program (201610361242) for their financial support. We would also like to thank Professor Song Wang from Jilin University (JLU) for his assistance with our calculations.Notes and references
- (a) G. M. Diamond, K. A. Hall, A. M. LaPointe, M. K. Leclerc, J. Longmire, J. A. W. Shoemaker and P. Sun, ACS Catal., 2011, 1, 887–900 CrossRef CAS; (b) K. Press, V. Venditto, I. Goldberg and M. Kol, Dalton Trans., 2013, 42, 9096–9103 RSC; (c) H. Makio, A. V. Prasad, H. Terao, J. Saito and T. Fujita, Dalton Trans., 2013, 42, 9112–9119 RSC; (d) I. E. Nifant'ev, P. V. Ivchenko, V. V. Bagrov, S. M. Nagy, L. N. Winslow, J. A. Merrick-Mack, S. Mihan and A. V. Churakov, Dalton Trans., 2013, 42, 1501–1511 RSC.
- (a) L. Rocchigiani, V. Busico, A. Pastore, G. Talarico and A. Macchioni, Angew. Chem., Int. Ed., 2014, 53, 2157–2161 CrossRef CAS PubMed; (b) I. Haas, T. Dietel, K. Press, M. Kol and R. Kempe, Chem.–Eur. J., 2013, 19, 14254–14262 CrossRef CAS PubMed; (c) S. Mehdiabadi and J. B. P. Soares, Macromolecules, 2013, 46, 1312–1324 CrossRef CAS; (d) N. Nakata, T. Toda, T. Matsuo and A. Ishii, Macromolecules, 2013, 46, 6758–6764 CrossRef CAS; (e) H. Hamaki, N. Takeda, M. Nabika and N. Tokitoh, Macromolecules, 2012, 45, 1758–1769 CrossRef CAS.
- (a) X. Wang, Y. Wang, X. Shi, J. Liu, C. Chen and Y. Li, Macromolecules, 2014, 47, 552–559 CrossRef CAS; (b) G. Li, M. Lamberti, G. Roviello and C. Pellecchia, Organometallics, 2012, 31, 6772–6778 CrossRef CAS; (c) E. Szuromi and J. Klosin, Organometallics, 2011, 30, 4589–4597 CrossRef CAS; (d) S. H. Jun, J. H. Park, C. S. Lee, S. Y. Park, M. J. Go, J. Lee and B. Y. Lee, Organometallics, 2013, 32, 7357–7365 CrossRef CAS; (e) P. P. Fontaine, R. Figueroa, S. D. McCann, D. Mort and J. Klosin, Organometallics, 2013, 32, 2963–2972 CrossRef CAS; (f) D. K. Steelman, P. D. Pletcher, J. M. Switzer, S. Xiong, G. A. Medvedev, W. N. Delgass, J. M. Caruthers and M. M. Abu-Omar, Organometallics, 2013, 32, 4862–4867 CrossRef CAS.
- (a) C. Zhang, H. Pan, J. Klosin, S. Tu and A. Jaganathan, Org. Process Res. Dev., 2015, 19, 1383–1391 CrossRef CAS; (b) L. He, B. Wang, F. Yang, Y. Li and Z. Ma, Macromolecules, 2016, 49, 6578–6589 CrossRef CAS; (c) Y. Gao, A. R. Mouat, A. Motta, A. Macchioni, C. Zuccaccia, M. Delferro and T. J. Marks, ACS Catal., 2015, 5, 5272–5282 CrossRef CAS; (d) D. Prema, Y. L. N. M. Arachchige, R. E. Murray and L. M. Slaughter, Chem. Commun., 2015, 51, 6753–6756 RSC; (e) E. Y. Hwang, G. H. Park, C. S. Lee, Y. Young Kang, J. Lee and B. Y. Lee, Dalton Trans., 2015, 44, 3845–3855 RSC.
- (a) G. Blay, I. Fernández, M. C. Muñoz, J. R. Pedro and C. Vila, Eur. J. Org. Chem., 2013, 2013, 1902–1907 CrossRef CAS; (b) H. Ishitani, H. Suzuki, Y. Saito, Y. Yamashita and S. Kobayashi, Eur. J. Org. Chem., 2015, 2015, 5485–5499 CrossRef CAS; (c) Y.-C. Wu, H.-J. Li, L. Liu, N. Demoulin, Z. Liu, D. Wang and Y.-J. Chen, Adv. Synth. Catal., 2011, 353, 907–912 CrossRef CAS.
