
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation behavior of silicon nitride fibers obtained from polycarbosilane fibers via electron beam irradiation curing
X. Sun,
H. T. Liu * and
H. F. Cheng
* and
H. F. Cheng
Science and Technology on Advanced Ceramic Fibers and Composites Laboratory, National University of Defense Technology, Changsha 410073, China. E-mail: htslht@163.com
First published on 11th October 2017
Abstract
Near-stoichiometric silicon nitride (Si3N4) fibers, which were successfully prepared from polycarbosilane fibers via electron beam irradiation curing, were heat-treated at elevated temperature for 2 h in the air atmosphere. The compositions and microstructures of silicon nitride fibers before and after heat treatment were investigated by XRD, XPS, NMR, SEM and TEM analyses. Tensile properties of the untreated and heat-treated fibers were also studied. The results show that the untreated fibers were mainly composed of amorphous silicon nitride and a few Si2N2O phases. During the heat treatment process, the oxidation of the fibers from silicon nitride to silicon dioxide initiated at 1100 °C. As the treatment temperature increased to 1400 °C, an oxidation layer with a thickness of ∼2 μm was formed on the fiber surface. Besides, when the treatment temperature was up to 1200 °C, the strength retention of the fibers still was 50.29%, which indicates that the fiber might possess a high serving life at a temperature lower than 1200 °C.
1. Introduction
Continuous silicon nitride (Si3N4) fibers are considered as one of the important ceramic fibers due to their ascendant properties, such as excellent mechanical properties, high oxidation resistance, superior electrical insulating property at elevated temperature and low dielectric constant. They can be regarded as one of the most suitable reinforcements for high-performance ceramic matrix composites (CMCs) employed at high temperatures.1–5 Continuous silicon nitride ceramic fibers, as a type of silicon nitride materials, are optimal candidates for the reinforcement of wave-transparent ceramic matrix composites (CMCs) applied in radome materials.6In the last two decades, several ceramic fibers, which are mainly composed of silicon and nitride, have been prepared by many researchers worldwide. For instance, Sinber fibers were produced by Japan Atomic Energy Research Institute,7 Si–C–N fibers were created by Domaine University,8 and silicon nitride fibers (SNF) were developed by Dow Corning Corporation.9 Among these fibers, only SNF fibers can be employed in the applications that require the materials with wave-transparent properties and contain 3 wt% of carbon and can be treated as high-purity silicon nitride fibers. However, the SNF fibers, which are fabricated by pyrolysis of hydridopolysilazane (HPZ), have strong reactivity, which leads to diffusion reaction with the ceramic matrix. Therefore, in this work, in order to satisfy the application in radome materials, near-stoichiometric silicon nitride ceramic fibers were prepared by pyrolysis of polycarbosilane (PCS) fibers via electron beam curing in the NH3 atmosphere. The PCS, which consists of a skeleton of alternate carbon and silicon atoms, is widely used as the precursor for preparation of silicon nitride ceramic materials, such as porous ceramics and ceramic fibers.10–14 Electron beam (EB) irradiation curing, other than thermal oxidation, can effectively reduce the oxygen content of the fiber and, sequentially, enhance the high-temperature performance of the fiber.15,16
Since Si3N4 fibers are the backbone in load-bearing CMCs, their strength and strength retention throughout the service life are critical to the performance of CMCs. Therefore, it is essential to acquire a comprehensive knowledge of the microstructure and the tensile behavior of Si3N4 fibers at elevated temperature in order to understand the degradation behavior and forecast the performance of CMCs at high temperatures. Moreover, the key thermal characteristic of Si3N4 fibers is their oxidation resistance at high temperatures for a long time.17 Furthermore, to the best of our knowledge, the oxidation behavior of the silicon nitride fibers produced by this method at high temperatures has rarely been reported before. Hence, the microstructures and the mechanical properties of Si3N4 fibers before and after heat treatment in the air were investigated in detail, and the relationship between their microstructures and mechanical properties was also established.
2. Experimental
2.1 Preparation of silicon nitride fibers and heat treatment
PCS (average molecular weight ≈ 1500, synthesized in our laboratory) was melt-spun into fibers at 300 °C in N2 with a lab scale melt-spinning apparatus. Bundles of PCS fibers were irradiated in He gas atmosphere by 2 MeV electron beams, and the dose was 15 MGy with a dosing rate of around 1 kGy s−1. After irradiation, the fibers underwent heat treatment up to 400 °C with a heating rate of 5 °C min−1. Then, the cured PCS fibers were set in an alumina tube furnace and heated up to 1000 °C in NH3 gas, followed by heat treatment under nitrogen gas (N2) flow at a temperature of 1300 °C, yielding silicon nitride fibers.Before the heat treatment in air, original fibers were first desized at 600 °C for 1 h in air. Then, the heat treatment was carried out at a temperature range of 1000 °C-1400 °C for a dwell time of 2 h in the air in a box furnace under a pressure of 105 Pa.
2.2 Characterizations
The bulk composition of the silicon nitride fibers was determined by chemical analysis. Nitrogen and oxygen contents were determined using an N/O analyzer (EMGA-820, Horiba). From our previous experience, we know that the content of silicon can be obtained by the subtraction method since hydrogen was released from the precursor upon annealing at 1300 °C. The surface morphology of the fibers was observed by a scanning electron microscope (SEM, Helios 600i FEI) equipped with an energy-dispersive X-ray spectroscopy. The crystalline state of the fibers was characterized by X-ray diffraction (XRD) analysis using a monochromatic Cu Kα radiation with a D8 ADVANCE diffractometer (Bruker, Germany). The compositions of the fibers were traced by solid-state 29Si NMR spectroscopy with a Varian Infinity Plus 300 spectrometer and X-ray photoelectron spectroscopy (XPS) analysis with a Thermo ESCALAB 250 apparatus using Al Kα radiation with the energy of 1486.6 eV. TEM lamellas with a thickness <100 nm were fabricated by the lift-out method in an FEI Helios 600i focused ion beam (FIB) system, and TEM imaging was performed using an FEI Tecnai F20 transmission electron microscope operating at 200 kV.18,19In the present study, tensile tests were performed in ambient atmosphere on a universal material testing machine (Testometric, M3505CT) at a gauge length of 25 mm and an extension rate of 5 mm min−1. Weibull modulus was calculated from 20 specimens for each heat-treated temperature.
3. Results and discussion
3.1 Composition and microstructure of the untreated fibers
The chemical composition analysis suggests that the main components of the ceramic fibers were Si, N, and O. The general properties of the fibers are shown in Table 1, and they have a molar composition of SiN1.27O0.09. Fig. 1 shows the elemental maps of the untreated fibers. The fiber was embedded in epoxy resin. Overall, the spatial distribution of Si, N and O elements in the fiber was uniform.| Properties | Element (wt%) | Density (g cm−3) | Diameter (μm) | Strength (GPa) | Modulus (GPa) | ||
|---|---|---|---|---|---|---|---|
| Si | N | O | |||||
| Si3N4 fiber | 59.24 | 37.85 | 2.91 | 2.3 | 12.2 | 1.81 | 213 |
 | ||
| Fig. 1 Elemental distribution of the untreated fibers. | ||
Fig. 2 shows the external appearance of the desized silicon nitride fiber. Overall, the fibers exhibited a relatively uniform diameter of 12 μm with smooth surfaces and rare defects. The fracture cross-section in Fig. 2(a) shows distinct mirror and hackle features, which are recognized as typical fracture traits of the ceramic fibers. Besides, no visible pores or defects were observed on the fracture surface in Fig. 2(b). From the results shown in Fig. 2(b) and 1, it can be seen that the fibers have high structural uniformity.
 | ||
| Fig. 2 SEM images of the untreated fibers. | ||
The crystalline state of the untreated fibers is shown in Fig. 3.
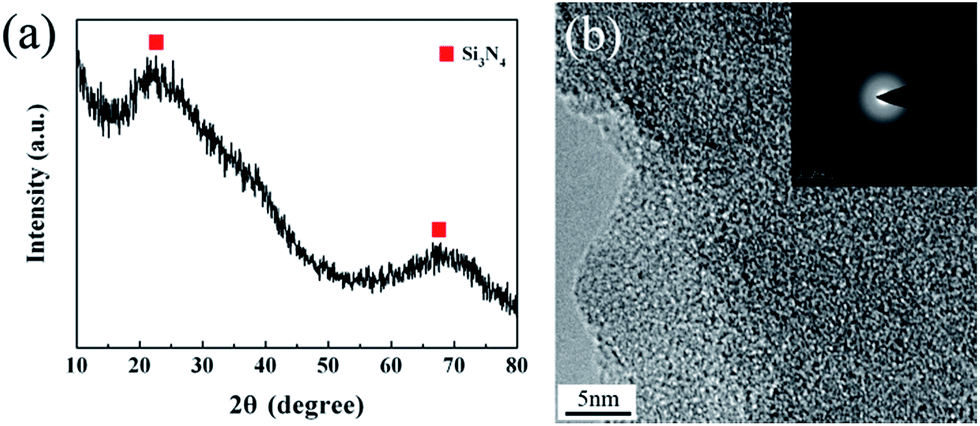 | ||
| Fig. 3 (a) XRD spectrum and (b) TEM pattern of the untreated fibers. | ||
The XRD pattern of the untreated Si3N4 fibers represents two broad diffraction peaks at around 23° and 69°, revealing the amorphous nature of the fibers. A detailed microstructure was observed by high-resolution transmission electron microscopy (HR-TEM) in conjunction with selected-area electron diffraction (SAED). The HR-TEM revealed a homogeneous amorphous structure for the untreated fibers, while the diffraction pattern demonstrated a diffuse ring pattern, being a character of the microstructure with short-range order. This is consistent with the XRD result.
The most powerful technique by far to understand the chemical bonding in the ceramic fibers is NMR spectroscopy. A 29Si NMR spectrum of the untreated fiber with a broad and continuous signal is shown in Fig. 4. It was noted that the strongest signal at δ = −49 ppm, representing the Si3N4 structure, was close to the reported values (δ = −46 ppm) for amorphous Si3N4 (a-Si3N4).20 The result showed that the fiber was mostly composed of the amorphous Si3N4 structure.
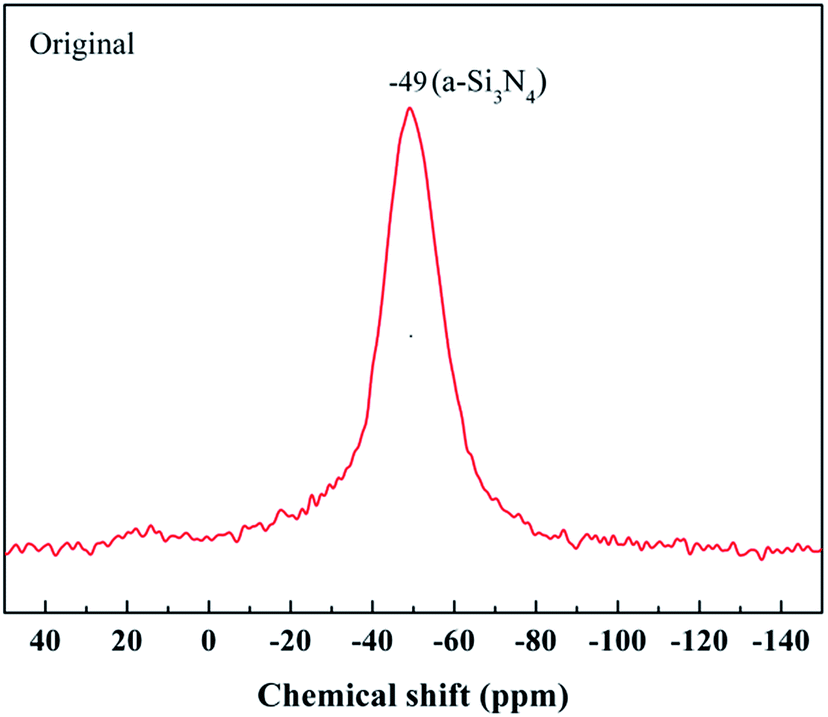 | ||
| Fig. 4 29Si NMR spectrum of the untreated fiber. | ||
Besides, the surface chemical environment of the untreated fiber is investigated by XPS. Fig. 5 shows the overall XPS spectrum and the resolved high-resolution spectra of Si 2p, N 1s, C 1s, and O 1s electrons. The C 1s component could be assigned as the standard for calibration. Before being examined by XPS, the fiber was etched by Ar in order to eliminate the surface contaminants on the fiber surface. Hence, the Ar component might be introduced during the etching process. As shown in Fig. 5(b), there were three peaks in the Si 2p spectrum. The peak located at 101.8 eV can be attributed to the bond in Si3N4. The other two peaks located at 102.2 eV and 103.3 eV were assigned to the Si–N and Si–O bonds in Si2N2O. The Si2N2O phase, whose chemical structure is silicon atoms linked with both nitrogen and oxygen, was likely derived from the nitridation of the cured PCS fiber.21 Similarly, the peaks at 398.0 eV and 398.6 eV in the N 1s spectrum were assigned to the N–Si and N–Si–O bonds, respectively. Furthermore, the single peak at 532.2 eV in the O 1s spectrum, which was shifted from 532.7 eV of the standard SiO2, was attributed to the O–Si bond in the Si–N–O structure. However, there was no Si–N–O structure observed in the above NMR spectrum. Thus, the content of Si–N–O structure may be too small to be able to be detected by NMR technique. Therefore, in conjunction with the NMR spectrum, it can be concluded that the fiber was composed of the amorphous Si3N4 and Si2N2O phase.
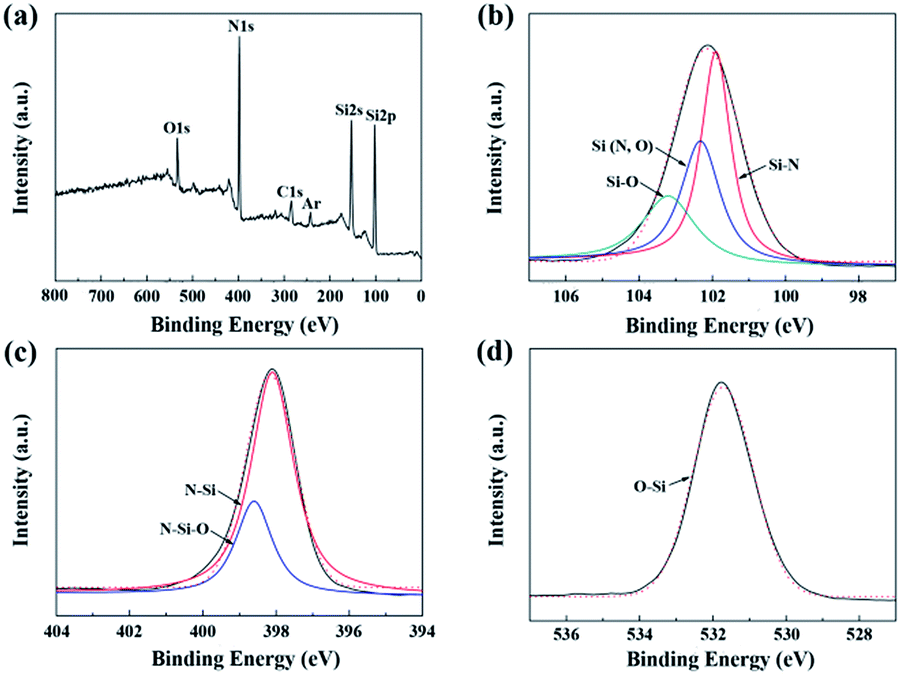 | ||
| Fig. 5 XPS spectra of the untreated fibers: (a) overall spectrum; (b) Si 2p; (c) N 1s; and (d) O 1s. | ||
3.2 Influence of heat treatment on microstructure and mechanical properties
The XRD patterns of the untreated fibers after the heat treatment are shown in Fig. 6. It can be seen that the fibers retained their amorphous structure in the range of room temperature-1000 °C. As the temperature increased, the degree of oxidation increased. Hence, the conspicuous cristobalite phase, which is one crystalline phase of SiO2, can be found in the XRD patterns when the treatment temperature exceeded 1200 °C, demonstrating the sufficient oxidation of the fibers. Moreover, three main diffraction peaks at 22.0°, 31.5° and 36.1°, which were respectively assigned to the (101), (102) and (200) lattice planes of the cristobalite phase, presented in the XRD spectrum of the sample treated at 1200 °C. Furthermore, the XRD peaks became sharper with the increase in the treatment temperature. The apparent crystallite sizes of cristobalite, which were calculated from the half-width value of three peaks above using Scherrer formula, are listed in Table 2. Obviously, the cristobalite grains grew into larger crystals with the increase in temperature and tended to grow rapidly at 1300 °C, resulting in a strength degradation of the fiber, as discussed later in Section 3.3.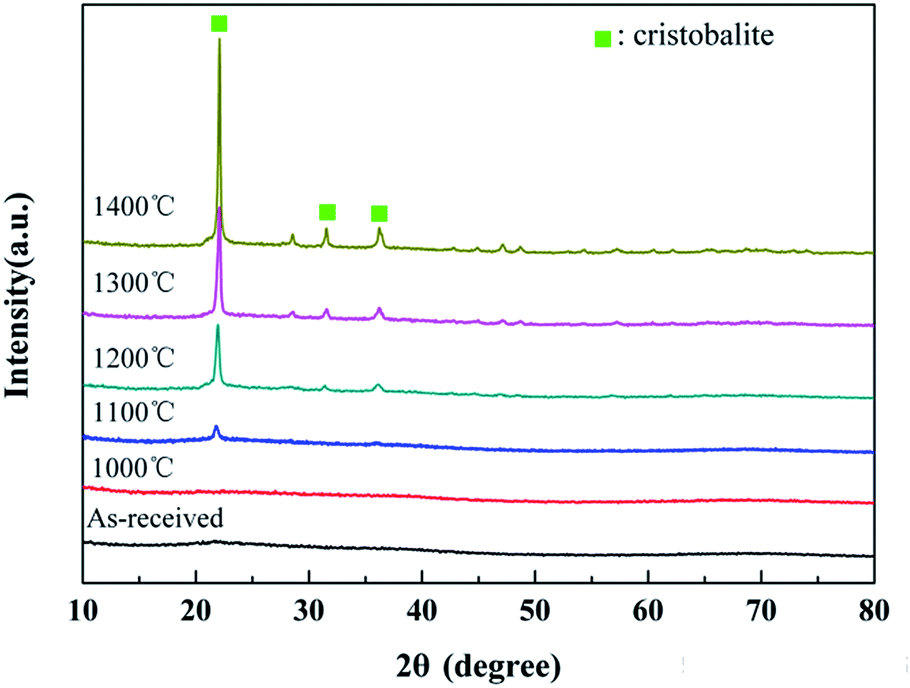 | ||
| Fig. 6 XRD patterns of the untreated fibers after the heat treatment. | ||
| Temperature | hkl | ||
|---|---|---|---|
| (101) | (102) | (200) | |
| 1200 °C | 158 | 190 | 101 |
| 1300 °C | 279 | 263 | 211 |
| 1400 °C | 335 | 297 | 233 |
The surface morphology of the untreated fibers after heat treatment is presented in Fig. 7. It can be found that the untreated fibers had relatively smooth and homogeneous surface, and this surface morphology was well retained even after the fibers were annealed at 1000 °C. The fibers were subjected to a mild oxidation at 1100 °C with a few crystalline grains on the surface. With temperature increasing up to 1200 °C, it was found that the size of crystalline grains increased. The EDS result verified that the crystalline grains were SiO2 particles. As the temperature rose to 1300 °C, some cracks appeared on the fiber surface which could cause damage to the fiber. When the treatment temperature was 1400 °C, the gap between cracks increased with the expansion of the cracks.
 | ||
| Fig. 7 Surface morphology of the untreated fibers before and after the heat treatment. | ||
The chemical composition of the fibers after the heat treatment is listed in Table 3. The weight ratio of N element in the fibers treated at 1400 °C was still 25.55%. Compared to that of the untreated fiber (37.85%), one can see that only a part of the fibers participated in the oxidation reaction. Hence, the changes in the composition and the structure of the fiber after the heat treatment at 1400 °C due to the oxidation were investigated by XPS and NMR. As shown in Fig. 8(a), the overall XPS spectrum of the fibers after the heat treatment at 1400 °C clearly revealed the presence of Si 2s, O 1s, C 1s, and Ar 2p electrons. Similarly, the C 1s component could be assigned as the standard for calibration and the Ar component might be introduced during the etching process. In Fig. 8(b), the Si 2p spectrum with the peak centered at 103.5 eV corresponded to the Si–O bond, and the peak centered at 532.7 eV in the O 1s spectrum in Fig. 8(c) was caused by the O–Si bond.22,23 Furthermore, the concentration of oxygen (60.1 at%) at the surface was higher compared to the bulk concentration of oxygen (3.4 at%), as determined by chemical analysis. Hence, the XPS spectra confirm that the oxidation layer was formed on the fiber surface. The 29Si NMR spectrum of the fiber after the heat treatment at 1400 °C is shown in Fig. 9. It is noticed that the strong signal at δ = −49 ppm, representing the Si3N4 structure, was the same as that in the 29Si NMR spectrum shown in Fig. 4 of the untreated fiber. The signal at δ = −111 ppm, representing the SiO4 structure, showed that the fiber contained SiO2.24,25 However, it should be noted that there was no Si–N structure found in the above XPS spectra, which is probably due to the fact that the internal Si–N structures were surrounded by the SiO2 oxidation layer.
| Samples | Si | N | O | |||
|---|---|---|---|---|---|---|
| wt% | at% | wt% | at% | wt% | at% | |
| Untreated | 59.24 | 42.20 | 37.85 | 54.20 | 2.91 | 3.60 |
| 1000 °C | 59.29 | 42.40 | 37.69 | 53.80 | 3.02 | 3.80 |
| 1100 °C | 57.39 | 40.76 | 35.38 | 50.29 | 7.23 | 8.95 |
| 1200 °C | 57.41 | 41.12 | 31.62 | 45.11 | 10.97 | 13.77 |
| 1300 °C | 55.88 | 39.92 | 28.12 | 40.12 | 16.00 | 19.95 |
| 1400 °C | 55.32 | 39.56 | 25.55 | 39.46 | 19.13 | 20.98 |
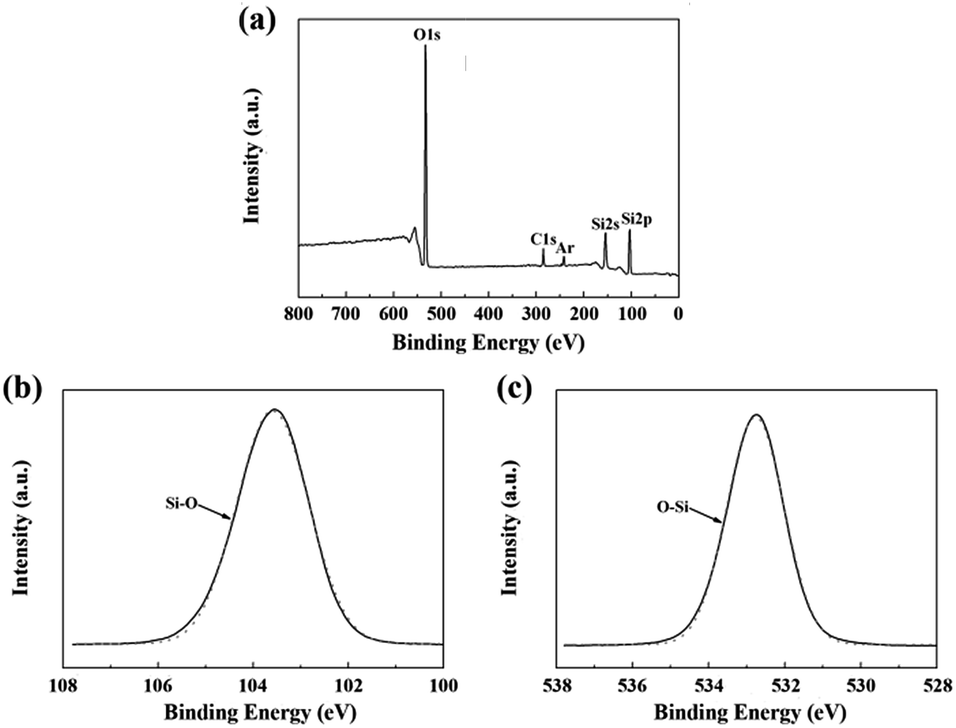 | ||
| Fig. 8 XPS spectra of the fiber after the heat treatment at 1400 °C: (a) overall spectrum; (b) Si 2p; and (c) O 1s. | ||
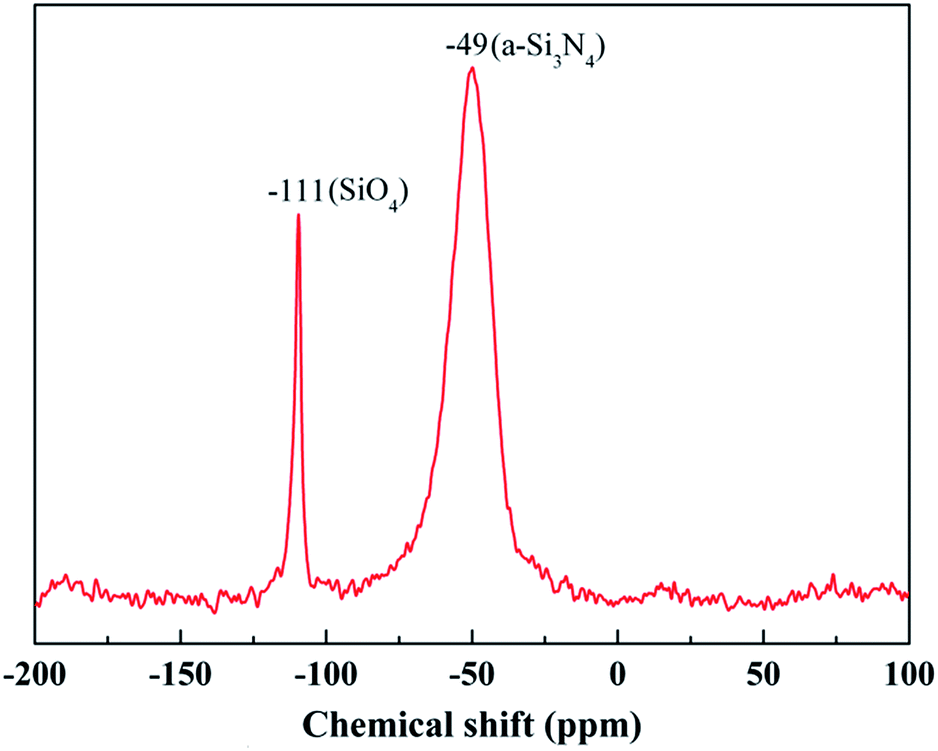 | ||
| Fig. 9 29Si NMR spectrum of the fiber after the heat treatment at 1400 °C. | ||
In order to further study the microstructure of the fiber after the heat treatment at 1400 °C, a cross-sectional thin foil was extracted out of the fiber by FIB and then was characterized by bright field TEM (BF-TEM), high-resolution TEM (HR-TEM), and selected area electron diffraction (SAED). The results are shown in Fig. 10(a–e). A Cr layer, covering the thin foil surface, was utilized to increase the electrical conductivity of the fiber. Apparently, upon the heat treatment at 1400 °C, a silica oxidation layer with a thickness ∼2 μm covered the internal fiber, whose composition was further certified by EDS in Fig. 10(d and e). The Mo element, shown in EDS, resulted from a holder used to support the thin foil. Contrary to the internal fiber, the cover layer displayed a high degree of crystallinity, as evidenced by the HR-TEM images in Fig. 10(c). According to the experimental results, the oxidation of the fiber begins at the surface. With heat-treatment temperatures increasing, the inside fiber is oxidized. Consequently, the oxidation was uniformly performed from the outside in due to the amorphous state of the internal fiber. The result indicated that although the treatment temperature was up to 1400 °C, only a part of the fiber participated in the oxidation reaction, and the silica oxidation layer was generated around the fiber surface due to the relatively short oxidation time.
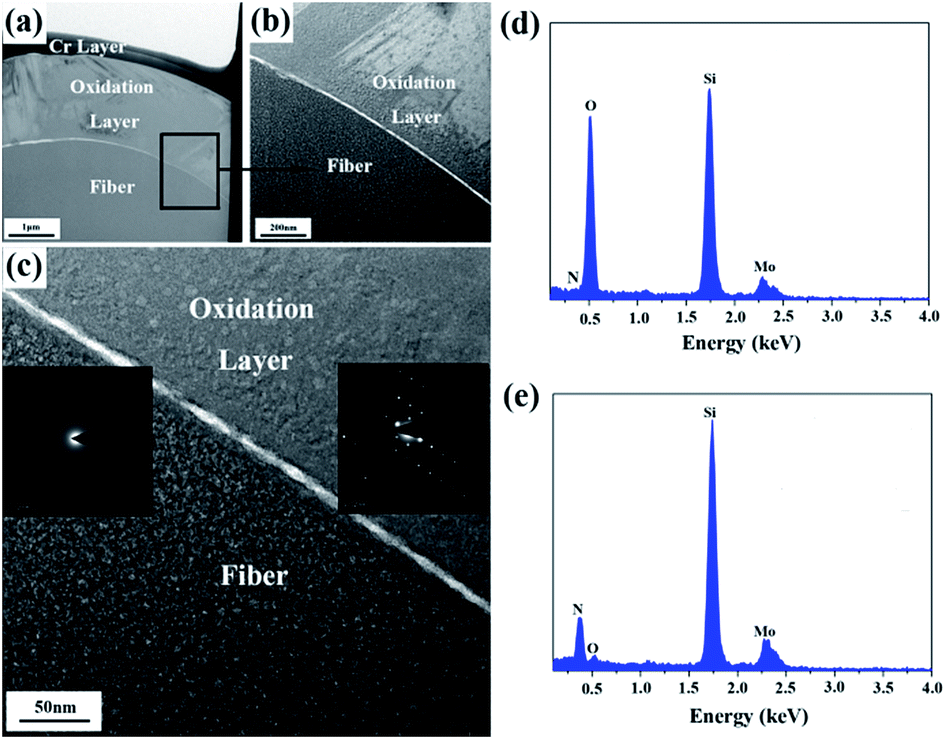 | ||
| Fig. 10 (a) BF-TEM of the fiber after the heat treatment at 1400 °C; (b) magnified BF-TEM and (c) HR-TEM of the chosen region, the insets correspond to the SAED patterns; (d) EDS of the oxidation layer; and (e) EDS of the internal fiber. | ||
According to the results above, the oxidation seemed to result from reactions (1) and (2) of Si3N4 and Si2N2O.26
| Si3N4(s) + 3O2(g) → 3SiO2(s) + 2N2(g) ΔrGθ = [−1859 × 103 + 268.6T] J mol−1 | (1) |
| Si2N2O(s) + 3/2O2(g) → 2SiO2(s) + N2(g) ΔrGθ = [−843.3 × 103 + 154.3T] J mol−1 | (2) |
Using thermodynamic data of the reactions (1) and (2), we can calculate ΔrGθ (J mol−1) < 0 in the temperature range of 1000–1400 °C. Hence, Si3N4 and Si2N2O can react with oxygen to produce SiO2, resulting in a weight gain. Hence, theoretically with the temperature increasing continuously, a dense outer oxide layer will be formed around the fiber and protect the internal fiber from further oxidation. However, in practice, as the oxidation reaction progresses, nitrogen released by the oxidation reaction continuously will blow up the dense outer oxide layer and generate cracks. Under these conditions, the micro-cracked oxide layer cannot protect the fiber and allows fast oxygen diffusion to the interface, resulting in a dramatic increase in the oxidation rate. A tentative schematic, describing the oxidation process, is illustrated in Fig. 11. Fig. 11(a) showed the oxidation reaction initiated at the fiber surface, corresponding to the observation on the fibers treated at the temperature of 1100 °C and 1200 °C as shown in Fig. 7(c) and (d). Finally, as shown in Fig. 11(b), N2 diffusion outwards through the oxide layer resulted in the formation of cracks on the fiber surface, corresponding to the phenomena observed in Fig. 7(e) and (f).
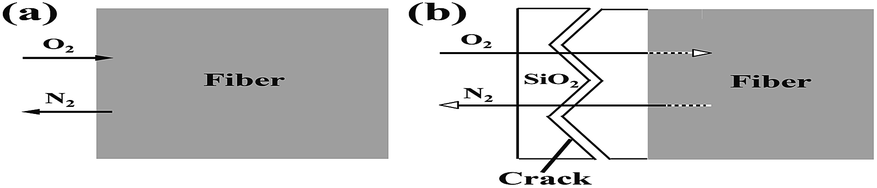 | ||
| Fig. 11 Tentative schematic description of the oxidation of silicon nitride fibers showing the mass transfer at reactant/product interface: (a) 1100 °C −1200 °C and (b) 1300−1400 °C. | ||
3.3 Influence of heat treatment on the mechanical property of Si3N4 fiber
In order to evaluate the influence of heat treatment on the mechanical properties of the Si3N4 fibers, the single filament tensile strength of the untreated fibers and those after the heat treatment was measured at room temperature.27 In this study, Weibull statistical model, which is commonly used for characterization of brittle materials, was used to characterize the tensile strength of the individual fiber. In this model, the failure probability Pf is estimated by eqn (3), where σ is the fracture strength, and σ0 and m are the Weibull parameters, i.e. the scale parameter and the Weibull modulus, respectively. It is well-known that a larger m will lead to a steeper function and then a lower dispersion of tensile strength.| Pf(V) = 1 − exp[−(σ/σ0)m] | (3) |
Taking the logarithm of eqn (3) twice gave a linear equation, eqn (4), with a slope equal to m and a y-intercept equal to −m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnσ0.
lnσ0.
|
lnln[1/(1 − Pf)] = mlnσ − mlnσ0
| (4) |
The σ values were the experimental tensile stresses. The test results were converted into an experimental probability distribution. This was done by ordering the results from the lowest to the highest rupture stress, which means σ1 < σ2 < … < σi… < σN−1 < σN (N was the sample number). Since the true value of Pfi for each σi was unknown, it had to be estimated. According to Riccardi,28 when N ≥ 20, Pf was in non-weighted analysis and could be defined as following (eqn (5)).
| Pfi = (i − 0.5)/N | (5) |
The Weibull plots of the untreated fibers before and after the heat treatment were summarized in Fig. 12, and the Weibull parameters of the individual fiber tensile test are listed in Table 4. As mentioned before, a bigger m value means a lower dispersion of tensile strength. The Weibull modulus decreased from 4.99 to 3.39 with the temperature increasing up to 1200 °C, which indicated that the dispersion of the tested tensile increased indistinctively with temperature. It was necessary to note that the Weibull statistics of the fibers after the heat treatment at 1300 °C and 1400 °C were not provided in this study because the fibers after the heat treatment at those temperatures were too brittle to prepare enough fiber samples for testing.
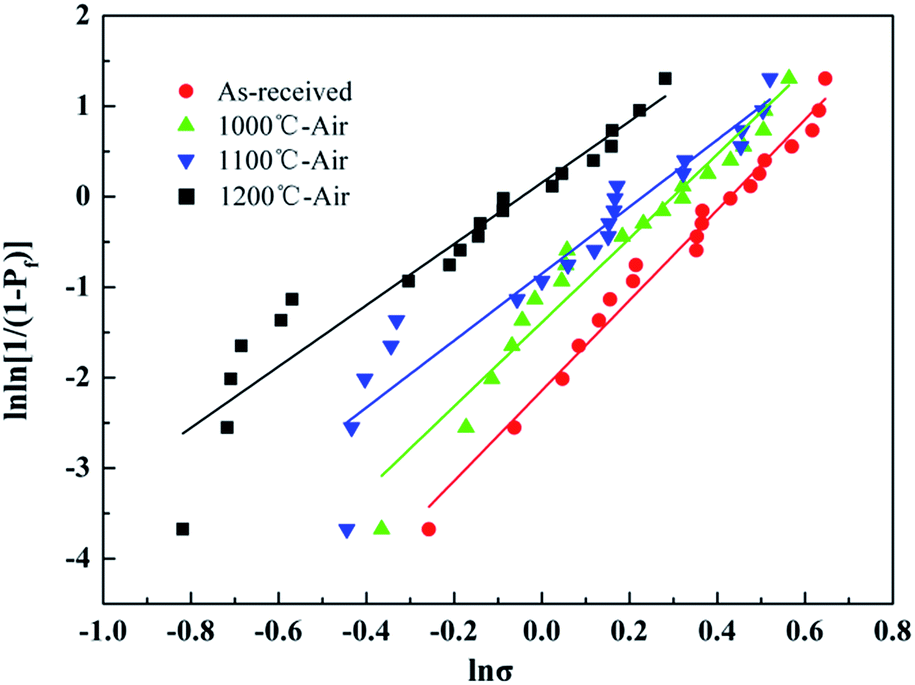 | ||
| Fig. 12 Weibull plots for the fiber tested before and after the heat treatment. | ||
| Temperature (°C) | Weibull modulus | Scale parameter (GPa) | Tensile strength (GPa) | Retention (%) |
|---|---|---|---|---|
| Untreated | 4.99 | 1.84 | 1.81 | — |
| 1000 | 4.65 | 1.55 | 1.57 | 87.23 |
| 1100 | 3.69 | 1.16 | 1.08 | 60.24 |
| 1200 | 3.39 | 0.96 | 0.91 | 50.29 |
As can be seen in Table 4, the fiber possessed a comparatively high tensile strength due to its excellent heat resistance when the treatment temperature was below 1200 °C. Although the tensile strength of the fiber decreased to 0.86 GPa after the heat treatment at 1200 °C, the strength retention was still about 50%. Such strength loss after high-temperature exposure was mainly attributed to the oxidation of silicon nitride as shown in Fig. 6. In conclusion, the silicon nitride fibers could be used in the air at a temperature lower than 1200 °C without considering some other factors, such as vapor, pressure, and serving time.
In general, the strength degradation of silicon nitride fibers after the heat treatment is believed to result from the flaw size on the fiber surface as well as the inside during the heat treatment because the mechanical properties of a brittle material are under control of the flaw. According to the research by Taylor,29 there is a relationship existing between the flaw type and the tensile strength of the fibers. The tensile strength of the fibers can remain 1.0–2.0 GPa with a few internal flaws and, in contrast, the tensile strength of the fibers decreases to 0.6–0.9 GPa with a rough surface, such as particles, holes, or cracks.
As shown in Fig. 7 and Table 4, the change of the internal flaw size resulted in the strength degradation of silicon nitride fibers after the heat treatment at 1000 °C. With the temperature increasing up to 1100 °C and 1200 °C, the strength degradation of silicon nitride fibers was dominated by the presence of SiO2 particles, which were derived from the oxidation reaction, on the fiber surface. When the temperature increased to 1300 °C, an oxide layer consisting mainly of pure silica was formed on the fiber surface, leading to the brittleness of the fiber. Consequently, the fiber had no tensile strength after the treatment at 1300 °C and 1400 °C.
4. Conclusions
The microstructure and the mechanical properties of silicon nitride fibers have been investigated before and after the heat treatment at elevated temperature in air. Microstructural analysis shows that the fibers are composed of amorphous silicon nitride and a few Si2N2O phases. The heat treatments above 1100 °C provoked the oxidation reaction from silicon nitride to silicon dioxide and the cracks, which were derived from the gas emission during the oxidation reaction with the temperature increasing to 1300 °C, were formed on the fiber surface. Ultimately, an oxidation layer with a thickness of ∼2 μm formed on the fiber surface upon the heat treatment at 1400 °C, which indicated that the fiber was not completely oxidized. This change was accompanied by a fall in the tensile strength of the individual fiber at room temperature. The relatively lower strength degradation indicates that the fiber would possess a high serving life at a serving temperature lower than 1200 °C. It demonstrates an excellent thermal resistance, which can be useful for a potential application in the ceramic matrix composites in the air environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the financial support from the National Natural Science Foundation of China (51202291), the Aid Program for Innovative Group of National University of Defense Technology, and the Aid program for Science and Technology Innovative Research Team in Higher Educational Institutions of Hunan Province.Notes and references
- M. Sato and K. Okamura, J. Ceram. Soc. Jpn., 1998, 106, 248–255 CrossRef CAS.
- V. Pavarajarn, R. Precharyutasin and P. Praserthdam, J. Am. Ceram. Soc., 2010, 93, 973–979 CrossRef CAS.
- C. Shi, Y. Zhu and H. Qian, Mater. Res. Bull., 2014, 51, 161–166 CrossRef CAS.
- A. L. Hector, Coord. Chem. Rev., 2016, 323, 120–127 CrossRef CAS.
- G. X. Zhu, S. M. Dong and D. W. Ni, RSC Adv., 2016, 6, 83482–83492 RSC.
- S. Kamimura, T. Seguchi and K. Okamura, Radiat. Phys. Chem., 1999, 54, 575–581 CrossRef CAS.
- D. Mocaer, R. Pailler and R. Naslain, J. Mater. Sci., 1993, 28, 2639–2653 CrossRef CAS.
- N. Bansal and M. Dickerson, Mater. Sci. Eng., A, 1997, 22, 149–157 CrossRef.
- O. Delverdier and M. Monthioux, J. Eur. Ceram. Soc., 1994, 14, 313–325 CrossRef CAS.
- K. Sato, H. Kakisawa and Y. Kagawa, J. Mater. Sci. Lett., 2000, 19, 1179–1183 CrossRef CAS.
- X. Zhao, K. Han and Y. Peng, Mater. Lett., 2011, 65, 2717–2720 CrossRef CAS.
- G. S. Bibbo, P. M. Benson and C. G. Pantano, J. Mater. Sci., 1991, 26, 5075–5080 CrossRef CAS.
- E. Bouillon, D. Mocaer and J. F. Villeneuve, J. Mater. Sci., 1991, 26, 1517–1530 CrossRef CAS.
- T. Seguchi, Radiat. Phys. Chem., 2000, 57, 367–371 CrossRef CAS.
- M. Sugimoto, T. Shimoo and K. Okamura, J. Am. Ceram. Soc., 1995, 78, 1013–1017 CrossRef CAS.
- G. Chollon, J. Eur. Ceram. Soc., 2000, 20, 1959–1974 CrossRef CAS.
- H. T. Liu, L. W. Yang and X. Sun, Carbon, 2016, 109, 435–443 CrossRef CAS.
- K. Okamura, M. Sato and Y. Hasegawa, Ceram. Int., 1987, 13, 55–61 CrossRef CAS.
- X. S. Zhang, L. W. Yang and H. T. Liu, RSC Adv., 2017, 7, 23334–23341 RSC.
- M. K. Cinibulk, G. Thomas and S. M. Johnson, J. Am. Ceram. Soc., 1992, 75, 2044–2049 CrossRef CAS.
- X. Hu, C. W. Shao and J. Wang, J. Mater. Sci., 2017, 52, 7555–7566 CrossRef CAS.
- F. Porz and F. Thommler, J. Mater. Sci., 1984, 19, 1283–1295 CrossRef CAS.
- S. Ichimura, K. Koike and A. Kurokawa, Surf. Interface Anal., 2000, 30, 497–501 CrossRef CAS.
- F. Liu, L. Qiu and L. Zhao, Chem, 2006, 69, 393–398 CAS.
- M. Casu, A. Lai and A. Musinu, J. Mater. Sci., 2001, 36, 3731–3735 CrossRef CAS.
- F. F. Lange, J. Am. Ceram. Soc., 1978, 61, 53–56 CrossRef CAS.
- Y. Wang and H. F. Cheng, Mater. Sci. Eng., A, 2013, 578, 287–293 CrossRef CAS.
- C. C. Riccardi and C. I. Vallo, Polym. Eng. Sci., 2002, 42, 1260–1273 CAS.
- S. T. Taylor, Y. T. Zhu and W. R. Blumenthal, J. Mater. Sci., 1998, 33, 1465–1473 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |