
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MiR-138-5p targeting LIMK1 suppresses breast cancer cell proliferation and motility
Dengfeng Li,
Hongming Song,
Tianqi Wu,
Dan Xie,
Jiashu Hu,
Junyong Zhao and
Lin Fang *
*
Department of Thyroid and Breast, General Surgery, Shanghai Tenth People's Hospital, Tongji University School of Medicine, No. 301 Yanchang Middle Road, Jing'an Area, Shanghai 200072, China. E-mail: fanglin2017@126.com; Tel: +86 18917683221
First published on 9th November 2017
Abstract
Breast cancer is the most frequently diagnosed female cancer. LIM domain kinase 1 (LIMK1) was reported to increase breast cancer progressive biological behavior and was associated with clinical breast cancer outcomes. MiRNAs are a kind of small non-coding RNA that binds the target mRNAs, leading to protein downregulation. The aim of this study was to find whether miR-138-5p could target LIMK1 in breast cancer to suppress breast cancer biological behavior. The TCGA database showed that LIMK1 was highly expressed in breast cancer. The dual-luciferase report showed that LIMK1 was a target gene of miR-138-5p. Over-expression of miR-138-5p could significantly reduce expression of LIMK1 in mRNA and the protein level. Colony formation and MTT assay showed that over-expression of miR-138-5p could suppress proliferation of breast cancer cells. Wound-healing assay and transwell assay showed that over-expression of miR-138-5p could suppress migration and invasion of breast cancer cells. In addition, over-expression of miR-138-5p could arrest breast cancer cells into the G0/G1 phase by down-regulating expression of CDK4, 6/Cyclin D1 and CDK2/Cyclin E1 compounds. In conclusion, our study showed that miR-138-5p could target LIMK1 and suppress the progressive biological behavior of breast cancer cells. Mi-138-5p targeting LIMK1 may be a new potential treatment for breast cancer.
Introduction
Nowadays, breast cancer has been a frequently diagnosed womens cancer and it was estimated that in 2015, there were over 2 million women diagnosed with breast cancer and more than 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 patients died of it.1 Different molecules decide different subtypes and therefore lead to different therapeutic strategies and clinical outcomes.2 Up to now, there have only been two biomarkers used to guide therapeutic strategies despite the stage of breast cancer: estrogen receptor (ER) and the Human Epidermal growth factor Receptor 2 (HER2).3 Although current therapies constitute a considerable improvement in breast cancer treatments, there are still some types of breast cancer existing with tumor progression and therapy-resistance.2,3 Therefore, to figure out the molecular mechanisms of breast cancer is an urgent requirement for the treatment of breast cancer.
000 patients died of it.1 Different molecules decide different subtypes and therefore lead to different therapeutic strategies and clinical outcomes.2 Up to now, there have only been two biomarkers used to guide therapeutic strategies despite the stage of breast cancer: estrogen receptor (ER) and the Human Epidermal growth factor Receptor 2 (HER2).3 Although current therapies constitute a considerable improvement in breast cancer treatments, there are still some types of breast cancer existing with tumor progression and therapy-resistance.2,3 Therefore, to figure out the molecular mechanisms of breast cancer is an urgent requirement for the treatment of breast cancer.
Some hypotheses about resistance to breast cancer treatments have been proposed to overcome this challenges, including transporters and enzymes for intracellular drug depletion, DNA repair, targets mutations, resistance to apoptosis, and miRNAs' role.4 MiRNAs are a kind of small non-coding 22 nucleotide-long RNAs that bind the target mRNAs, leading to protein downregulation.5 Since miRNAs dysregulated in breast cancer in 2005, more and more studies about miRNAs in breast cancer have been reported, including onco-miRNAs (suppressing tumor suppressor genes) and tumor-suppressor miRNAs (downregulating oncogenic genes).2,6
LIM domain kinase 1 (LIMK1) is a serine/threonine kinase that regulates actin polymerization through phosphorylation and inactivation cofilin (CFL1).7 It was reported that LIMK1 could increase breast cancer cells' metastasis via regulation of the urokinase-type plasminogen activator system, indicating that LIMK1 signaling played a major role in breast cancer in tumor growth, angiogenesis and motility.8 Several studies found that miR-143-3p, miR-200b-3p and miR-429-5p could suppress LIMK1 expression in breast cancer, inhibiting breast cancer cells' biological behaviors, such as proliferation, migration and invasion.9,10 According to miRNA target databases, one miRNA may have more than one target genes, meanwhile one gene may be regulated by different miRNAs.11
In this study, we found that LIMK1 was highly expressed in breast cancer tissues and miR-138-5p targeted LIMK1 and suppressed the proliferation, migration, and invasion of breast cancer cells. We hope our results could broaden the views of miRNAs mechanisms in breast cancer and in future miR-138-5p could be a potential target for breast cancer treatment.
Materials and methods
Cell culture and transfection
MDA-MB-231 and HCC1937 TNBC cells were purchased from the Chinese Academy of Science at Shanghai (Shanghai, China). HEK 293T human embryonic kidney cells were a gift from the Department of Laboratory Medicine, Shanghai Tenth People's Hospital. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco Life Technologies, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS, Gibco) and penicillin-streptomycin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) in an incubator at 37 °C in 5% CO2.MiR-138-5p mimics and negative-control (NC) mimics were purchased from Ibsbio (Shanghai Integrated Biotech Co., Ltd., Shanghai, China). For transfection, cells (2.5 × 105/well) were cultured in a 6-well plate. After cells reached to 40–50% confluence, the mimics were transfected into cells with Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. The concentration of mimics used was 100 nM L−1, and the ratio of mimics to Lipofectamine 2000 was 2.5:2 (volume). The sequence of the miR-138-5p mimics was: sense, 5′-AGCUGGUGUUGUGAAUCAGGCCG-3'; antisense, 5′-GCCUGAUUCACAACACCAGCUUU-3'. The NC mimic sequence was: sense, 5′-UCACAACCUCCUAGAAAGAGUAGA-3′; antisense, 5′-UACUCUUUCUAGGAGGUUGUGAUU-3′.
Cell proliferation assays
After MDA-MB-231 cells were transfected with miR-138-5p, or NC mimics for 24 h, single-cell suspensions of these transfected cells were prepared, and then replated in a 12-well plate with 600 cells per well. The culture medium was changed every 3 days. After 14 days, the plated cells were washed with phosphate-buffered saline (PBS) twice and then treated with 4% polyformaldehyde for 10 min, stained with 0.1% crystal violet for 10 min, and washed with double-distilled water 3 times. Photos of the culture surfaces on dried plates were taken.For a 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) proliferation assay, MDA-MB-231 cells (2 × 103/well) were seeded in a 96-well plate. The cell proliferation rate was determined by measuring the absorbance of MTT according to the manufacturer's instructions (Thermo Fisher Scientific, Inc., Waltham, MA, USA) at d1, d2, d3. A microplate spectrophotometer (BioTek, Winooski, VT, USA) was used to measure the absorbance of each sample at 490 nm.
Cell migration and invasion assays
Wound-healing and Transwell-cup assays were performed to evaluate the motility of MDA-MB-231 cells transfected with miR-138-5p mimics and NC mimics. Cells (2.5 × 105/well) were cultured in a 6-well plate. When cells reached 90% confluence, the plate containing the cells was scratched with a 200 μL pipette tip. Photos of plate containing the cells in culture were taken at 0 and 24 h at the same position to assess cell motility. Transwell assays, with or without Matrigel, was also used to evaluate migration and invasion. After MDA-MB-231 cells' treatment with miR-138-5p mimics, or NC mimics, 5 × 104 cells per well were seeded in the upper chambers of a 24-well Transwell cup plate (Corning, NY, USA) with 200 μL of DMEM supplemented with 0.1% bovine serum albumin. The lower chambers were filled with 500 μL of DMEM with 10% bovine serum albumin. The cells were cultured for 12 h to 16 h, and then the outer surface of cup was washed with PBST 3 times, then fixed with 4% polyformaldehyde 10 min, and stained with 0.1% crystal violet for 10 min. Photos of the bottoms of the inserts were captured when the surface dried.Quantitative reverse transcription-polymerase chain reaction
MDA-MB-231 and HCC1937 TNBC cells were treated with miR-138-5p mimics, or NC mimics in 6-well plates. After 36 h, cells were collected for total RNA isolation with TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and cDNA was generated using a real-time polymerase chain reaction (PCR) kit (Takara, Shiga, Japan). Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) was performed on a 7900HT Fast real-time PCR system (Applied Biosystems, Singapore). We followed our previously published amplification procedure: 5 min at 95 °C, followed by 40 cycles at 95 °C for 30 s and 65 °C for 45 s.12 Expression of mRNA was assessed by evaluating threshold cycle values. The threshold cycle values of LIMK1 were normalized to the level of GAPDH expression. The primer sequences used were as follows:10 LIMK1-forward: 5′-CAAGGGACTGGTTATGGTGGC-3′; LIMK1-reverse: 5′-CCCCGTCACCGATAAAGGTC-3′; GAPDH-forward: 5′-CATGAGAAGTATGACAACAGCCT-3′; and GAPDH-reverse: 5′-AGTCCTTCCACGATACCAAAGT-3′. The qRT-PCR results were analyzed using the 2−ΔΔt method.12Western blot analysis
MDA-MB-231 and HCC1937 TNBC cells were collected after 72 h of treatment with miR-138-5p and NC mimics, and total protein samples were extracted from them using a RIPA buffer (Beyotime Biotechnology, Shanghai, China). Protein concentrations of the samples were measured with a BCA Protein Assay Kit (Beyotime Biotechnology) according to the manufacturer's instructions. The protein samples (60 μg) were separated on a 10% SDS polyacrylamide gel using electrophoresis and transferred onto 0.45 μm nitrocellulose membranes (Beyotime Biotechnology). The membranes were blocked for 60 min with a 5% nonfat milk solution and then incubated with primary antibodies overnight at 4 °C. The blots were washed with TBST and incubated for 1 h with an anti-rabbit or anti-mouse secondary antibody (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). After 3 washes with TBST, immunoreactive protein bands were detected using an Odyssey Scanning System (LI-COR Biosciences, Lincoln, NE, USA). Primary antibodies against the following were used:10 PCNA (1:2000; Cell Signaling Technology, Danvers, MA, USA), MMP2 (1:1000; Arigo Biolaboratories, Hsinchu City, Taiwan, China), MMP9 (1:1000; Arigo Biolaboratories), LIMK1 (1:500; Arigo Biolaboratories), CFL1 (1:500; Arigo Biolaboratories), phospho-CFL1 (1:500; Arigo Biolaboratories), CDK2 (1:2000; Bioworld, Shanghai, China), CDK4 (1:2000; Abcam, Cambridge, MA, USA), CDK6 (1:2000; Abcam), cyclin D1 (1:10000; Abcam), cyclin E1 (1:2000; Abcam), and β-actin (1:2000; Santa Cruz Biotechnology, CA, USA).
Cell cycle assay
MDA-MB-231 cells were treated with miR-138-5p mimics, or NC mimics for 36 h. The cells were then collected, washed, suspended in cold PBS, and fixed in cold 70% ethanol overnight at 4 °C. After a 30 min incubation with RNase (0.1 g L−1), 300 μL (0.05 g L−1) of propidium iodide, staining solution was added to each cell sample. The stained samples were incubated for 30 min at room temperature in the dark. The cell-cycle progression was then analyzed by flow cytometry.Dual-luciferase reporter assay
HEK 293T cells were seeded in 48-well plates and cultured until they reached 70% confluence. The psiCHECK-2/LIMK1 3′-untranslated region (UTR) wild reporter and psiCHECK-2/LIMK1 3′UTR mutant reporter plasmids were purchased from Ibsbio (Shanghai, China). HEK 293T cells were transiently co-transfected with 0.2 μg psiCHECK-2/LIMK1 3′-UTR wild reporter or psiCHECK-2/LIMK1 3′-UTR mutant reporter plasmids and 100 nM L−1 miR-138-5p mimics, or NC mimics by Lipofectamine 2000. After 72 h, cell lysates were collected to determine firefly and Renilla luciferase activity using a Dual Luciferase Assay Kit (Beyotime Biotechnology). Firefly luciferase activity values were normalized to the values for Renilla luciferase, and the results are presented as the ratio of firefly to Renilla activity values.Statistical analysis
GraphPad Prism version 6.0 (GraphPad, San Diego, CA, USA) was used for all statistical analyses. Data are presented as the mean ± standard (mean ± SD) with at least three separate experiments. The student t-test and were used for comparisons between groups. Differences were considered significant for a p-values less than 0.05.Results
Expression of LIMK1 was up-regulated in TCGA breast cancer database
The expression of LIMK1 in breast cancer was checked in oncomine website (https://www.oncomine.org/). Data from the TCGA database (Fig. 1) showed that LIMK1 was highly expressed in different breast cancer type, including intraductal cribriform breast adenocarcinoma, invasive breast carcinoma, invasive lobular breast carcinoma and mixed lobular and ductal breast carcinoma. These results indicated that LIMK1 could be an oncogene in breast cancer.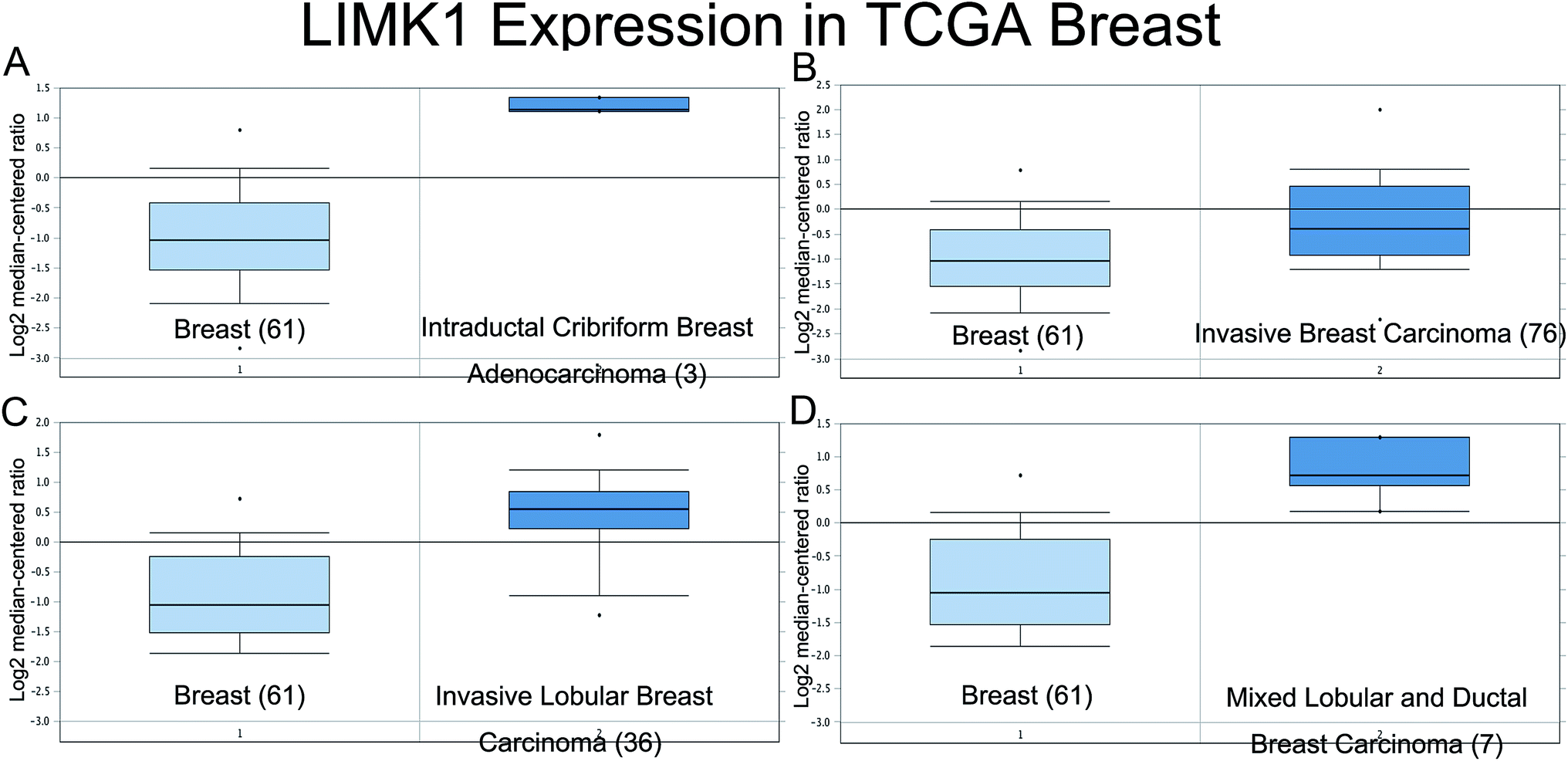 | ||
| Fig. 1 LIMK1 is highly expressed in different breast cancer subtypes. (A) Intraductal Cribriform Breast Adenocarcinoma vs. Normal (fold change: 4.559, p-value: 3.71 × 10−12); (B) Invasive Breast Carcinoma vs. Normal (fold change: 1.682, p-value: 4.43 × 10−7); (C) Intraductal Cribriform Breast Adenocarcinoma vs. Normal (fold change: 2.531, p-value: 2.29 × 10−13); (D) Mixed Lobular and Ductal Breast Carcinoma vs. Normal (fold change: 3.196, p-value: 4.99 × 10−12). | ||
Mir-138-5p targeted LIMK1 and inhibited the expression of LIMK1
Several databases (such as TargetScan, PicTar and miRwalk) indicated that LIMK1 was a target gene of miR-138-5p, regulating LIMK1/CFL1 signal pathway to function as a tumor suppressor in breast cancer. To confirm this prediction, two types of plasmids that contained the binding sites of miR-138-5p and LIMK1 (wild- and mutant-type, Fig. 2A) were constructed and transferred into HEK293T cells. As shown in Fig. 2B, miR-138-5p significantly decreased the luciferase activity when compared to NC group in wild-type group. More importantly, in mutant-type group, miR-138-5p seemed to have no effect compared with NC group. These results indicated LIMK1 was a target gene of miR-138-5p. After transfection of miR-138-5p mimics into MDA-MB-231 and HCC1937 breast cancer cells, miR-138-5p significantly inhibited expression of LIMK1 in RNA level (Fig. 3A and B). Moreover, miR-138-5p not only inhibited protein level of LIMK1 but also down-regulated the expression of CFL1 and phosphorylation of CFL1 in both two breast cancer cellines (Fig. 3C and D). Taken all together into consideration, it indicated that miR-138-5p could target LIMK1 and inhibited LIMK1/CFL1 expression in breast cancer cells.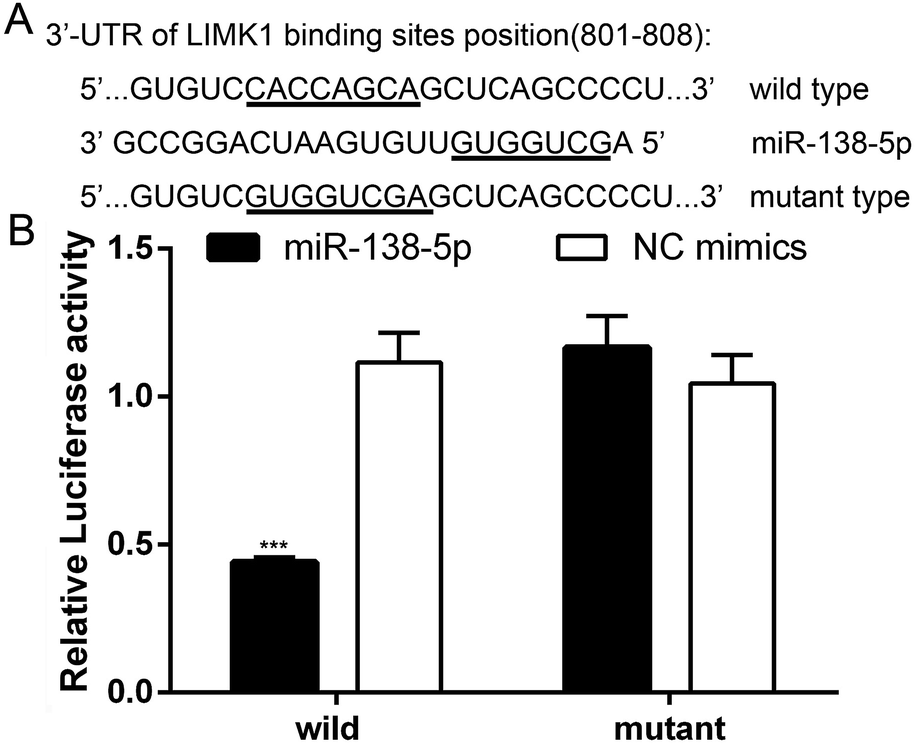 | ||
| Fig. 2 LIMK1 is a target gene of miR-138-5p. (A) Binding sites of miR-138-5p and LIMK1 3′-UTR. (B) MiR-138-5p decreased luciferase activity in wild-type group (student t-test, *** means p value < 0.001). | ||
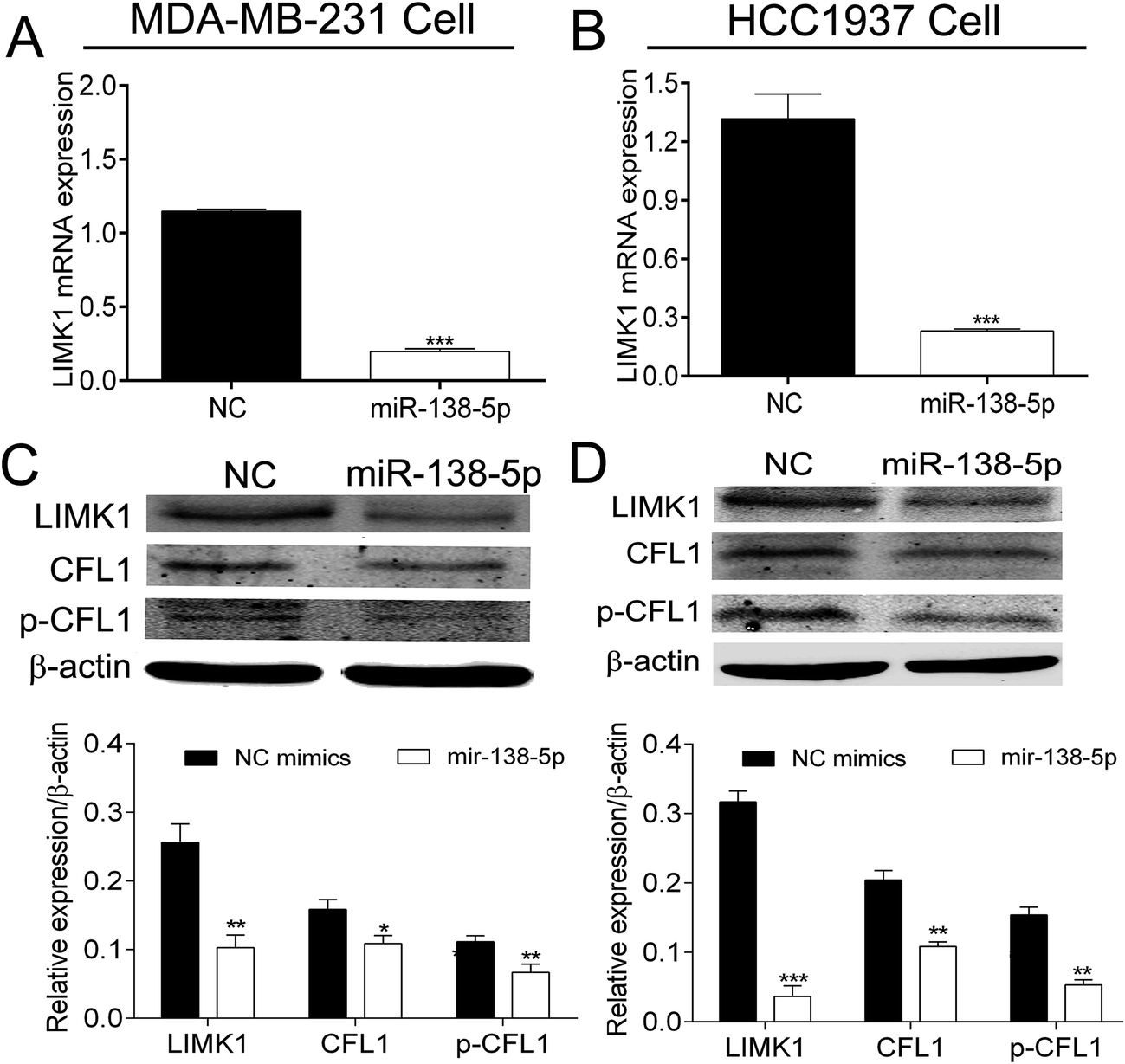 | ||
| Fig. 3 MiRNA-138-5p suppresses expression of LIMK1 and its substrate CFL1. (A and B) MiR-138-5p suppressed mRNA expression of LIMK1 in MDA-MB-231 and HCC1937 cells (student t-test, *** means p value < 0.001); (C and D) MiR-138-5p suppressed protein expression of LIMK1 and reduced expression and phosphorylation of CFL1 in MDA-MB-231 and HCC1937 cells (student t-test, * means p value < 0.05; ** means p value < 0.01; *** means p value < 0.001). | ||
Mir-138-5p inhibited proliferation of breast cancer cells
Colony formation assay and MTT showed that over-expression of miR-138-5p in MDA-MB-231 breast cancers significantly inhibited the proliferation of MDA-MB-231 breast cancer cells (Fig. 4A and B). Western blot showed that miR-138-5p could also inhibit the expression of PCNA, which was thought to be a proliferation biological marker.13 These results showed that miR-138-5p inhibited the proliferation of breast cancer cells.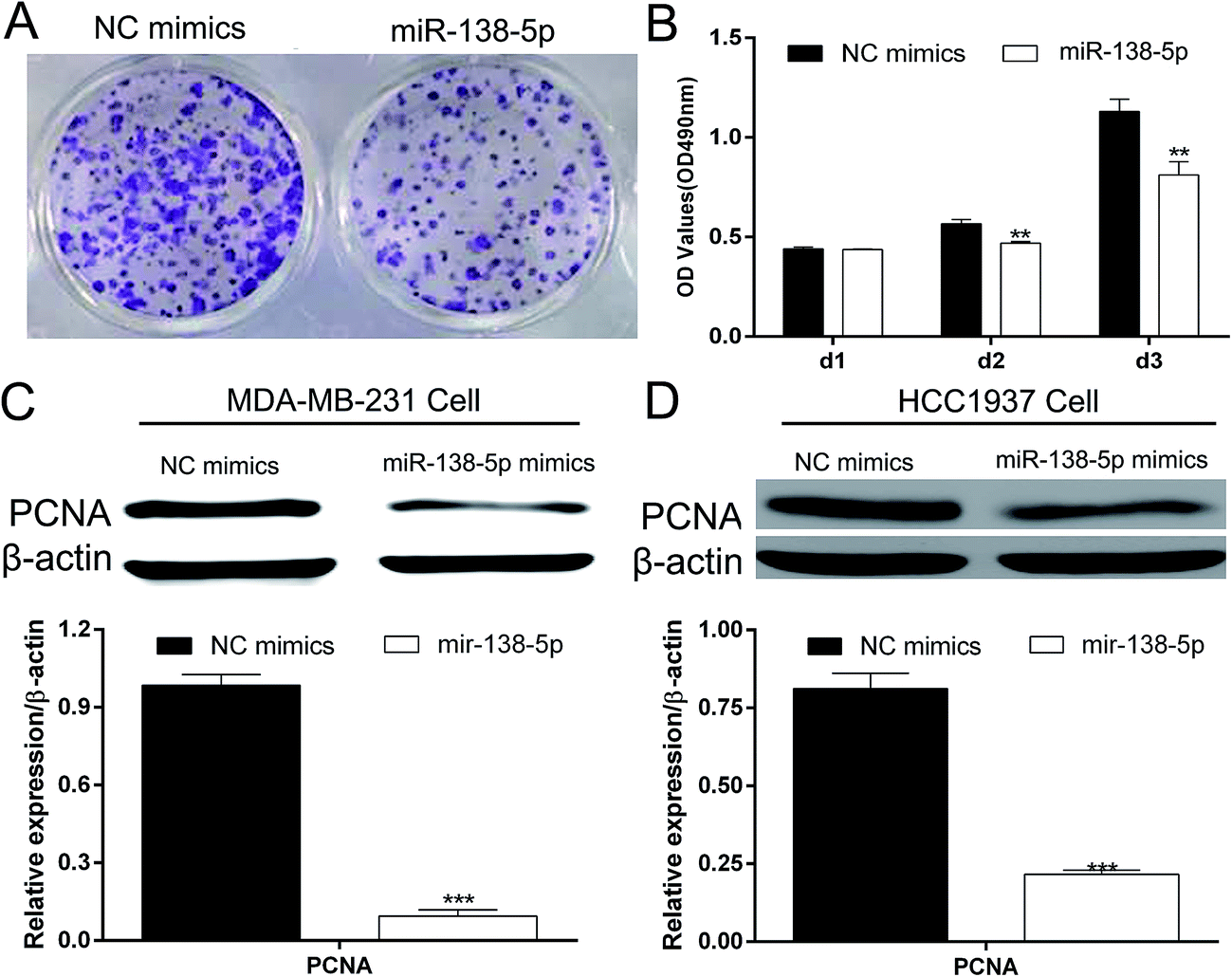 | ||
| Fig. 4 MiRNA-138-5p suppresses proliferation of breast cancer cells. (A) Colony formation assat showed that miR-138-5p inhibited proliferation of MDA-MB-231 cells; (B) MTT assay showed miR-138-5p inhibited proliferation of MDA-MB-231 cells (student t-test, ** means p value < 0.01); (C) over-expression suppressed PCNA protein level in MDA-MB-231 cells (student t-test, *** means p value < 0.001); (D) over-expression suppressed PCNA protein level in HCC1937 cells (student t-test, *** means p value < 0.001). | ||
Mir-138-5p inhibited migration and invasion of breast cancer cells
As shown in Fig. 5A, the wound healing assay showed that over-expression of miR-138-5p in MDA-MB-231 breast cancer cells healed more slowly compared to NC group. At 24 h, in miR-138-5p treatment group, there was still 50% space not migrated by MDA-MB-231 cells while in NC group, the MDA-MB-231 cells have already healed already. Besides, the transwell assay showed over-expression of miR-138-5p also decreased the migrating and invasive cells (Fig. 5B and C). It is well known that MMP2 and MMP9 are usually used as indicators of metastasis because they are important for metastatic cancer cell invading the basement membrane.14,15 Therefore we chose MMP2 and MMP9 as the biomarkers to reflect migration and invasion of metastatic breast cancer cells. As shown in Fig. 5D and E, we found that miR-138-5p could significantly inhibit the protein level of MMP2 and MMP9 both in MDA-MB-231 and HCC1937 two breast cancer cellines. These results showed that over-expression of miR-138-5p could inhibit migration and invasion of breast cancer cells.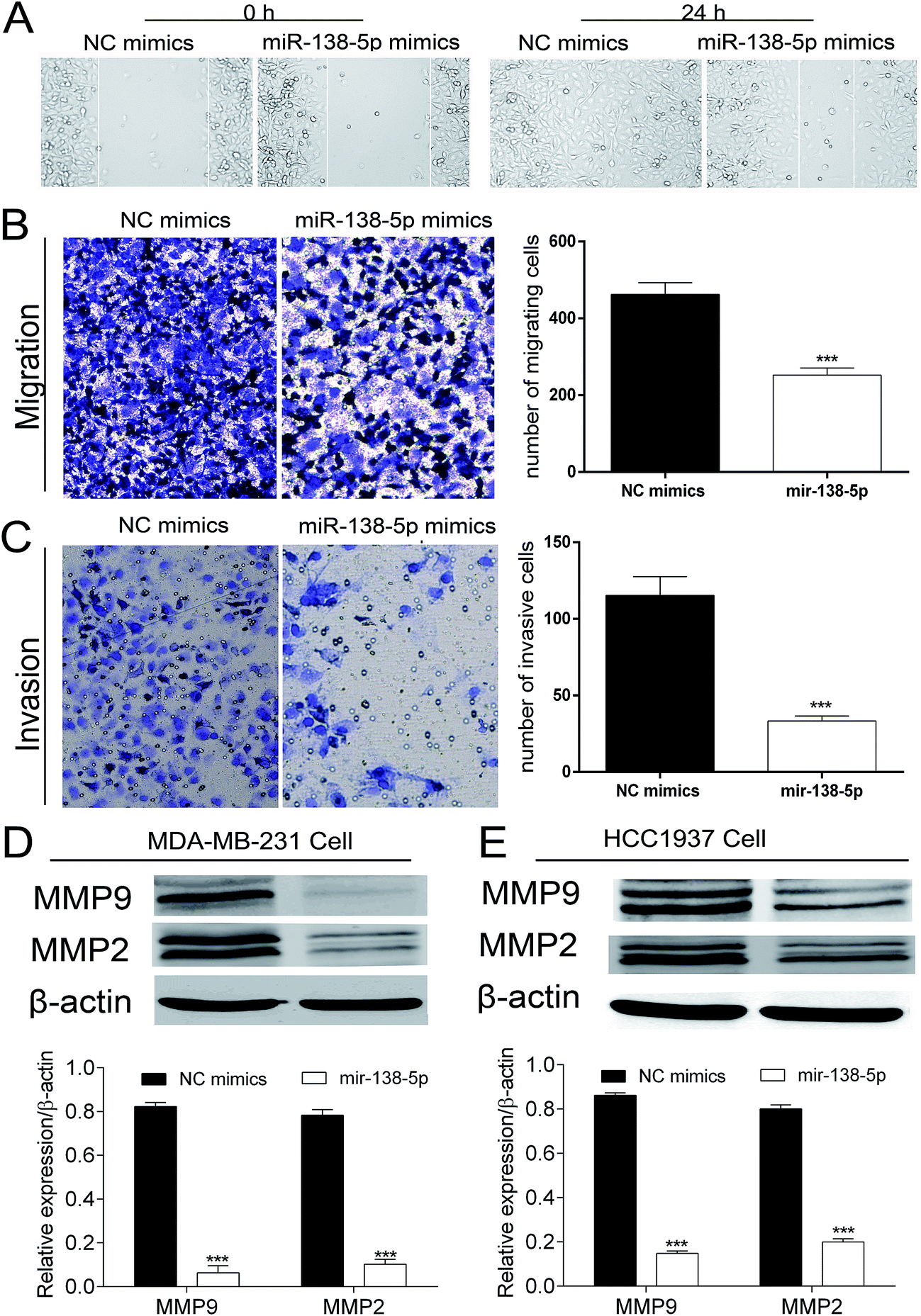 | ||
| Fig. 5 MiRNA-138-5p suppresses motility ability of breast cancer cells. (A) Wound-healing assay showed that miR-138-5p inhibited migration of MDA-MB-231 cells. (B and C) Transwell assay showed that miR-138-5p inhibited migration and invasion of MDA-MB-231 cells (student t-test, ** means p value < 0.01); (D) miR-138-5p inhibited expression of MMP2 and MMP9 in MDA-MB-231 and HCC1937 breast cancer cells (student t-test, *** means p value < 0.001). | ||
Mir-138-5p arrested breast cancer cells into G0/G1 phase
After over-expression of miR-138-5p, MDA-MB-231 cells were collected for flow cytometry analysis. The results showed that compared to NC group, over-expression of miR-138-5p could significantly arrest MDA-MB-231 cells into G0/G1 cell phase (Fig. 6A). Then we checked expression status of CDK4, 6/Cyclin D1 and CDK2/Cyclin E after over-expression of miR-138-5p, which were thought as the main regulators for cells from G0/G1 phase to S phase.16 Our results showed that over-expression of miR-138-5p significantly decreased the expression of CDK4, 6/Cyclin D1 and CDK2/Cyclin E in both MDA-MB-231 and HCC1937 breast cancer cellines. These results revealed that miR-138-5p decreased the expression of CDK4, 6/Cyclin D1 and CDK2/Cyclin E, thus arrested cells into G0/G1 phase.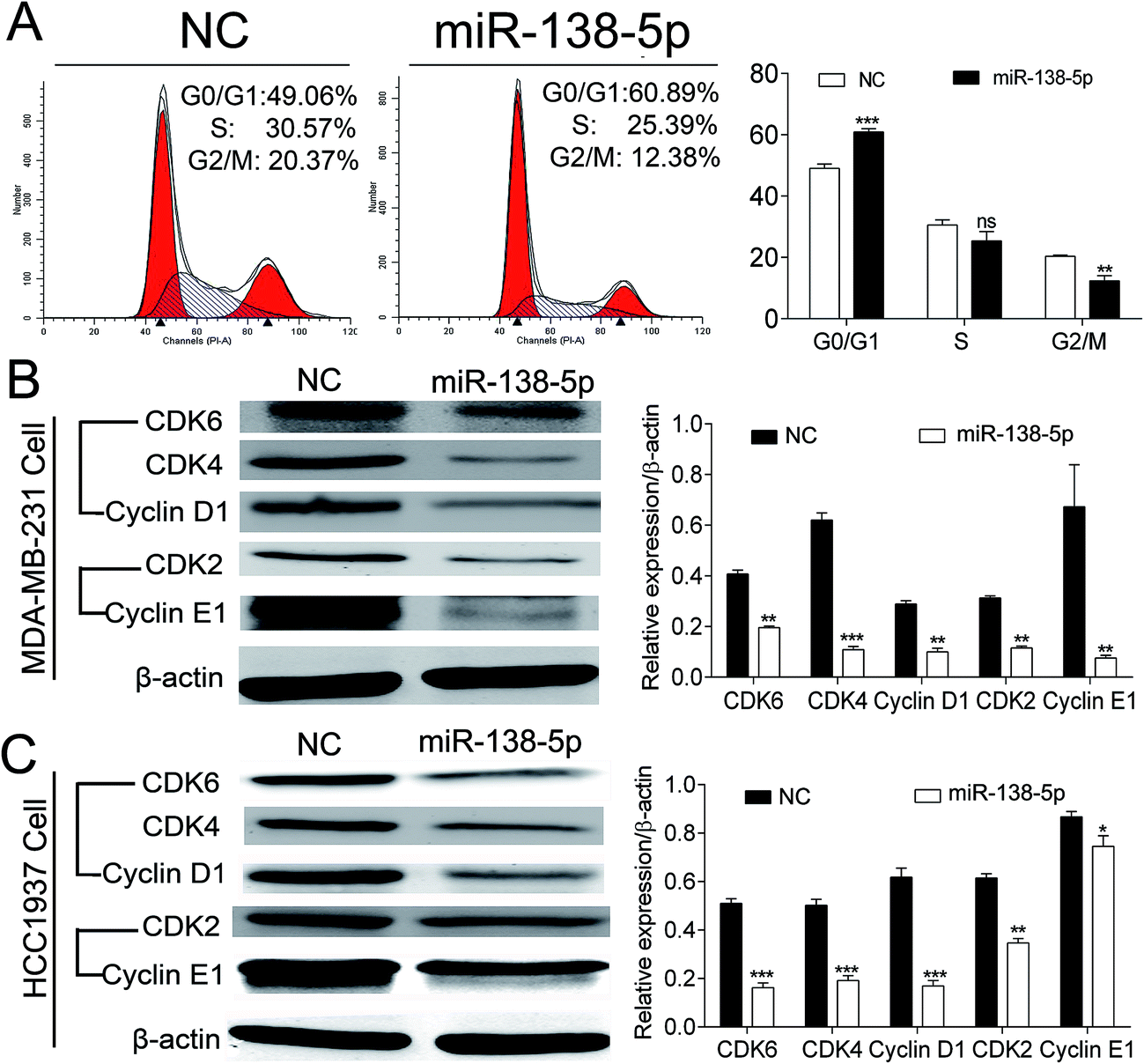 | ||
| Fig. 6 MiRNA-138-5p arrested breast cancer cells into G0/G1 phase. (A) Over-expression of miR-138-5p arrested MDA-MB-231 cells into G0/G1 phase (miR-138-5p: 60.89% vs. NC: 49.06%); (B and C) over-expression of miR-138-5p inhibited expression of CDK4, 6/Cyclin D1 and CDK2/Cyclin E1 Compounds in MDA-MB-231 and HCC1937 breast cancer cells (student t-test, * means p value < 0.05; ** means p value < 0.01; *** means p value < 0.001). | ||
Discussion
Breast cancer now is the No. 1 diagnosed female cancer in the world and just in the USA along, there are over 2.5 million new cases of in 2016–2017.17,18 It was reported that high expression of LIMK1 was found in breast cancer and the status of LIMK1 in breast cancer was associated with breast cancer progression, angiogenesis, and metastasis.19 We searched the expression of LIMK1 in breast cancer from oncomine website (https://www.oncomine.org/). It confirmed that LIMK1 was highly expressed in different breast cancer type (Fig. 1). One study revealed Inhibition of LIM kinase can reduce tumor growth and invasiveness of breast cancer.20 Therefore, targeting LIMK1 may be a potential treatment for breast cancer. A series of studies were designed and performed to explore potential LIMK1 inhibitors to treat cancers. For example, bis-aryl urea derivatives were selected to be the potential LIMK inhibitors21,22 5,6-substituted 4-aminothieno[2,3-d]pyrimidines was identified as LIMK1 inhibitors;23 computer-based identification of a novel LIMK1/2 Inhibitor that synergizes with salirasib to destabilize the actin cytoskeleton;24 transcriptional profiling of GBM invasion genes identifies effective inhibitors of the LIM kinase-cofilin pathway.25 In breast cancer, a series of aminothiazole inhibitors were discovered by high throughput screening and subsequent SAR directed development led to a series of low nanomolar inhibitors of LIMK1 and LIMK2 that also inhibited the direct biomarker p-cofilin in cells and inhibited the invasion of MDA-MB-231 cells.26 CRT0105446 and CRT0105950 were screened and identified as significantly sensitive to both LIMK inhibitors to be against 656 cancer cell lines, and rhabdomyosarcoma, neuroblastoma and kidney cancer cells; therefore CRT0105446 and CRT0105950 will enable further development of LIMK-targeted cancer therapy.27MiRNAs are a kind of 20–22 nt non-coding RNAs that can bind mRNA of target genes and therefore down-regulate the expression of target genes, function as oncogenes or tumor suppressors.28 In non-small cell lung cancer (NSCLC), miR-27b and miR-143 were reported to target LIMK1 to inhibit tumor growth, migration and invasion of NSCLC cells.29,30 In addition, miR-20a was also found to target LIMK1 to counteract tumor progression in anaplastic thyroid31 and inhibited metastasis and proliferation in cutaneous squamous cell carcinoma.32 These studies indicated that miRNAs targeting LIMK1 maybe a potential treatment for cancer therapies. In our study, we found that miR-138-5p could bind 3′-UTR of LIMK1 at the site 801–808 (Fig. 2A). To confirm this, two types of plasmids containing the binding (wild-type binding sites and/or mutant binding sites) were constructed to be co-transfected with miR-138-5p mimics together into HEK293T cells. The luciferase activity assay showed that in miR-138-5p could significantly decrease the luciferase activity, compared to NC group (Fig. 2B). Furthermore, after overexpression of miRNA-138-5p in MDA-MB-231 and HCC1937 breast cancer cells, the expression of LIMK1 decreased in both mRNA and protein level. Meanwhile, overexpression of miR-138-5p inhibits the expression and phosphorylation of CFL1 (Fig. 3).
The colony formation assay and MTT assay showed that over-expression of miR-138-5p in breast cancer could significantly inhibit the proliferation of MDA-MB-231 cells (Fig. 4A and B). Given that PCNA protein is a common biomarker for assessing cell proliferation and prognosis,33 we analyzed PCNA protein expression levels after over-expression of miR-138-5p to reflect the trend of proliferation in MDA-MB-231 and HCC1937 breast cancer cells. In both two cancer cells, PCNA expression was significantly reduced upon treatment with the miR-138-5p (Fig. 4C and D). In addition, we performed wound-healing assay and transwell assay to explore whether miR-138-5p could inhibit the motility of breast cancer cell. The results showed that over-expression of miR-138-5p could significantly reduce the motility ability of MDA-MB-231 cells. Given that MMP2 expression is highly associated with risk of metastasis and MMP-9 expression is also significantly correlated with BC grades and overall survival.14,15 Therefore, MMP2 and MMP9 were chosen as the representative biomarkers to reflect metastatic trend of BC cells. The expression of MMP2 and MMP9 were significantly decreased by miR-138-5p, which was in accordance with another study that miR-138-5p could inhibit the metastasis of breast cancer cells by suppressing EMT process.34
Flow cytometry analysis showed that overexpression of miR-138-5p arrested MDA-MB-231 cells into G0/G1 phase. Cyclin D1 is an important regulator for the G1/S transition, through activating CDK4/6, which phosphorylate and inactivate pRb to release the transcription factor E2F to initiate a series of genes that required for cell cycle progression through the G1/S transition.35CDK2/Cyclin E1 compound also played a major role in inducing cell cycle from G1 phase to S phase.36 After transfected with miR-138-5p mimics into MDA-MB-231 and HCC1937 cells, CDK4, 6/Cyclin D1 and CDK2/Cyclin E1 compounds expression all reduced in both two cellines. One study reported during cell cycle process, activation of cyclin-dependent kinases (CDKs) directly or indirectly activated LIMK1 and in turn LIMK1 may also play participate in cell cycle progress.37 Overexpression of LIMK1 in MDA-MB-435 breast cancer also increased cell cycle progression.38 In another study, it was reported that via cofilin phosphorylation, LIMK1 regulates cytokinesis to be involved in the actin cytoskeletal reorganization.39 Our results indicated that miR-138-5p suppressed breast cancer growth by regulating the expression of CDK4, 6/Cyclin D1 and CDK2/Cyclin E1 through targeting LIMK1.
It was reported that in breast cancer, miR-451 could reverse the chemo-resistance in chemo-resistant breast cancer cells by targeting YWHAZ, which indicated that targeting miRNA therapy may be a potential treatment for resistance breast cancer.40 In addition, it was exciting that using miRNAs as therapeutic entities and it was reported that US and European has published almost 500 patent applications and issued patents.41 For example, in hepatocellular carcinoma, up-regulation miR-34a via NOV340 liposome showed a great effect on treating hepatocellular carcinoma, including significant tumor reduction, life extension, and disease protection in animals; therefore clinical trials in 2013 for miR-34 has been initiated and miR-34a may be one of the first miRNA mimics to reach the clinic.42 Therefore, we hope one day miR-138-5p targeting LIMK1 could be a potential therapy for breast cancer.
Conclusion
Our results indicated that miR-138-5p could suppress breast cancer proliferation, motility and arrested breast cancer cells into G0/G1 phase by targeting LIMK1. However, more work still needs to be performed to explore miR-138-5p function in vivo experiments and further mechanisms.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by grant no. 201640097 from the Shanghai Municipal Health Bureau of Shanghai, China and grant no. 82172240 from the National Natural Science Foundation of China (to L. Fang).References
- T. G. Steenbruggen, M. S. van Ramshorst, M. Kok, S. C. Linn, C. H. Smorenburg and G. S. Sonke, Drugs, 2017, 77, 1313–1336 CrossRef CAS PubMed.
- A. D. Campos-Parra, G. C. Mitznahuatl, A. Pedroza-Torres, R. V. Romo, F. I. P. Reyes, E. Lopez-Urrutia and C. Perez-Plasencia, Int. J. Mol. Sci., 2017, 18, 1182 CrossRef PubMed.
- J. Leignadier, F. Dalenc, M. Poirot and S. Silvente-Poirot, Biochem. Pharmacol., 2017, 144, 18–28 CrossRef CAS PubMed.
- C. Marquette and L. Nabell, Curr. Treat. Options Oncol., 2012, 13, 263–275 CrossRef PubMed.
- S. Jonas and E. Izaurralde, Nat. Rev. Genet., 2015, 16, 421–433 CrossRef CAS PubMed.
- E. O'Day and A. Lal, Breast Cancer Res., 2010, 12, 201 CrossRef PubMed.
- T. You, W. Gao, J. Wei, X. Jin, Z. Zhao, C. Wang and Y. Li, Biomed. Pharmacother., 2015, 69, 96–101 CrossRef CAS PubMed.
- R. Bagheri-Yarmand, A. Mazumdar, A. A. Sahin and R. Kumar, Int. J. Cancer, 2006, 118, 2703–2710 CrossRef CAS PubMed.
- D. Li, J. Hu, H. Song, H. Xu, C. Wu, B. Zhao, D. Xie, T. Wu, J. Zhao and L. Fang, Am. J. Transl. Res., 2017, 9, 2276–2285 Search PubMed.
- D. Li, H. Wang, H. Song, H. Xu, B. Zhao, C. Wu, J. Hu, T. Wu, D. Xie, J. Zhao, Q. Shen and L. Fang, Oncotarget, 2017, 8, 85276–85289 Search PubMed.
- Y. Hashimoto, Y. Akiyama and Y. Yuasa, PLoS One, 2013, 8, e62589 CAS.
- J. Zhang, X. Kong, J. Li, Q. Luo, X. Li, L. Shen, L. Chen and L. Fang, Oncol. Rep., 2014, 31, 1357–1363 CrossRef CAS PubMed.
- R. Bologna-Molina, A. Mosqueda-Taylor, N. Molina-Frechero, A. D. Mori-Estevez and G. Sanchez-Acuna, Med. Oral Patol. Oral Cir. Bucal, 2013, 18, e174–179 CrossRef PubMed.
- A. Jezierska and T. Motyl, Med. Sci. Monit., 2009, 15, RA32–40 CAS.
- A. Merdad, S. Karim, H. J. Schulten, A. Dallol, A. Buhmeida, F. Al-Thubaity, M. A. Gari, A. G. Chaudhary, A. M. Abuzenadah and M. H. Al-Qahtani, Anticancer Res., 2014, 34, 1355–1366 CAS.
- R. P. Fisher, F1000Research, 2016, 5, 2374 CrossRef PubMed.
- Y. Li, S. Li, X. Meng, R. Y. Gan, J. J. Zhang and H. B. Li, Nutrients, 2017, 9, 728 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 7–30 CrossRef PubMed.
- F. Manetti, Med. Res. Rev., 2012, 32, 968–998 CrossRef CAS PubMed.
- R. Li, J. Doherty, J. Antonipillai, S. Chen, M. Devlin, K. Visser, J. Baell, I. Street, R. L. Anderson and O. Bernard, Clin. Exp. Metastasis, 2013, 30, 483–495 CrossRef CAS PubMed.
- J. Cui, M. Ding, W. Deng, Y. Yin, Z. Wang, H. Zhou, G. Sun, Y. Jiang and Y. Feng, Bioorg. Med. Chem., 2015, 23, 7464–7477 CrossRef CAS PubMed.
- Y. Yin, K. Zheng, N. Eid, S. Howard, J. H. Jeong, F. Yi, J. Guo, C. M. Park, M. Bibian, W. Wu, P. Hernandez, H. Park, Y. Wu, J. L. Luo, P. V. LoGrasso and Y. Feng, J. Med. Chem., 2015, 58, 1846–1861 CrossRef CAS PubMed.
- B. E. Sleebs, G. Nikolakopoulos, I. P. Street, H. Falk and J. B. Baell, Bioorg. Med. Chem. Lett., 2011, 21, 5992–5994 CrossRef CAS PubMed.
- E. Mashiach-Farkash, R. Rak, G. Elad-Sfadia, R. Haklai, S. Carmeli, Y. Kloog and H. J. Wolfson, Oncotarget, 2012, 3, 629–639 Search PubMed.
- J. B. Park, S. Agnihotri, B. Golbourn, K. C. Bertrand, A. Luck, N. Sabha, C. A. Smith, S. Byron, G. Zadeh, S. Croul, M. Berens and J. T. Rutka, Oncotarget, 2014, 5, 9382–9395 Search PubMed.
- M. D. Charles, J. L. Brookfield, T. C. Ekwuru, M. Stockley, J. Dunn, M. Riddick, T. Hammonds, E. Trivier, G. Greenland, A. C. Wong, A. Cheasty, S. Boyd, D. Crighton and M. F. Olson, J. Med. Chem., 2015, 58, 8309–8313 CrossRef CAS PubMed.
- K. Mardilovich, M. Baugh, D. Crighton, D. Kowalczyk, M. Gabrielsen, J. Munro, D. R. Croft, F. Lourenco, D. James, G. Kalna, L. McGarry, O. Rath, E. Shanks, M. J. Garnett, U. McDermott, J. Brookfield, M. Charles, T. Hammonds and M. F. Olson, Oncotarget, 2015, 6, 38469–38486 Search PubMed.
- M. Zare, M. Bastami, S. Solali and M. Alivand, J. Cell. Physiol., 2017, 1–16 CAS.
- L. Wan, L. Zhang, K. Fan and J. Wang, Mol. Cell. Biochem., 2014, 390, 85–91 CrossRef CAS PubMed.
- H. Xia, S. Sun, B. Wang, T. Wang, C. Liang, G. Li, C. Huang, D. Qi and X. Chu, Int. J. Mol. Sci., 2014, 15, 11973–11983 CrossRef PubMed.
- Y. Xiong, L. Zhang and E. Kebebew, PLoS One, 2014, 9, e96103 Search PubMed.
- J. Zhou, R. Liu, C. Luo, X. Zhou, K. Xia, X. Chen, M. Zhou, Q. Zou, P. Cao and K. Cao, Cancer Biol. Ther., 2014, 15, 1340–1349 CrossRef CAS PubMed.
- M. Jurikova, L. Danihel, S. Polak and I. Varga, Acta Histochem., 2016, 118, 544–552 CrossRef CAS PubMed.
- J. Zhang, D. Liu, Z. Feng, J. Mao, C. Zhang, Y. Lu, J. Li, Q. Zhang, Q. Li and L. Li, Biomed. Pharmacother., 2016, 77, 135–141 CrossRef CAS PubMed.
- A. P. Trape, S. Liu, A. C. Cortes, N. T. Ueno and A. M. Gonzalez-Angulo, J. Cancer, 2016, 7, 947–956 CrossRef PubMed.
- J. Kanska, M. Zakhour, B. Taylor-Harding, B. Y. Karlan and W. R. Wiedemeyer, Gynecol. Oncol., 2016, 143, 152–158 CrossRef CAS PubMed.
- T. Sumi, K. Matsumoto and T. Nakamura, Biochem. Biophys. Res. Commun., 2002, 290, 1315–1320 CrossRef CAS PubMed.
- O. Bernard, Int. J. Biochem. Cell Biol., 2007, 39, 1071–1076 CrossRef CAS PubMed.
- T. Sumi, A. Hashigasako, K. Matsumoto and T. Nakamura, Exp. Cell Res., 2006, 312, 1021–1030 CrossRef CAS PubMed.
- W. Wang, L. Zhang, Y. Wang, Y. Ding, T. Chen, Y. Wang, H. Wang, Y. Li, K. Duan, S. Chen, Q. Yang and C. Chen, Cell Death Dis., 2017, 8, e3071 CrossRef PubMed.
- A. F. Christopher, R. P. Kaur, G. Kaur, A. Kaur, V. Gupta and P. Bansal, Perspect. Clin. Res., 2016, 7, 68–74 CrossRef PubMed.
- A. G. Bader, Front. Genet., 2012, 3, 120 CAS.
| This journal is © The Royal Society of Chemistry 2017 |