- (a) S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 11373–11383 CrossRef CAS PubMed; (b) S. P. Semproni and P. J. Chirik, Angew. Chem., Int. Ed., 2013, 52, 12965–12969 CrossRef CAS PubMed.
- (a) F. M. Chadwick, R. T. Cooper and D. O'Hare, Organometallics, 2016, 35, 2092–2100 CrossRef CAS; (b) M. J. Sgro and D. W. Stephan, Chem. Commun., 2013, 49, 2610–2612 RSC.
- (a) T. K. Saha, V. Ramkumar and D. Chakraborty, Inorg. Chem., 2011, 50, 2720–2722 CrossRef CAS PubMed; (b) S. L. Hancock, M. F. Mahon, G. Kociok-Kőhn and M. D. Jones, Eur. J. Inorg. Chem., 2011, 2011, 4596–4602 CrossRef CAS; (c) L.-C. Liang, S.-T. Lin, C.-C. Chien and M.-T. Chen, Dalton Trans., 2013, 42, 9286–9293 RSC.
- J. L. Olivares-Romero, Z. Li and H. Yamamoto, J. Am. Chem. Soc., 2012, 134, 5440–5443 CrossRef CAS PubMed.
- (a) L. Rocchigiani, V. Busico, A. Pastore and A. Macchioni, Organometallics, 2016, 35, 1241–1250 CrossRef CAS; (b) C. Adler, N. Frerichs, M. Schmidtmann and R. Beckhaus, Organometallics, 2016, 35, 3728–3733 CrossRef CAS; (c) M. Kamitani, B. Pinter, C.-H. Chen, M. Pink and D. J. Mindiola, Angew. Chem., Int. Ed., 2014, 53, 10913–10915 CrossRef CAS PubMed; (d) Y. Wang, J. D. Lewis and Y. Román-Leshkov, ACS Catal., 2016, 6, 2739–2744 CrossRef CAS.
- (a) R. Waterman and T. D. Tilley, Chem. Sci., 2011, 2, 1320–1325 RSC; (b) T. Beweries, M. Haehnel and U. Rosenthal, Catal. Sci. Technol., 2013, 3, 18–28 RSC; (c) M. de Léséleuc and S. K. Collins, Chem. Commun., 2015, 51, 10471–10474 RSC; (d) M. de Léséleuc and S. K. Collins, ACS Catal., 2015, 5, 1462–1467 CrossRef; (e) T. De Baerdemaeker, M. Feyen, U. Müller, B. Yilmaz, F.-S. Xiao, W. Zhang, T. Yokoi, X. Bao, H. Gies and D. E. De Vos, ACS Catal., 2015, 5, 3393–3397 CrossRef CAS.
- (a) H. Lundberg and H. Adolfsson, ACS Catal., 2015, 5, 3271–3277 CrossRef CAS; (b) D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC; (c) Y.-X. Yang, Y. Li, R. Ganguly and C.-W. So, Dalton Trans., 2015, 44, 12633–12639 RSC; (d) H.-J. Chuang, B.-H. Wu, C.-Y. Li and B.-T. Ko, Eur. J. Inorg. Chem., 2014, 2014, 1239–1248 CrossRef CAS.
- (a) S. G. Minasian, K. S. Boland, R. K. Feller, A. J. Gaunt, S. A. Kozimor, I. May, S. D. Reilly, B. L. Scott and D. K. Shuh, Inorg. Chem., 2012, 51, 5728–5736 CrossRef CAS PubMed; (b) A. C. Bowman, J. England, S. Sproules, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2013, 52, 2242–2256 CrossRef CAS PubMed; (c) L.-C. Liang, C.-C. Chien, M.-T. Chen and S.-T. Lin, Inorg. Chem., 2013, 52, 7709–7716 CrossRef CAS PubMed; (d) S. Manabe and Y. J. Ito, J. Org. Chem., 2013, 78, 4568–4572 CrossRef CAS PubMed; (e) T. Gehrmann, G. T. Plundrich, H. Wadepohl and L. H. Gade, Organometallics, 2012, 31, 3346–3354 CrossRef CAS.
- (a) P. D. Schweizer, H. Wadepohl and L. H. Gade, Organometallics, 2014, 33, 1726–1739 CrossRef CAS; (b) N. Nakata, S. Aoki, V. Y. Lee and A. Sekiguchi, Organometallics, 2015, 34, 2699–2702 CrossRef CAS; (c) P. P. Fontaine, Organometallics, 2012, 31, 6244–6251 CrossRef CAS.
- (a) S. E. Gibson and A. Stevenazzi, Angew. Chem., Int. Ed., 2003, 42, 1800–1810 CrossRef CAS PubMed; (b) E. J. Corey and S. W. Walinsky, J. Am. Chem. Soc., 1972, 94, 8932–8933 CrossRef CAS.
- (a) S. K. Guchhait, V. Chaudhary and C. Madaan, Org. Biomol. Chem., 2012, 10, 9271–9277 RSC; (b) W. Sha, J.-T. Yu, Y. Jiang, H. Yang and J. Cheng, Chem. Commun., 2014, 50, 9179–9181 RSC; (c) Y. Li, A. Chao and F. F. Fleming, Chem. Commun., 2016, 52, 2111–2113 RSC; (d) G. Lesma, I. Bassanini, R. Bortolozzi, C. Colletto, R. Bai, E. Hamel, F. Meneghetti, G. Rainoldi, M. Stucchi, A. Sacchetti, A. Silvani and G. Viola, Org. Biomol. Chem., 2015, 13, 11633–11644 RSC; (e) M. Tobisu and N. Chatani, Chem. Lett., 2011, 40, 330–340 CrossRef CAS.
- (a) T. Buyck, D. Pasche, Q. Wang and J. Zhu, Chem.–Eur. J., 2016, 22, 2278–2281 CrossRef CAS PubMed; (b) B. Zhang and A. Studer, Org. Lett., 2014, 16, 1216–1219 CrossRef CAS PubMed; (c) B. Zhang and A. Studer, Org. Lett., 2014, 16, 3990–3993 CrossRef CAS PubMed; (d) Q. Dai, J.-T. Yu, X. Feng, Y. Jiang, H. Yang and J. Cheng, Adv. Synth. Catal., 2014, 356, 3341–3346 CrossRef CAS.
- (a) A. Dewanji, C. Mück-Lichtenfeld, K. Bergander, C. G. Daniliuc and A. Studer, Chem.–Eur. J., 2015, 21, 12295–12298 CrossRef CAS PubMed; (b) A. Hinz, A. Schulz and A. Villinger, J. Am. Chem. Soc., 2015, 137, 9953–9962 CrossRef CAS PubMed; (c) N. Shao, G.-X. Pang, C.-X. Yan, G.-F. Shi and Y. Cheng, J. Org. Chem., 2011, 76, 7458–7465 CrossRef CAS PubMed; (d) S. U. Dighe, A. K. K. S., S. Srivastava, P. Shukla, S. Singh, M. Dikshit and S. Batra, J. Org. Chem., 2015, 80, 99–108 CrossRef CAS PubMed.
- (a) Y. Li and F. F. Fleming, Angew. Chem., Int. Ed., 2016, 55, 14770–14773 CrossRef CAS PubMed; (b) D. Riedel, T. Wurm, K. Graf, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2015, 357, 1515–1523 CrossRef CAS; (c) M. Seidl, M. Schiffer, M. Bodensteiner, A. Y. Timoshkin and M. Scheer, Chem.–Eur. J., 2013, 19, 13787–13791 CrossRef PubMed; (d) S. Tong, Q. Wang, M.-X. Wang and J. Zhu, Angew. Chem., Int. Ed., 2015, 54, 1293–1297 CrossRef CAS PubMed.
- (a) S. Tong, Q. Wang, M.-X. Wang and J. Zhu, Chem.–Eur. J., 2016, 22, 8332–8338 CrossRef CAS PubMed; (b) R. M. Wilson, J. L. Stockdill, X. Wu, X. Li, P. A. Vadola, P. K. Park, P. Wang and S. J. Danishefsky, Angew. Chem., Int. Ed., 2012, 51, 2834–2848 CrossRef CAS PubMed; (c) B. Zhang, C. Mück-Lichtenfeld, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2013, 52, 10792–10795 CrossRef CAS PubMed; (d) A. S. K. Hashmi, C. Lothschütz, C. Böhling and F. Rominger, Organometallics, 2011, 30, 2411–2417 CrossRef CAS.
- (a) H. Zhang, D. Shi, S. Ren, H. Jin and Y. Liu, Eur. J. Org. Chem., 2016, 2016, 4224–4229 CrossRef CAS; (b) R. S. Chay, K. V. Luzyanin, V. Y. Kukushkin, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Organometallics, 2012, 31, 2379–2387 CrossRef CAS; (c) H. V. Le and B. Ganem, Org. Lett., 2011, 13, 2584 CrossRef CAS PubMed; (d) M. Maj, C. Ahn, D. Kossowska, K. Park, K. Kwak, H. Han and M. Cho, Phys. Chem. Chem. Phys., 2015, 17, 11770–11778 RSC.
- (a) M. Adib, S. Feizi, M. S. Shirazi, L.-G. Zhu and H. R. Bijanzadeh, Helv. Chim. Acta, 2014, 97, 524–531 CrossRef CAS; (b) H. Stöckmann, A. A. Neves, S. Stairs, K. M. Brindle and F. J. Leeper, Org. Biomol. Chem., 2011, 9, 7303–7305 RSC; (c) G. Bianchini, G. L. Sorella, N. Canever, A. Scarso and G. Strukul, Chem. Commun., 2013, 49, 5322–5324 RSC.
- (a) A. Zeiler, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem.–Eur. J., 2015, 21, 11065–11071 CrossRef CAS PubMed; (b) G. Pendecy and S. Batra, RSC Adv., 2015, 5, 28875–28878 RSC; (c) H. Gu, Z. Qiu, Z. Zhang, J. Li and B. Yan, Dalton Trans., 2015, 44, 9839–9846 RSC; (d) H. Jiang, H. Gao, B. Liu and W. Wu, Chem. Commun., 2014, 50, 15348–15351 RSC.
- (a) L. Xiang and Z. Xie, Organometallics, 2016, 35, 233–241 CrossRef CAS; (b) L. Xiang and Z. Xie, Organometallics, 2016, 35, 1430–1439 CrossRef CAS; (c) K. Takamatsu, K. Hirano and M. Miura, Org. Lett., 2015, 17, 4066–4069 CrossRef CAS PubMed; (d) T. N. Valadez, J. R. Norton, M. C. Neary and P. J. Quinlivan, Organometallics, 2016, 35, 3163–3169 CrossRef CAS.
- (a) F. Foschi, T. Roth, H. Wadepohl and L. H. Gade, Org. Lett., 2016, 18, 5182–5185 CrossRef CAS PubMed; (b) H. Braunschweig, M. A. Celik, R. D. Dewhurst, K. Ferkinghoff, A. Hermann, J. O. C. Jimenez-Halla, T. Kramer, K. Radacki, R. Shang, E. Siedler, F. Weiβenberger and C. Werner, Chem.–Eur. J., 2016, 22, 11736–11744 CrossRef CAS PubMed; (c) C.-H. Lei, D.-X. Wang, L. Zhao, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2013, 135, 4708–4711 CrossRef CAS PubMed; (d) W. Lu, H. Lu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650–6661 CrossRef CAS PubMed.
- (a) H.-Y. Tu, Y.-R. Liu, J.-J. Chu, B.-L. Hu and X.-G. Zhang, J. Org. Chem., 2014, 79, 9907–9912 CrossRef CAS PubMed; (b) M. Ma, A. Stasch and C. Jones, Chem.–Eur. J., 2012, 18, 10669–10676 CrossRef CAS PubMed; (c) Y. Odabachian, S. Tong, Q. Wang, M.-X. Wang and J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 10878–10882 CrossRef CAS PubMed; (d) A. G. Tskhovrebov, K. V. Luzyanin, F. M. Dolgushin, M. F. C. Guedes da Silva, A. J. L. Pombeiro and V. Y. Kukushkin, Organometallics, 2011, 30, 3362–3370 CrossRef CAS; (e) M. Okazaki, K. Suto, N. Kudo, M. Takano and F. Ozawa, Organometallics, 2012, 31, 4110–4113 CrossRef CAS; (f) G. Greidanus-Strom, C. A. G. Carter and J. M. Stryker, Organometallics, 2002, 21, 1011–1013 CrossRef CAS.
- T. N. Valadez, J. R. Norton and M. C. Neary, J. Am. Chem. Soc., 2015, 137, 10152–10155 CrossRef CAS PubMed.
- S. Batke, M. Sietzen, L. Merz, H. Wadepohl and J. Ballmann, Organometallics, 2016, 35, 2294–2308 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- A. Höllwarth, M. Böhme, S. Dapprich, A. W. Ehlers, A. Gobbi, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 237–240 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1972, 72, 650–654 CrossRef.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS.
- (a) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS; (b) C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- D. D. Devore, F. J. Timmers and D. L. Hasha, Organometallics, 1995, 14, 3132–3134 CrossRef CAS.
- B. Hessen, H. Blenkers and J. H. Teuben, Organometallics, 1989, 8, 830–835 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Choice of calculated method, Fig. S1–S5, calculated Cartesian coordinates and electronic energies (E) for the involved species in the paper. See DOI: 10.1039/c7ra08981c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |