
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulating molecular structures and dielectric transitions in organic–inorganic hybrid crystals†
Xiao-Fen Sun,
Zhongxia Wang *,
Wei-Qiang Liao,
Peng-Fei Li,
Jixing Gao,
Yi-Yang Huang,
Hai-Peng Chen,
Heng-Yun Ye and
Yi Zhang*
*,
Wei-Qiang Liao,
Peng-Fei Li,
Jixing Gao,
Yi-Yang Huang,
Hai-Peng Chen,
Heng-Yun Ye and
Yi Zhang*
Ordered Matter Science Research Center, Jiangsu Key Laboratory for Science and Applications of Molecular Ferroelectrics, College of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, PR China. E-mail: yizhang1980@seu.edu.cn; zx880376@126.com
First published on 8th November 2017
Abstract
In this paper, three new hybrid phase transition compounds, [Hmpy]CdBr3 (1, Hmpy = N-methylpyrrolidinuium cation), [Hmpy]2CdBr4 (2) and [Hmpy]3CdBr3·CdBr4 (3), were synthetized by means of regulating the ratio of reactants. Systematic characterizations consisting of variable temperature X-ray single crystal diffraction, differential scanning calorimetry (DSC) and dielectric measurements reveal that 1 with infinite one-dimensional (1D) [CdBr3]n− chains undergoes a phase transition around 278 K; 2 with isolated [CdBr4]2− tetrahedrons exhibits a high-temperature phase transition close to 367 K, accompanied by prominent switchable dielectric behavior. Interestingly, 1D [CdBr3]n− chains and isolated [CdBr4]2− tetrahedrons both exist in 3, associated with a phase transition at 320 K. The phase transitions in the three compounds are originated from the order-disorder transitions of the Hmpy cation. It is expected that our finding would promote the development of hybrid dielectric transition materials with adjustable properties.
Introduction
The macroscopic properties of functional materials are generally closely related to their microstructure. Consequently, the control of material properties, such as dielectric, optical and magnetic properties could be realized through the regulation of molecular structure.1 Recently, much interest has been devoted to the dielectric materials (switchable dielectric and ferroelectric, etc.) for their promising uses as the modern electrical and electronic devices.2 Theoretically, dielectric behavior is always companied by the changes in molecular dipolar moments, as a result, phase transition compounds associated with the variable dipolar moments have been mostly reported to be dielectric materials.3–5 However, only a small number of dielectric transition materials could be considered as promising dielectric candidates regarding their phase transition temperature (above/or near the room temperature) and dielectric behavior (e.g. switchable dielectric). Therefore, great efforts have been paid into controlling the dielectric properties by means of adjusting the molecular structures.6–9Organic–inorganic hybrid strategy has been proven to be one of the most feasible approaches to design diverse structures due to their readily self-assembling.10,11 At the same time, studies of organic–inorganic hybrid has also been extended to phase transition materials.12–14 Structurally, the organic components located in the gaps of the inorganic metal skeletons possess large freedom of motion (e.g. reorientation/frozen transitions).15 The dynamic components are responsive to the external stimuli such as temperature, light, pressure, electric and magnetic fields, etc.,16 resulting in structural phase transition accompanied by some interesting physical properties17–21 In this case, the dielectric properties might be tuned through the modulation of organic cations in organic–inorganic hybrid phase transition materials.22–25 For example, a series of metal-formate A[Mg(HCOO)3] (A = organic ammonium cation), [NH4][Mg(HCOO)3], [CH3CH2NH3][Mg(HCOO)3], and [NH3(CH2)4NH3][Mg2(HCOO)6] display dielectric transitions at 255, 374, and 412 K, respectively.26 The approaches of replacing organic cations achieve the regulation of dielectric transitions from low temperature to high temperature, as well as the dielectric responsive characteristics. Besides, the regulation of dielectric properties can also be achieved by the control of inorganic frameworks.5,27 For instance, B-site of metal-formate system [NH2NH3][B(HCOO)3] (B = Mn2+, Zn2+, Co2+ and Mg2+) undergo dielectric responses at 350 K (Mn and Zn), 380 K (Co) and 348 K (Mg), respectively.5 Therefore, some practical and promising dielectric properties could be obtained by substituting components within the similar structures. But the reports of controlling dielectric responses in completely different inorganic frameworks resulted from the modulation of ratio of the reactants are very scarce.
Herein, the present work has been reported structural phase transitions and dielectric responses in three hybrids, [Hmpy]CdBr3 (1), [Hmpy]2CdBr4 (2) and [Hmpy]3CdBr3·CdBr4 (3), which were synthetized by the stoichiometric regulation of same reactants (Scheme 1). These three organic–inorganic hybrid compounds with completely different molecular structures display controllable structural phase transitions and dielectric responses: 1 with 1D chains experiences a dielectric phase transition at 278 K, 2 with isolated tetrahedrons shows a switchable dielectric anomaly at about 367 K and 3 with integrated geometry undergoes a phase transition at 320 K, achieving the regulation of phase transition temperatures and dielectric behaviors. Systematic characterization reveals that the order-disorder transitions of Hmpy cations lead to the phase transitions coupled with striking dielectric behavior in the three compounds.
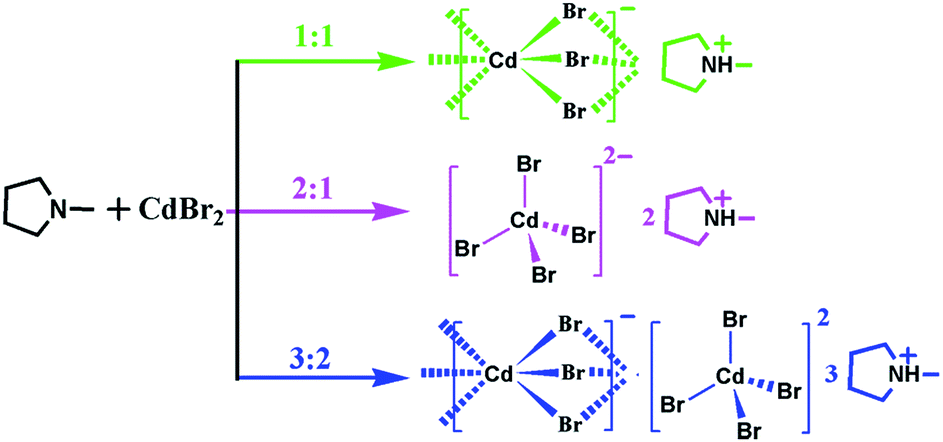 | ||
| Scheme 1 The synthetic routes of 1, 2 and 3. | ||
Experimental
Synthesis
All chemicals were purchased from Aladdin and without further purification. Clear colorless crystals of 1, 2 and 3 were synthesized by slow evaporation of the HBr/H2O (40%, w/w) solution containing Hmpy hydrobromide and CdBr2·4H2O with the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1 and 3:2 after a few weeks, respectively (Fig. S1, ESI†). Phase purities of 1, 2 and 3 in the form of polycrystalline samples were confirmed by powder X-ray diffraction (PXRD) measurements, in which the experimental patterns match well with the simulated ones (Fig. S2–S4, ESI†). Infrared (IR) spectra show the strong vibration peak in the three compounds at around 3100 cm−1, indicating the presence of R3N+H (Fig. S5, ESI†). TGA measurements reveal that the samples of 1, 2 and 3 are stable below about 560 K (Fig. S6, ESI†).
:1, 2:1 and 3:2 after a few weeks, respectively (Fig. S1, ESI†). Phase purities of 1, 2 and 3 in the form of polycrystalline samples were confirmed by powder X-ray diffraction (PXRD) measurements, in which the experimental patterns match well with the simulated ones (Fig. S2–S4, ESI†). Infrared (IR) spectra show the strong vibration peak in the three compounds at around 3100 cm−1, indicating the presence of R3N+H (Fig. S5, ESI†). TGA measurements reveal that the samples of 1, 2 and 3 are stable below about 560 K (Fig. S6, ESI†).
Single-crystal X-ray crystallography
Variable-temperature X-ray single-crystal diffraction data for the single crystals of 1, 2 and 3 were collected on a Rigaku Saturn 724+ diffractometer with Mo-Kα radiation (λ = 0.71073 Å). All the structures were solved using direct methods and refined by the full-matrix method based on F2 by means of the SHELXLTL software package.28 The non-hydrogen atoms were refined anisotropically using all reflections with I > 2σ(I). The carbon-bound H atoms were added geometrically and refined using the riding model with Uiso = 1.2Ueq(C and N). As to 2, the structure at 373 K was refined in P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) with high R factors, resulting from the poor crystal quality after long exposure at high temperature. Crystallographic data and details of the three compounds are summarized in Table S1, (ESI†).
with high R factors, resulting from the poor crystal quality after long exposure at high temperature. Crystallographic data and details of the three compounds are summarized in Table S1, (ESI†).
Thermal measurements
Thermogravimetric analysis was recorded on a DSC/DTA-TG SDT-Q600 instrument at the rate of 10 K min−1 under nitrogen atmosphere from room temperature to 800 K. Differential scanning calorimetry (DSC) was performed on a Perkin-Elmer Diamond DSC instrument by heating and cooling the polycrystalline samples with a rate of 10 K min−1 in aluminum crucibles under nitrogen at atmospheric pressure.Dielectric measurements
The dielectric permittivity toward 1, 2 and 3 were measured on a Tonghui TH2828A instrument at selected frequencies in the corresponding temperature range. The measuring AC voltage was 1 V. The powder-pressed pellets and single crystal sample deposited with silver conducting glue were used in dielectric measurements.Results and discussion
Thermal properties
DSC measurements were performed to study the phase transitions in 1, 2 and 3. As plotted in Fig. 1, thermal anomaly peaks were detected at 273/278 K for 1, 340/367 K for 2 and 315/320 K for 3 in cooling and heating runs. Such results demonstrate a reversible phase transition in 1 at 273 K (below the room temperature), in 2 at 367 K and in 3 at 320 K (above the room temperature), respectively. Moreover, the observed thermal hysteresis (5 K for 1 and 27 K for 2) and obvious λ-type peak reveal the feature of discontinuous first-order in the process of phase transitions.4,7–10 The broad peaks in 3 with a hysteresis of 5 K indicates a weak first-order type of phase transition. It is noted that the phase transition point (Tc) of 2 is approximately 97 K higher than 1 and about 47 K higher than 3. The obvious changes of Tc in the three compounds illustrate that their phase transition properties have been adjusted through the modification of molecular structures (Scheme 1), achieving the regulation of Tc in a wide temperature range. In addition, the ΔS (J mol−1 K−1) for phase transitions in the heating process of 1, 2 and 3 were estimated approximately 3.146, 0.813 and 0.472, respectively. Based on the Boltzmann equation ΔS = Rln(N), R being the gas constant, the ratio of respective geometrically distinguishable orientations N (1), N (2) and N (3) are calculated to be 1.46, 1.10 and 1.06, respectively. For convenience, the phases above 273 K (1), 367 K (2) and 320 K (3) are assigned as the high-temperature phase (HTP), and the phase below 273 K (1), 340 K (2) and 315 K (3) as the low-temperature phase (LTP).
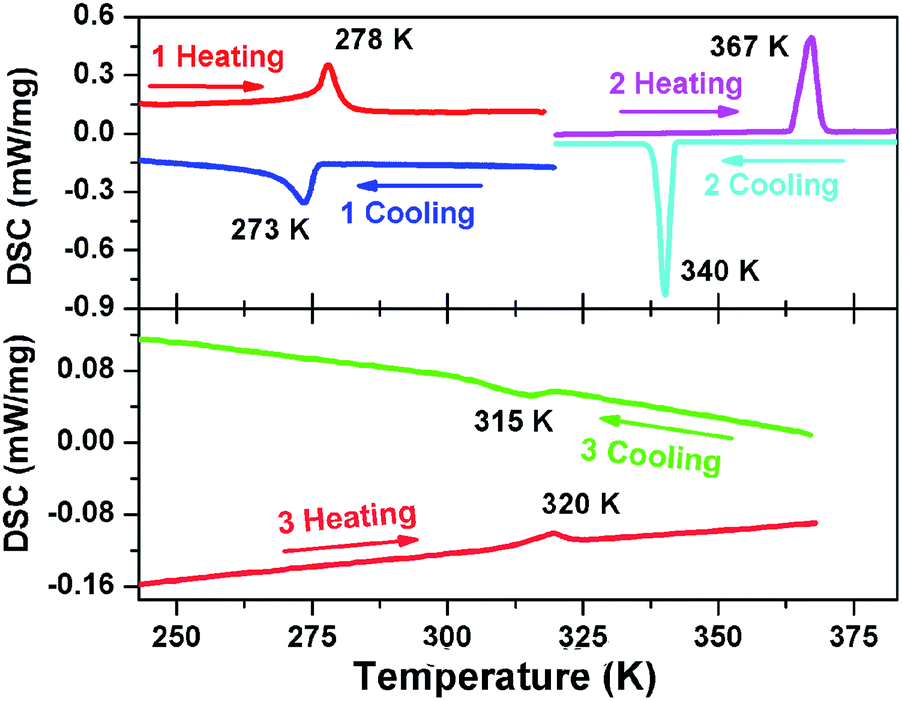 | ||
| Fig. 1 DSC curves of 1 (a), 2 (b) and 3 (c). | ||
Crystal structures
The crystal structure of 1 has a hexagonal perovskite-type structure, containing infinite chains of face-sharing CdBr6 octahedrons, which are comparable to some reported one dimensional perovskite-type hybrids, such as [C3H4NS][CdBr3].29 1 crystallizes in the orthorhombic space group Pnma (no. 62) at 293 K (HTP). The asymmetric unit in the HTP consists of one independent Hmpy cation, three Cd atoms, and six Br atoms, where the Hmpy cation is divided equally by the m plane (101) (Fig. 2a). The face-sharing Cd octahedrons with Cd–Br distances ranging from 2.771 (1) to 2.806 (1) Å and adjacent Br–Cd–Br angles varying from 83.63 (5)° to 180° (Table S2, ESI†) form one-dimensional chains along the crystallographic b-axis, in which the Hmpy cations occupy the cavities between the adjacent chains (Fig. S7, ESI†). Upon cooling to 263 K (LTP), the inorganic configuration of 1 is similar to that in the HTP. But the crystal structure was solved in the space group P21/c (no. 14). The relationship of the cell unit is b263 K ≈ a293 K and a263 K ≈ b293 K (Table S1, ESI†). The molecular structure of LTP is composed of an independent Hmpy cation, three Cd atoms, and nine Br atoms, accompanied by the absence of mirror plane. Moreover, the Hmpy cation is clearly ordered than that in the HTP (Fig. 2b).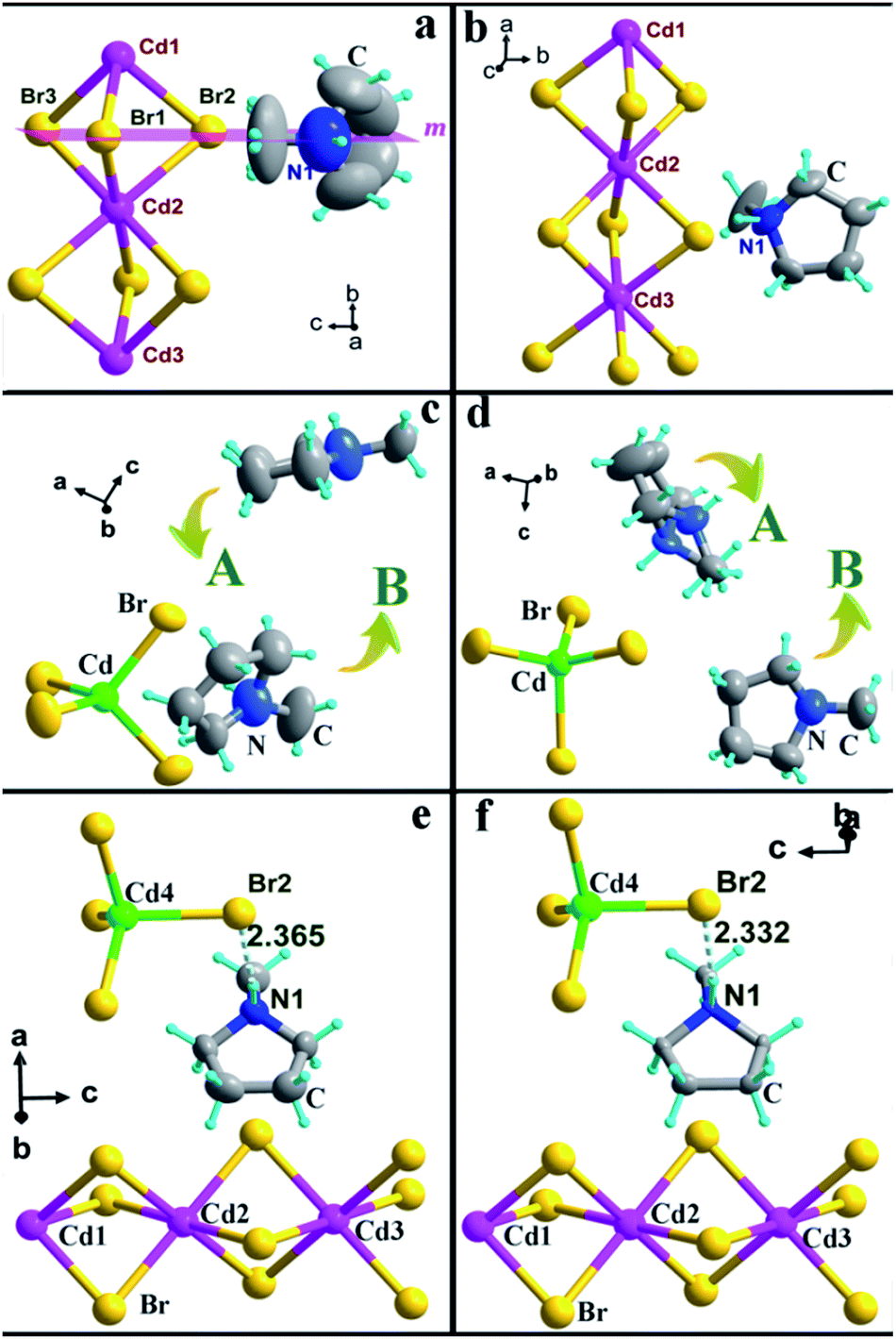 | ||
| Fig. 2 Molecular structures of 1 at 293 K (a) and 263 K (b). Molecular structures of 2 at 373 K (c) and 293 K (d). Molecular structures of 3 at 343 K (e) and 253 K (f). The dashed lines denote the N–H⋯Br interactions. Thermal ellipsoids for all atoms are shown at 30% probability level. | ||
Compound 2 contains an independent monomeric [CdBr4]2− anion and two crystallographically independent Hmpy cations (Fig. 2c and d). In the HTP, 2 crystallizes in the centrosymmetric orthorhombic space group Pbca (no. 61). The value of Br–Cd–Br angles belonging to [CdBr4]2− anion vary from 104.74(2)° to 113.11(2)° and the Cd–Br distances range from 2.554 (4) to 2.615 (5) Å (Table S3, ESI†), leading to a weak distortion of tetrahedrons. As the temperature decreased to the LTP, the inorganic skeleton of 2 is almost unchanged compared to that in the HTP (Table S3, ESI†). It is noted that the Hmpy cations can be described in two parts (A and B). In the HTP, A and B molecules are all strongly disordered. Moreover, the atoms of A molecule are almost in a plane (Fig. 2c). Whereas, the Hmpy cation experiences great changes in the LTP, in which the N atom of A part occupies two symmetric sites and the two Hmpy cations are more ordered than those in the HTP (Fig. 2d). In addition, the organic cations have hydrogen-bonding interactions with the inorganic chains through N–H⋯Br hydrogen bonds with the donor–acceptor distances of 3.491(1) Å, contributing to a three-dimensional hydrogen bond network (Table S4 and Fig. S8, ESI†).
Interestingly, compound 3 consists of monomeric [CdBr4]2− anions, infinite [CdBr3]n− chains and independent Hmpy cations (Fig. 2e and f). The crystal structures of 3 in the HTP and LTP both crystallize in trigonal space group P and the inorganic frameworks regarding [CdBr3]− and [CdBr4]2− anions are comparable to those in 1 and 2 (Table S5, ESI†). The cell parameters of HTP and LTP show slightly different (Table S1 ESI†), but the thermal ellipsoids of Hmpy are obviously larger than those in the LTP (Fig. 2e and f). As shown in Fig. S9 (ESI†), each N atom of the Hmpy bearing an extra proton that forms hydrogen-bonding interaction toward a Br atom belonging to a [CdBr4]2− anion with the distance of N–H⋯Br 2.332 Å (HTP) and 2.365 Å (LTP), respectively (Table S6, ESI†). Then, it seems like that three Hmpys and an independent [CdBr4]2− anion as a whole fills in the blank between the inorganic chains along the c-axis.
Dielectric properties
The temperature dependence of the real part (ε′) of the complex dielectric permittivity at several selected frequencies is shown in Fig. 3. For 1, there is a pair of anomalies at 1000 kHz in the vicinity of 273 K in a cooling and heating run, indicating a reversible dielectric transition (Fig. 3a). Upon cooling, the ε′ slowly decreases to ca. 14 with the decreasing of temperature from 310 to 275 K. Then ε′ occurs a turning point at 273 K and goes on decreasing slowly to offer a inconspicuous step-like type dielectric anomaly. The unstable dielectric value before and after the phase transition point demonstrates the competition between external electric field induced orientational polarization and temperature triggered dynamic motion of the Hmpy cation.30 Moreover, as the frequency decreases, the anomaly around 273 K is gradually obvious with the larger changes of the ε′, revealing the ε′ is strongly frequency dependent (Fig. 3b). As to the imaginary part ε′′, there is a pair of wide peaks close to 273 K in a cooling and heating run (1000 kHz), confirming the reversible phase transition in 1 (Fig. S10a, ESI†). Also, the ε′′ demonstrates slightly frequency dependence in that the maximum value of 800 kHz is greater than that of 1000 kHz. In combination of structural analyses, the dielectric transition in 1 is in virtue of the order-disorder transition of the Hmpy cation.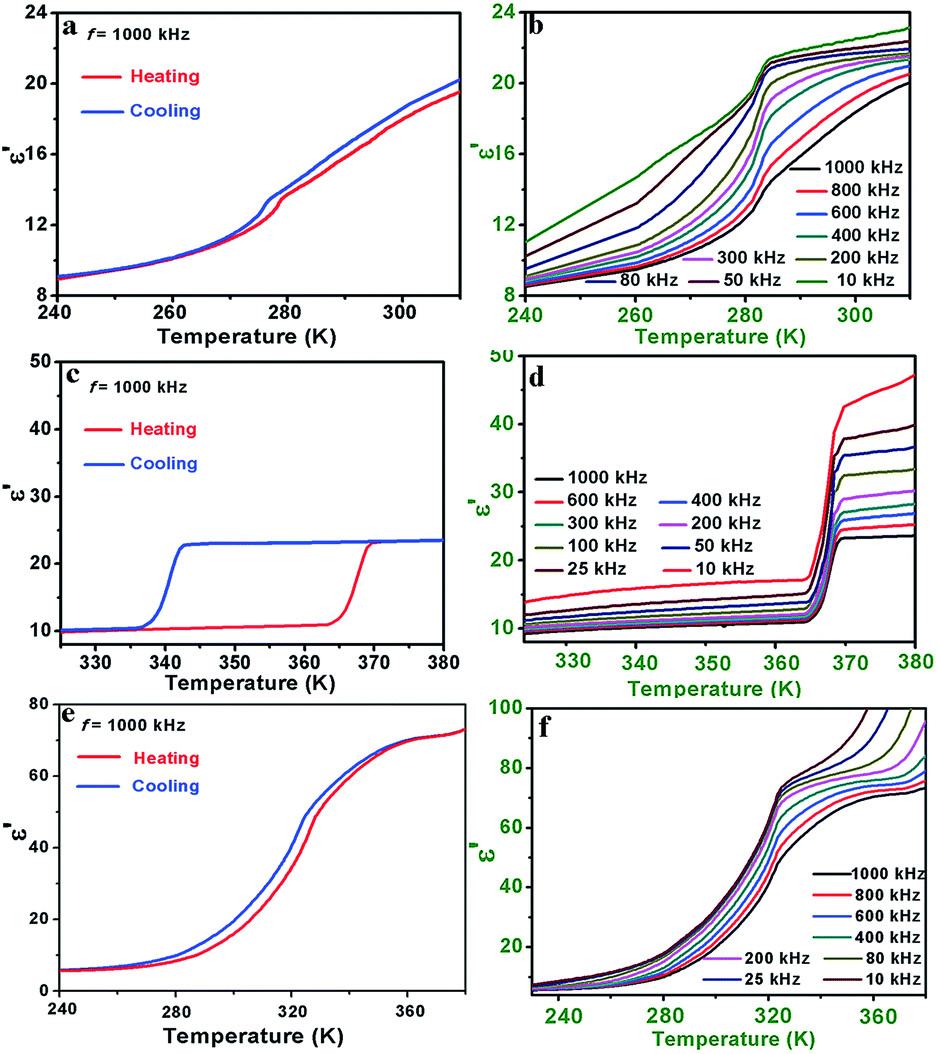 | ||
| Fig. 3 The temperature-dependence of the real part (ε′) measured at 1000 kHz and selected frequencies of the polycrystalline sample of 1 (a and b), 2 (c and d) and 3 (e and f). | ||
For 2, temperature dependent ε′ was measured in the temperature range of 325–380 K at 1000 kHz are drawn in Fig. 3c, where ε′ exhibits an remarkable step-like dielectric behavior. In the heating process, the ε′ remains stable at ca. 10 (low-dielectric state) below 364 K. Then the ε′ shows a sharp rise close to 23.5 (high-dielectric state) followed by an approximately 2 times as high as that in the LTP. The large thermal hysteresis during the heating and cooling process (ca. 25 K) and the remarkable dielectric switching make 2 a promising energy storage and switchable dielectric material. Also, the ε′ of 2 shows the prominent frequency dependence. The ε′ at lower frequencies increases quickly accompanied by the decreasing of measured frequencies (Fig. 3d), and the ε′′ is similar to ε′ with strongly frequency dependence (Fig. S10b, ESI†). The thermal hysteresis observed in the curve of ε′′ (ca. 27 K) is accordance with the real part and the result of DSC measurement. Based on the single crystal structures of 2, the Hmpy cations are strongly disordered in the HTP. As the temperature decreased to the LTP, the Hmpy cations change to be relative ordered.31,32 Therefore, the dielectric responses of 2 are originated from the dynamic motions of the organic cations.
For 3, there is a general change in the curve of ε′ around 320 K (Fig. 3e). In the HTP above 360 K, the ε′ keeps a value of ca. 70. And then, the ε′ slowly decreases to 6 with the temperature cooling to around 240 K in the LTP. Moreover, the dependence of frequency also can be observed in the curve of ε′ in the range of 10 kHz–1000 kHz (Fig. 3f). The unstable dielectric behaviors in the HTP and LTP are similar to those in 1, indicating the competition between the thermal motion and polarization of organic cations. The imaginary part ε′′ shows broad peaks in the process of phase transition around 320 K (Fig. S10c, ESI†).
Therefore, the features of the dielectric responses of 1, 2 and 3 can be addressed by the orientational transitions of the Hmpy cations between the HTP and LTP. In the HTP, the organic cations demonstrate dynamic motions. As the temperature decreased in the LTP, the motions of cations are frozen.33 In addition, the difference in phase transition temperature and the dielectric behaviors in the three compounds is due to the various inorganic anionic frameworks, leading to the different dynamic motions of the Hmpy cations.11c
The dielectric anisotropy measurements of 1, 2 and 3 were performed on the crystal samples, respectively. As shown in Fig. 4a, the ε′ of 1 in the HTP along the b- and a-axes are larger than that in the powdered samples, but the ε′ along the c-axis is the smallest one. Based on the structural analyses, the rotation motions of the Hmpy cations in the ab-plane may be responsible for the dielectric anisotropy in 1. In 2, step-like dielectric anomalies are observed around 367 K along the different crystallographic directions. Moreover, the ε′ along the a- and c-axes are larger than that in the direction of b-axis. The anisotropy can be explained that the reorientationally disordered Hmpy cations in 2 settled in the ac-plane will result in the motions along a-and c-axes. In 3, the ε′ along the c-axis is remarkably greater than those along the a-and b-axes. It can be illustrated that the Hmpy cations in 3 are located in the planes parallel to c-axis and the reorientational motions of Hmpy cations will give the largest contribution to the dielectric anomaly along the c-axis.
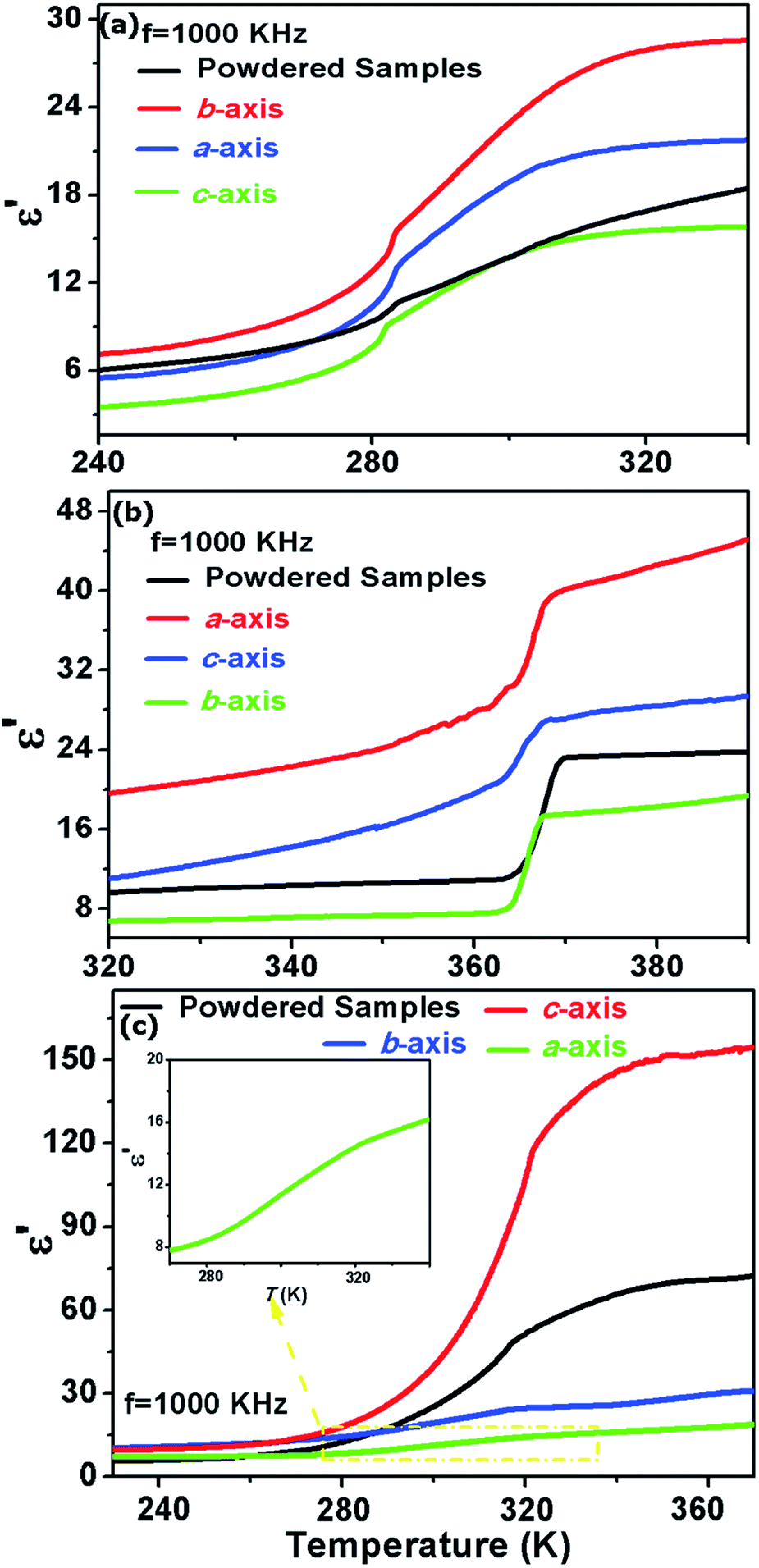 | ||
| Fig. 4 The temperature-dependence of the real part (ε′) of 1 (a), 2 (b) and 3 (c) measured on crystal samples along the a-, b- and c-axes. | ||
Conclusions
In this work, three new controllable structural cadmium–bromate compounds have successfully synthesized by regulating the ratio of reactants. Compound 1 is comprised of the [CdBr3]n− chains and the Hmpy cations occupied the space of the adjacent chains. The isolated [CdBr4]2− tetrahedrons and the Hmpy cations are alternated in 2. Interestingly, 3 shows novel structure, containing [CdBr3]n− chains, isolated [CdBr4]2− tetrahedrons and Hmpy cations filled in the interval of inorganic frameworks. In the diverse molecular structures of the three hybrid compounds, the order-disorder transitions of the Hmpy cations, which are responsive to the temperature, contribute to the adjustable dielectric responses. 1 displays a phase transition at ca. 273 K. 2 shows a promising switchable dielectric behavior approximately at 367 K. 3 undergoes a phase transition ca. 320 K. Given that the reported hybrid structures here are easily prepared and tuned, our finding is expected to provide new impetus to construct new functional materials showing controllable structures with adjustable properties based on organic–inorganic hybrids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (Grant 2017YFA0204803), the National Natural Science Foundation of China (21522101, 21371032), the Outstanding Young Teachers of Southeast University Research Fund (Grant 2242015R30025, 2242017K41027), and the Scientific Research Foundation of Graduate School of Southeast University (YBJJ1629).Notes and references
- (a) Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed; (b) P. P. Shi, Y. Y. Tang, P. F. Li, W. Q. Liao, Z. X. Wang, Q. Ye and R. G. Xiong, Chem. Soc. Rev., 2016, 45, 3811–3827 RSC; (c) Y. Morita, T. Murata and K. Nakasuji, Bull. Chem. Soc. Jpn., 2013, 86, 183–197 CrossRef CAS.
- (a) W. Zhang, H. Y. Ye, R. Graf, H. W. Spiess, Y. F. Yao, R. Q. Zhu and R. G. Xiong, J. Am. Chem. Soc., 2013, 135, 5230–5533 CrossRef CAS PubMed; (b) H. Y. Ye, S. H. Li, Y. Zhang, L. Zhou, F. Deng and R. G.Xiong, J. Am. Chem. Soc., 2014, 136, 10033–10040 CrossRef CAS PubMed; (c) Y. Imai, B. Zhou, Y. Ito, H. Fijimori, A. Kobayashi, Z. M. Wang and H. Kobayashi, Chem.–Asian J., 2012, 7, 2786–2790 CrossRef CAS PubMed.
- (a) X.-H. Lv, W.-Q. Liao, Z.-X. Wang, P.-F. Li, C.-Y. Mao and H.-Y. Ye, Cryst. Growth Des., 2016, 16, 3912–3916 CrossRef CAS; (b) P. Li, X. Yang, T. W. Mass, J. Hanss, M. Lewin, A. K. Michel, M. Wuttig and T. Taubner, Nat. Mater., 2016, 15, 870–875 CrossRef CAS PubMed.
- Y. Zhang, H.-Y. Ye, W. Zhang and R.-G. Xiong, Inorg. Chem. Front., 2014, 1, 118 RSC.
- S. Chen, R. Shang, K. L. Hu, Z. M. Wang and S. Gao, Inorg. Chem. Front., 2014, 1, 83–98 RSC.
- B. Orayech, L. Ortega-San-Martin, I. Urcelay-Olabarria, L. Lezama, T. Rojo, M. I. Arriortua and J. M. Igartua, Dalton Trans., 2016, 45, 14378–14393 RSC.
- Y.-L. Liu, Y.-F. Wang and W. Zhang, CrystEngComm, 2016, 18, 1958–1963 RSC.
- W. Zhang, Y. Cai, Y. R. G. Xiong, H. Yoshikawa and K. Awaga, Angew. Chem., Int. Ed., 2010, 49, 6608–6610 CrossRef CAS PubMed.
- Z. X. Wang, W. Q. Liao, H. Y. Ye and Y. Zhang, Dalton Trans., 2015, 44, 20406–20412 RSC.
- (a) H. P. Chen, Z. X. Wang, C. Chen, Y. Lu, Z. Yin, X. F. Sun and D. W. Fu, Dalton Trans., 2017, 46, 4711–4716 RSC; (b) A. Sieradzki, S. Pawlus, S. N. Tripathy, A. Gagor, M. Ptak, M. Paluch and M. Maczka, Dalton Trans., 2017, 46, 3681–3687 RSC; (c) R. Valiente, M. C. Marco de Lucas, J. I. Espeso and F. Rodriguez, Solid State Commun., 1993, 86, 663–666 CrossRef CAS.
- (a) Y. Zhang, W. Q. Liao, D. W. Fu, H. Y. Ye, Z. N. Chen and R. G. Xiong, J. Am. Chem. Soc., 2015, 137, 4928–4931 CrossRef CAS PubMed; (b) Y. Zhang, W. Q. Liao, D. W. Fu, H. Y. Ye, C. M. Liu, Z. N. Chen and R. G. Xiong, Adv. Mater., 2015, 27, 3942–3946 CrossRef CAS PubMed; (c) X. F. Sun, Z. Wang, P. F. Li, W. Q. Liao, H. Y. Ye and Y. Zhang, Inorg. Chem., 2017, 56, 3506–3511 CrossRef CAS PubMed.
- (a) P. Jain, N. S. Dalal, B. H. Toby, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2008, 130, 10450–10451 CrossRef CAS PubMed; (b) P. Jain, V. Ramachandran, R. J. Clark, H. D. Zhou, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625–13627 CrossRef CAS PubMed.
- A. Piecha, A. Białońska, R. Jakubas and W. Medycki, Solid State Sci., 2008, 10, 1469–1479 CrossRef CAS.
- (a) Z. Cui, K. Gao, C. Liu, Y. Yin, D.-W. Fu, H.-L. Cai and X. S. Wu, J. Phys. Chem. C, 2016, 120, 2925–2931 CrossRef CAS; (b) T. Khan, M. A. Asghar, Z. Sun, A. Zeb, L. Li, L. Sijie, S. Zhao, C. Ji and J. Luo, Chem.–Asian J., 2016, 11, 2876–2881 CrossRef PubMed.
- (a) W. Zhang, H.-Y. Ye and R.-G. Xiong, Coord. Chem. Rev., 2009, 253, 2980–2997 CrossRef CAS; (b) Z. X. Wang, P. F. Li, W. Q. Liao, Y. Tang, H. Y. Ye and Y. Zhang, Chem.–Asian J., 2016, 11, 981–985 CrossRef CAS PubMed.
- (a) R. Ramesh, Nature, 2009, 461, 1218–1219 CrossRef CAS PubMed; (b) M. Wuttig and N. Yamada, Nat. Mater., 2007, 6, 824–832 CrossRef CAS PubMed.
- (a) M. Salinga and M. Wuttig, Science, 2011, 332, 543–544 CrossRef CAS PubMed; (b) S. Zhang, J. Zhao, P. Ma, J. Niu and J. Wang, Chem.–Asian J., 2012, 7, 966–974 CrossRef CAS PubMed.
- F. M. Raymo, Adv. Mater., 2002, 14, 401–414 CrossRef CAS.
- C. Ji, Z. Sun, S. Q. Zhang, T. Chen, P. Zhou, Y. Tang, S. Zhao and J. Luo, J. Mater. Chem. C, 2014, 2, 6134–6139 RSC.
- M. N. Tchoul, S. P. Fillery, H. Koerner, L. F. Drummy, F. T. Oyerokun, P. A. Mirau, M. F. Durstock and R. A. Vaia, Chem. Mater., 2010, 22, 1749–1759 CrossRef CAS.
- W. Fu and T. Nakamura, Biosci., Biotechnol., Biochem., 2017, 81, 839–847 CrossRef CAS PubMed.
- K. Gao, M. Gu, X. Qiu, X. N. Ying, H. Y. Ye, Y. Zhang, J. Sun, X. Meng, F. M. Zhang and D. Wu, J. Mater. Chem. C, 2014, 2, 9957–9963 RSC.
- J.-Z. Ge, X.-Q. Fu, T. Hang, Q. Ye and R.-G. Xiong, Cryst. Growth Des., 2010, 10, 3632–3637 CAS.
- P. Zhou, Z. Sun, S. Zhang, C. Ji, S. Zhao, R. G. Xiong and J. Luo, J. Mater. Chem. C, 2014, 2, 2341–2345 RSC.
- Y.-Z. Tang, Z.-F. Gu, J.-B. Xiong, J.-X. Gao, Y. Liu, B. Wang, Y.-H. Tan and Q. Xu, Chem. Mater., 2016, 28, 4476–4482 CrossRef CAS.
- R. Shang, G. C. Xu, Z. M. Wang and S. Gao, Chem.–Eur. J., 2014, 20, 1146–1158 CrossRef CAS PubMed.
- (a) W.-J. Xu, K.-P. Xie, Z.-F. Xiao, W.-X. Zhang and X.-M. Chen, Cryst. Growth Des., 2016, 16, 7212–7217 CrossRef CAS; (b) Y. Zhang, W.-Q. Liao, H.-Y. Ye, D.-W. Fu and R.-G. Xiong, Cryst. Growth Des., 2013, 13, 4025–4030 CrossRef CAS; (c) Q. Ye, T. Akutagawa, N. Hoshino, T. Kikuchi, S. Noro, R. G. Xiong and T. Nakamura, Cryst. Growth Des., 2011, 11, 4175–4182 CrossRef CAS; (d) Z. Sun, J. Luo, T. Chen, L. Li, R. G. Xiong, M. L. Tong and M. Hong, Adv. Funct. Mater., 2012, 22, 4855–4861 CrossRef CAS.
- G. M. Sheldrick, SHELX-97, Program for Crystal Structure Determination, University of Göttingen, Germany, 1997 Search PubMed.
- W. Q. Liao, H. Y. Ye, Y. Zhang and R. G. Xiong, Dalton Trans., 2015, 44, 10614–10620 RSC.
- X. D. Shao, X. Zhang, C. Shi, Y. F. Yao and W. Zhang, Adv. Sci., 2015, 1500029 CrossRef PubMed.
- S. Thies, H. Sell, C. Schütt, C. Bornholdt, C. Näther, F. Tuczek and R. Herges, J. Am. Chem. Soc., 2011, 133, 16243–16250 CrossRef CAS PubMed.
- A. Piecha, A. Gągor, M. Węcławik, R. Jakubas and W. Medycki, Mater. Res. Bull., 2013, 48, 151–157 CrossRef CAS.
- (a) W. Q. Liao, G. Q. Mei, H. Y. Ye, Y. X. Mei and Y. Zhang, Inorg. Chem., 2014, 53, 8913–8918 CrossRef CAS PubMed; (b) D. W. Fu, W. Zhang, H. L. Cai, Y. Zhang, J. Z. Ge, R. G. Xiong, S. D. Huang and T. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 11947–11951 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal pictures, IR spectrums, PXRD patterns and TGA results. CCDC 1535112 (263 K) and 1535113 (293 K) for 1, 1535114 (293 K) and 1535115 (373 K) for 2, as well as 1535116 (253 K) and 1535117 (343 K) for 3. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra10616e |
| This journal is © The Royal Society of Chemistry 2017 |