
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient synthesis of benzene-fused 6/7-membered amides via Xphos Pd G2 catalyzed intramolecular C–N bond formation†
Zhou Xu *ab,
Ke Lib,
Rongliang Zhaiab,
Ting Liangab,
Xiaodie Guia and
Rongli Zhangb
*ab,
Ke Lib,
Rongliang Zhaiab,
Ting Liangab,
Xiaodie Guia and
Rongli Zhangb
aDepartment of Chemistry, Xuzhou Medical University, Xuzhou 221004, China. E-mail: xuzhou@xzhmu.edu.cn
bJiangsu Key Laboratory of New Drug Research and Clinical Pharmacy, Xuzhou Medical University, Xuzhou 221004, China
First published on 8th November 2017
Abstract
An efficient approach for benzene-fused 6/7-membered amides via intramolecular amidation of aryl chlorides catalyzed by a Buchwald–Hartwig second generation Pd catalyst (Xphos Pd G2) has been successfully developed. This catalyst system allows the primary amides which have only modest nucleophilicity to be coupled successfully even with electron rich aryl chlorides in short reaction time. The intramolecular amidation reaction also has good chemoselectivity and excellent functional group compatibility.
Introduction
Benzene-fused amides are important organic compounds commonly encountered in biologically relevant natural or synthetic pharmaceutical compounds.1–3 Some methods were developed towards the synthesis of these scaffolds which are mainly benzene-fused 6-membered amides known as 2H-1,4-benzoxazin-3-(4H)-one derivatives.4–6 However, the substituent diversity in the products is limited and more importantly, it is difficult to synthesize benzene-fused seven-membered amides.Organic halides are some of the most common industrially used compounds and widely used as starting materials for many chemical transformations. The efficient cleavage of C–X (X = halogen atom) bonds has been actively studied by organic chemists. Among them, palladium catalysts developed by Buchwald and Hartwig proved to be the most useful ones for the C–N bond formation.7,8 The amination of aryl halogens by transition metal catalyzed coupling methodology has been the subject of significant interest during past few decades, but the amidation reaction has not yet fully studied. Electron-deficient amides have been difficult coupling partners due to their modest nucleophilicity and intolerance of strong base. Although, amidation of aryl bromides and iodides have become the well-established processes in the reaction,9–12 however, largely absent from the literature on Pd-catalyzed amidation reactions is the description of an efficient method for coupling aryl chlorides.
To the best of our knowledge, there are limited literature reports the intramolecular amidation of aryl chlorides which can construct the valuable molecular structure of benzene-fused amides. Laha et al. reported the synthesis of dibenzoazepinones via the intramolecular amination of aryl bromide (not aryl chlorides) in 18 h with yield of 68–94%.13 While, Lautens et al. reported the synthesis of 3,4-dihydroquinolinones via the reaction between arylboronic acid and acrylamide (Scheme 1).14
 | ||
| Scheme 1 Benzene-fused amides: prior arts and design. | ||
As part of our continuing efforts for accessing biologically hetero compounds by metal-catalyzed reactions,15 herein, we report a facile, efficient, convenient and generally applicable method for the intramolecular amidation of organic halides to synthesize oxo-containing 6/7 membered benzene-fused amides with excellent yield using commerical available XPhos Pd G2 as the catalyst.
Results and discussion
At the outset of our investigation, we employed 1a as the substrate for the preparation of the desired benzene-fused 7-membered amide 2a. After considerable amount of efforts on condition discovery and optimization, we were able to obtain 2a with 97% isolated yield in 5 h.The reaction conditions, as shown in the equation in Table 1, including the use of Xphos Pd G2 as the most effective catalyst, 1,4-dioxane as the optimal solvent, and Cs2CO3 as the base. Indeed, other catalysts, such as Ph3PPdCl, JohnPhos Pd G2, Xphos PdCl2 all fared much worse than Xphos Pd G2 in terms of reaction rate and efficiency (entries 1–3 vs. 5). We also performed the reaction with catalysts Xphos Pd G1, Xphos Pd G3 and Xphos Pd G4. All of them gave almost the same result with Xphos Pd G2 (entries 4–7). However, Xphos Pd G2 is the cheapest one. Thus, we chose it as catalyst for further investigation. The combination of Pd(OAc)2 and ligand Xphos cannot proceed the transformation either (entry 8). The reaction can be faster and more efficiency when Cs2CO3 was used as the base instead of K2CO3 (entry 5 vs. entry 9). The best solvent for the reaction is 1,4-dioxane. Toluene is equal efficiency, but DMSO or DMF doesn't work for the reaction (entries 10, 12 and 13). The substrate 1a can also convert smoothly to product 2a at 80 °C, but requires longer reaction time (entry 14). Lowering the reaction temperature to 50 °C, the reaction could hardly happen and only trace product observed even prolonging the reaction time to 36 h (entry 15). Substrate concentration has limited effect on the reaction. When the concentration of the substrate was 0.25 M, it gave the best result with 99% yield. Increasing or decreasing the concentration of 1a, both gave slightly lower yield (entries 16 and 17). Higher the catalyst loading can faster the reaction with same yield (entry 18). Lowering the catalyst loading to 2.5% makes the reaction much difficult to proceed (entry 19). Small amount of water could hinder the reaction and slow down the reaction rate and yield (entry 20).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Time | Yieldb |
| a The reactions were carried in Schlenk flasks. 1a (0.5 mmol), Pd* (5 mol%), solvent (2 ml), base (1.5 equiv.), 110 °C.b Isolated yield.c The reaction temperature is 80 °C.d The reaction temperature is 50 °C.e Substrate concentration is 0.25 M.f Substrate concentration is 0.5 M.g The catalyst loading is 7.5 mol%.h The catalyst loading is 2.5 mol%.i H2O (5 μl) was added. | |||||
| 1 | PPh3PdCl | K2CO3 | 1,4-Dioxane | 24 h | NR |
| 2 | Johnphos Pd | K2CO3 | 1,4-Dioxane | 24 h | 18% |
| 3 | Xphos PdCl2 | K2CO3 | 1,4-Dioxane | 24 h | 84% |
| 4 | Xphos Pd G1 | K2CO3 | 1,4-Dioxane | 5 h | 95% |
| 5 | Xphos Pd G2 | K2CO3 | 1,4-Dioxane | 5 h | 97% |
| 6 | Xphos Pd G3 | K2CO3 | 1,4-Dioxane | 5 h | 94% |
| 7 | Xphos Pd G4 | K2CO3 | 1,4-Dioxane | 5 h | 97% |
| 8 | Pd(OAc)2/Xphos | K2CO3 | 1,4-Dioxane | 24 h | NR |
| 9 | XPhos Pd G2 | Cs2CO3 | 1,4-Dioxane | 2 h | 99% |
| 10 | XPhos Pd G2 | Cs2CO3 | DMSO | 24 h | Trace |
| 11 | XPhos Pd G2 | Cs2CO3 | H2O | 24 h | 14% |
| 12 | XPhos Pd G2 | Cs2CO3 | MePh | 8 h | 96% |
| 13 | XPhos Pd G2 | Cs2CO3 | DMF | 24 h | NR |
| 14c | XPhos Pd G2 | Cs2CO3 | 1,4-Dioxane | 8 h | 98% |
| 15d | XPhos Pd G2 | Cs2CO3 | 1,4-Dioxane | 36 h | Trace |
| 16e | XPhos Pd G2 | Cs2CO3 | 1,4-Dioxane | 4 h | 97% |
| 17f | XPhos Pd G2 | Cs2CO3 | 1,4-Dioxane | 4 h | 96% |
| 18g | XPhos Pd G2 | Cs2CO3 | 1,4-Dioxane | 2.5 h | 97% |
| 19h | XPhos Pd G2 | Cs2CO3 | 1,4-Dioxane | 12 h | Trace |
| 20i | XPhos Pd G2 | Cs2CO3 | 1,4-Dioxane | 24 h | 84% |

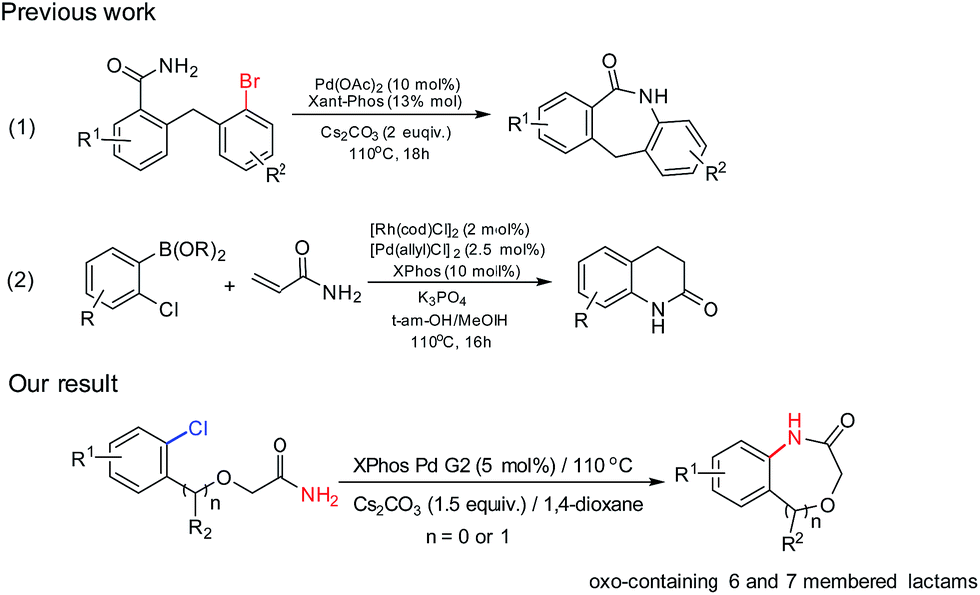
With the optimal conditions (Table 1, entry 9) in hand, we then set out to explore the substrate scope of the reaction for constructing benzene-fused 7-membered amides. As shown in Table 2, even electron-rich aryl chlorides can furnish the transformation in short time. A methyl (entry 1), a fluoro (entry 2), a trifluoromethyl (entries 3 and 4) or methoxyls (entry 5) substitution at the benzene ring were readily allowed and these reactions proceeded smoothly to afford the corresponding benzene-fused amides in good to excellent yields. In the case of 2-(1-phenylethoxy)acetamide as the substrate, 2g could be obtained with 89% yield in 5.5 h, which the reaction time was longer than that of 2a.

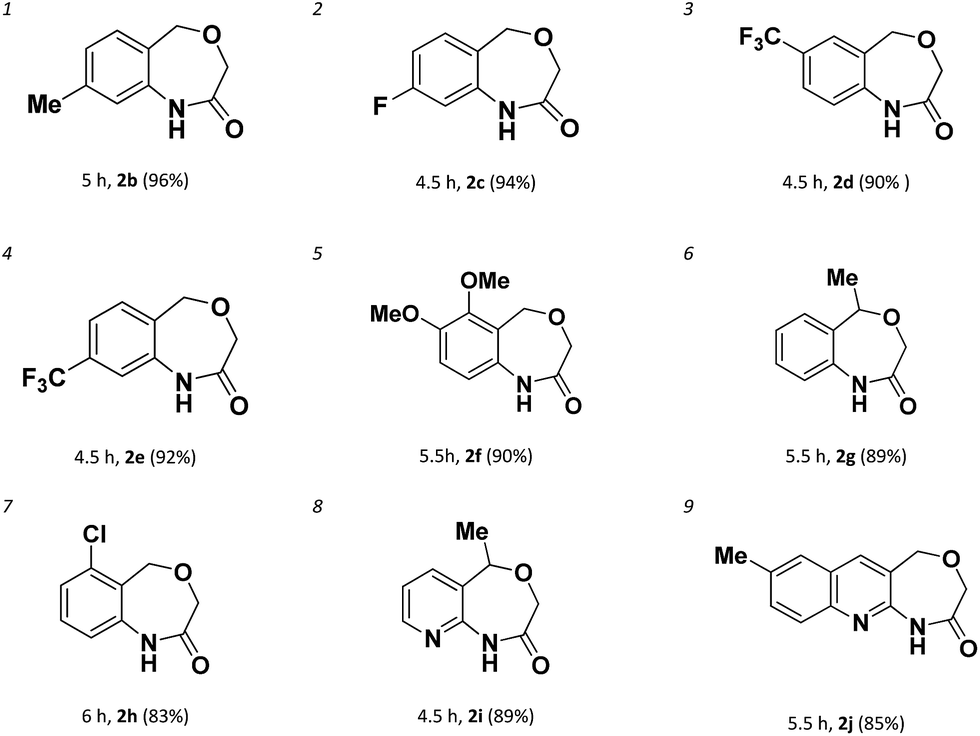
The chemoselectivity of the system was also briefly explored. When there are more than one chloro-groups on the ring, the reaction can hardly proceed. For example, as shown in Scheme 2, when 1h′ was used as the substrate, only trace of 2h was observed by NMR. Fortunately, when 1h was used instead of 1h′, 2h could be achieved successfully with 83% yield (entry 7) which indicated that the reaction had good chemoselectivity.
 | ||
| Scheme 2 (a) Chemoselectivity of the reaction. (b) Unsuccessful example. | ||
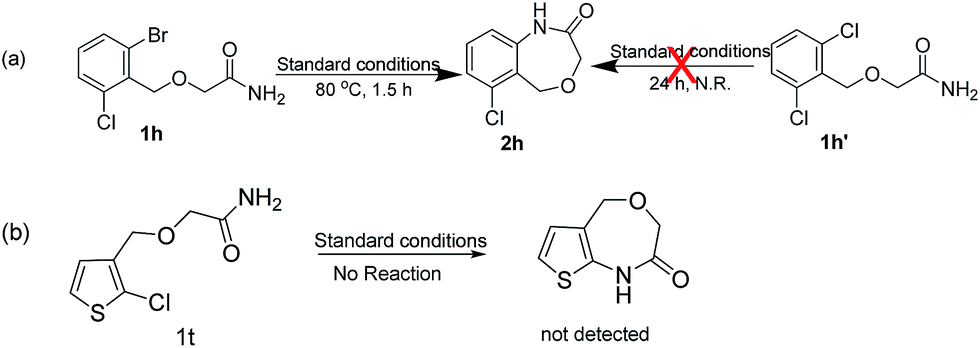
Unfortunately, when we explored this chemistry with thiophene substrate, no expected product was observed (Scheme 2b). For other heteroaromatic amides 1i-1j, the amidation reaction could proceed smoothly. When (1-(pyridin-3-yl)ethoxy)acetamide 1i was used, 2i could be obtained with 89% yield in 4.5 h (entry 8). Oxazepino[5,6-b]quinolin-2(3H)-one 2j was also obtained with good yield (entry 9).
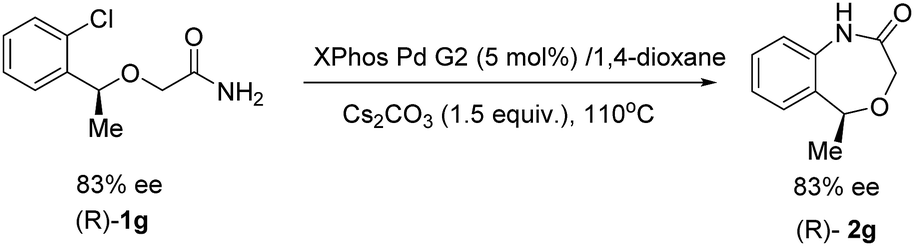
When Table 2, entry 6 was performed using the (R)-substrate (83% ee) under the optimal conditions, (R)-2g was formed without detectable ee erosion (Scheme 3).
 | ||
| Scheme 3 (R)-2g was formed without detectable ee erosion. | ||
With the success in the formations of benzene-fused seven membered rings, we subsequently turned to explore the 6-membered ring formation. As expected, the benzene-fused 6-membered rings are much easier formed than the seven membered ones both in efficiency and yields. We found a broad range of 6-membered ring amides possessing various functional groups could be achieved with high yields in short reaction times (Table 3).

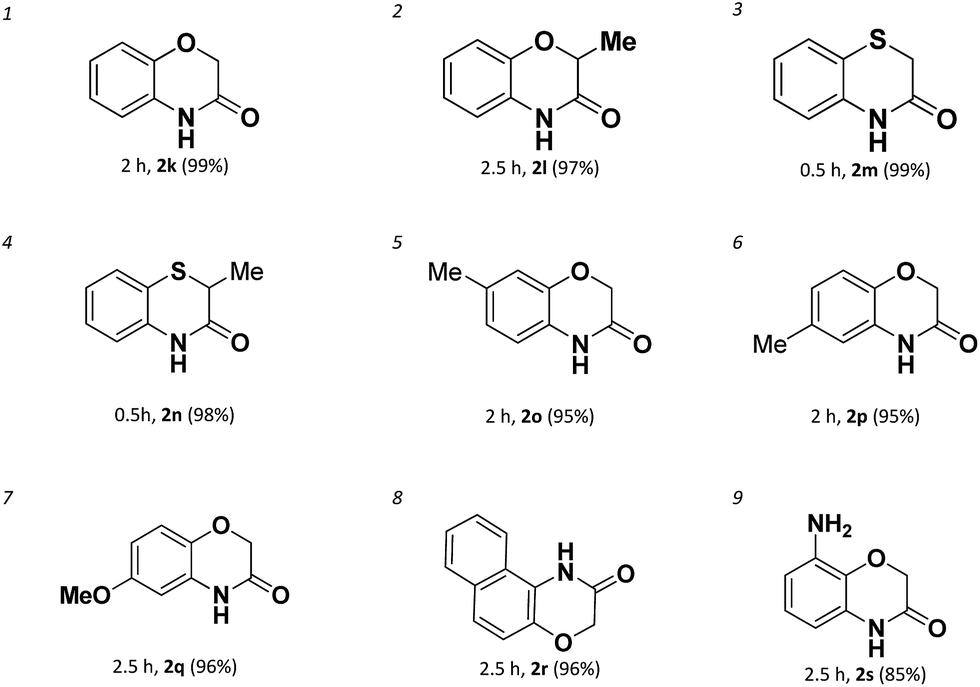
However, it needs to point out that the substrate used for the formation of 2n is aryl bromide (entry 4). The intramolecular amidation reaction could not proceed when the corresponding aryl chloride was used. Electron-rich aryl chlorides can also couple with amides successfully in short time affording the products in excellent yields (entries 5–7). For the sterically hindered aryl chloride, the amidation reaction was successfully proceeded affording 2r in 96% yield (entry 8). Even for 2-(2-aminophenoxy)acetamide which has a very strong electron donating amino-group which deactivated the C–Cl bond could also undergo the intramolecular amidation and gives product 2s in 85% yield (entry 9).
Conclusions
In conclusion, an efficient protocol for C–N cross-coupling reactions has been described. This catalyst system allows the primary amides which have only modest nucleophilicity could couple successfully even with electron rich aryl chlorides which can afford valuable benzene-fused 6/7-membered amides with high yield in short reaction time. The catalyst system also has good chemoselectivity and excellent functional group compatibility which supplies an important alternative way to the synthesis of benzene-fused 6/7-membered amides.Experimental section
General experimental methods
Commercially available reagents were used without further purification. The solvents used for experiment research were all through pretreatment on condition of anaerobic and without water. Reactions were monitored by thin layer chromatography (TLC) using silicycle precoated silica gel plates. Flash column chromatography was performed over silicycle silica gel (300–400 mesh). 1H NMR and 13C NMR spectra were recorded on JMTC-400/54/SS 400 MHz spectrometers using residue solvent peaks as internal standards (CHCl3, 1H: 7.26 ppm; 13C: 77.00 ppm). Infrared spectra were recorded with a PerkinElmer Spectrum Two FT-IR spectrometer and are reported in reciprocal centimeter (cm−1). Mass spectra were recorded with MicroTof-II using electron spray ionization (MeOH or CH3CN as solvent) or Waters GCT Premier time-of-flight mass spectrometer with a field ionization (FI) ion source.General procedure for the synthesis of benzene-fused 6/7-membered amides
2-((2-chlorobenzyl)oxy)acetyl chloride 1 (0.25 mmol), Cs2CO3 (122.65 mg, 0.375 mmol) and Pd Xphos G2 (9.8 mg, 0.0125 mmol) were added into Schlenk tube. The mixture was stirred under argon and dry 1,4-dioxane (1 ml) was added and heated to 110 °C. The reaction was monitored by TLC. After the reaction finished, the product was isolated with column chromatography.3,5-Dihydrobenzo[e][1,4]oxazepin-2(H)-one 2a
99% yield, m.p. 156–157 °C; 1H NMR (400 MHz, CDCl3): δ ppm 4.58 (s, 2H), 4.75 (s, 2H), 6.87 (d, J = 8.0 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 7.14 (d, J = 7.2 Hz, 1H), 7.26–7.27 (m, 1H), 7.72 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 72.8, 73.5, 119.2, 123.7, 128.6, 128.7, 129.2, 135.7, 173.2; IR v (cm−1): 3357.3, 3171.4, 1635.8, 1431.5, 1377.8; HRMS calcd for C9H9NO2 (M − H)+ 162.0555, found 162.0559.8-Methyl-3,5-dihydrobenzo[e][1,4]oxazepin-2(H)-one 2b
96% yield, m.p. 154–155 °C; 1H NMR (400 MHz, CDCl3): δ ppm 2.33 (s, 3H), 4.57 (s, 2H), 4.70 (s, 2H), 6.79 (s, 1H), 6.87 (d, J = 7.6 Hz, 1H), 7.02 (d, J = 7.6 Hz, 1H), 8.75 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 21.0, 72.5, 73.3, 119.9, 124.5, 125.9, 128.4, 135.7, 139.3, 173.5; IR v (cm−1): 3377.0, 3184.2, 2962.5, 1656.5, 1259.5; HRMS calcd for C10H11NO2 (M + Na)+ 200.0687, found 200.0673.8-Fluoro-3,5-dihydrobenzo[e][1,4]oxazepin-2(H)-one 2c
94% yield, m.p. 166–167 °C; 1H NMR (400 MHz, d6-DMSO): δ ppm 4.43 (s, 2H), 4.65 (s, 2H), 6.79 (dt, J1 = 8.0 Hz, J2 = 2.4 Hz, 1H), 6.92 (dd, J1 = 10.4 Hz, J2 = 2.4 Hz, 1H), 7.17 (d, J = 8.4 Hz, 1H), 10.30 (s, 1H); 13C NMR (100 MHz, d6-DMSO): δ ppm 71.7, 73.8, 106.2 (d, J = 25.8 Hz), 109.4 (d, J = 21.1 Hz), 126.0 (d, J = 2.8 Hz), 130.6 (d, J = 9.6 Hz), 139.1 (d, J = 10.5 Hz), 162.3 (d, J = 41.4 Hz), 173.4; 19F NMR: −113.9; IR v (cm−1): 3194.0, 3066.2, 1661.0, 1603.5, 1372.5; HRMS calcd for C9H8FNO2 (M + Na)+ 204.0437, found 204.0430.7-(Trifluoromethyl)-3,5-dihydrobenzo[e][1,4]oxazepin-2(1H)-one 2d
90% yield, 1H NMR (400 MHz, CDCl3): δ ppm 4.62 (s, 2H), 4.78 (s, 2H), 7.24–7.31 (m, 3H), 8.91 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 70.1, 70.2, 123.9 (q, J = 3.9 Hz), 124.5 (q, J = 267.2 Hz), 126.2 (q, J = 3.9 Hz), 129.3, 131.8 (q, J = 32.6 Hz), 133.5, 138.6, 171.6; 19F NMR: δ ppm 78.8; IR v (cm−1): 2979.8, 1663.7, 1334.6, 1121.5; HRMS calcd for C10H8F3NO2 (M − H) 230.0427, found 230.0425.8-(Trifluoromethyl)-3,5-dihydrobenzo[e][1,4]oxazepin-2(H)-one 2e
92% yield, m.p. 128–129 °C; 1H NMR (400 MHz, CDCl3): δ ppm 4.56 (s, 2H), 4.77 (s, 2H), 7.09 (d, J = 8.4 Hz, 1H), 7.37 (s, 1H), 7.53 (d, J = 8.4 Hz, 1H), 9.44 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 72.9, 74.0, 119.6, 123.9 (q, J = 270.2 Hz), 125.7 (q, J = 32.6 Hz), 125.7 (q, J = 3.9 Hz), 126.4 (q, J = 3.9 Hz), 129.2, 138.9, 174.5; 19F NMR: δ ppm 62.1; IR v (cm−1): 2919.9, 1658.6, 1447.8, 1084.8, 656.7; HRMS calcd for C10H8F3NO2 (M − H)+ 230.0427, found 230.0429.8,9-Dimethoxy-3,5-dihydrobenzo[e][1,4]oxazepin-2(H)-one 2f
90% yield, m.p. 148–149 °C; 1H NMR (400 MHz, CDCl3): δ ppm 3.87 (s, 3H), 3.88 (s, 3H), 4.61 (s, 2H), 4.67 (s, 2H), 6.58 (d, J = 8.8 Hz, 1H), 6.78 (d, J = 8.8 Hz, 1H), 8.30 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 55.9, 60.8, 72.9, 74.1, 106.6, 121.8, 122.8, 129.5, 136.8, 152.3, 172.4; IR: v (cm−1): 3195.3, 1661.6, 1391.6, 1112.3. HRMS calcd for C11H13NO4 (M + Na)+ 246.0742, found 246.0736.5-Methyl-3,5-dihydrobenzo[e][1,4]oxazepin-2(H)-one 2g
89% yield, m.p. 120–121 °C; 1H NMR (400 MHz, CDCl3): δ ppm 1.64 (d, J = 6.4 Hz, 3H), 4.54 (d, J = 2.4 Hz, 2H), 4.78 (q, J = 6.4 Hz, 1H), 6.99 (dd, J1 = 8.4H, J2 = 0.4 Hz, 1H), 7.01 (td, J1 = 7.6 Hz, J2 = 0.4 Hz, 1H), 7.23–7.29 (m, 2H), 8.68 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 19.0, 71.1, 75.2, 120.0, 123.9, 125.9, 128.8, 132.7, 136.0, 173.7; IR v (cm−1): 3196.7, 3065.5, 2997.0, 2903.1, 1656.9; HRMS calcd for C10H11NO2 (M − H)+ 176.0721, found 176.0725. For corresponding chiral R-2g: −78° (c 0.10, acetone), 83% ee (determined by a chiral AD-H column, iPrOH/hexane = 10/90, tmajor = 14.26 min, tminor = 15.40 min).6-Chloro-3,5-dihydrobenzo[e][1,4]oxazepin-2(H)-one 2h
83% yield, m.p. 172–173 °C; 1H NMR (400 MHz, CDCl3): δ ppm 4.52 (s, 2H), 4.99 (s, 2H), 6.78 (d, J = 7.6 Hz, 1H), 7.11–7.19 (m, 2H), 7.70 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 69.8, 73.1, 118.2, 124.9, 126.6, 129.2, 133.7, 137.5, 173.0; IR v (cm−1): 3246.6, 2918.9, 1671.0, 1480.4, 1258.1; HRMS calcd for C9H8ClNO2 (M + Na)+ 220.0141, found 220.0143.5-Methyl-3,5-dihydropyrido[2,3-e][1,4]oxazepin-2(H)-one 2i
89% yield, m.p. 106–106.5 °C; 1H NMR (400 MHz, CDCl3): δ ppm 1.64 (d, J = 6.4 Hz, 3H), 4.64 (d, J = 6.0 Hz, 2H), 4.73 (q, J = 6.4 Hz, 1H), 7.04 (dd, J1 = 7.6 Hz, J2 = 4.8 Hz, 1H), 7.53 (d, J = 7.8 Hz, 1H), 8.40 (d, J = 4.8 Hz, 1H), 9.38 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 18.9, 72.2, 75.0, 118.7, 127.4, 134.7, 147.6, 149.1, 173.0; IR v (cm−1): 3180.4, 1662.7, 1634.3, 1401.8, 1315.7; HRMS calcd for C9H10N2O2 (M + Na)+ 201.0640, found 201.0623.8-Methyl-3,5-dihydro-[1,4]oxazepino[5,6-b]quinolin-2(H)-one 2j
85% yield, m.p. 134–135 °C; 1H NMR (400 MHz, CDCl3): δ ppm 2.51 (s, 3H), 4.69 (s, 2H), 4.85 (s, 2H), 7.51 (d, J = 7.6 Hz, 2H), 7.77 (d, J = 8.4 Hz, 1H), 7.80 (s, 1H), 8.47 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 21.5, 72.2, 74.3, 123.3, 125.0, 126.1, 127.2, 132.8, 135.7, 145.0, 147.6, 172.3; IR v (cm−1): 2919.9, 1658.6, 1447.8, 1084.8, 656.7; HRMS calcd for C13H12N2O2 (M − H)+ 227.0821, found 227.0848.2H-Benzo[b][1,4]oxazin-3(4H)-one 2k6a,16,17
Known compound, 99% yield; 1H NMR (400 MHz, CDCl3): δ ppm 4.63 (s, 2H), 6.82–6.98 (m, 4H), 8.07 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 67.2, 116.0, 116.8, 122.7, 124.3, 126.0, 143.6, 165.9.2-Methyl-2H-benzo[b][1,4]oxazin-3(4H)-one 2l17
Known compound, 97% yield; 1H NMR (400 MHz, CDCl3): δ ppm 1.59 (d, J = 6.8 Hz, 3H), 4.67 (q, J = 6.8 Hz, 1H), 6.88–6.89 (m, 1H), 6.96–6.97 (m, 3H), 9.68 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 16.2, 73.2, 115.9, 117.0, 122.6, 124.1, 126.4, 143.1, 168.9.2H-Benzo[b][1,4]thiazin-3(4H)-one 2m18,19
Known compound, 99% yield; 1H NMR (400 MHz, CDCl3): δ ppm 3.44 (s, 2H), 6.90 (d, J = 8.0 Hz, 1H), 7.01 (dt, J1 = 8.0 Hz, J2 = 1.2 Hz, 1H), 7.17 (dt, J1 = 7.6 Hz, J2 = 1.2 Hz, 1H), 7.31 (d, J = 7.8 Hz, 1H), 9.11 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 29.9, 117.4, 119.9, 123.9, 127.2, 127.8, 136.3, 166.4;2-Methyl-2H-benzo[b][1,4]thiazin-3(4H)-one 2n18,19
Known compound, 98% yield; 1H NMR (400 MHz, CDCl3): δ ppm 1.50 (d, J = 7.2 Hz, 3H), 3.56 (q, J = 7.2 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 7.02 (t, J = 6.8 Hz, 1H), 7.18 (t, J = 8.0 Hz, 1H), 7.30–7.31 (m, 1H), 8.86 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 15.4, 37.0, 116.9, 119.4, 123.8, 127.1, 128.1, 136.0, 168.9.7-Methyl-2H-benzo[b][1,4]oxazin-3(4H)-one 2o18
Known compound, 95% yield; 1H NMR (400 MHz, CDCl3): δ ppm 2.28 (s, 3H), 4.60 (s, 2H), 6.67 (s, 1H), 6.78 (d, J = 8.0 Hz, 1H), 6.87 (d, J = 8.4 Hz, 1H), 9.41 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 20.7, 67.2, 116.5, 116.7, 120.3, 124.7, 125.8, 132.5, 166.6.6-Methyl-2H-benzo[b][1,4]oxazin-3(4H)-one 2p18,19
Known compound, 95% yield; 1H NMR (400 MHz, CDCl3): δ ppm 2.28 (s, 3H), 4.60 (s, 2H), 6.67 (s, 1H), 6.78 (dd, J1 = 8.4 Hz, J2 = 1.2 Hz, 1H), 6.86 (d, J = 7.6 Hz, 1H), 9.36 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 20.6, 67.2, 116.4, 116.5, 124.7, 125.7, 132.5, 141.4, 166.7.6-Methoxy-2H-benzo[b][1,4]oxazin-3(4H)-one 2q18,19
Known compound, 96% yield; 1H NMR (400 MHz, CDCl3): δ ppm 3.76 (s, 3H), 4.57 (s, 2H), 6.40 (d, J = 2.4 Hz, 1H), 6.50 (dd, J1 = 8.6 Hz, J2 = 3.2 Hz, 1H), 6.85 (d, J = 8.8 Hz, 1H), 8.93 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 55.8, 67.4, 102.1, 108.8, 117.2, 126.8, 137.6, 155.3, 166.6.1H-Naphtho[2,1-b][1,4]oxazin-2(3H)-one 2r
96% yield, m.p. 207–208 °C; 1H NMR (400 MHz, CDCl3): δ ppm 4.76 (s, 2H), 7.22 (d, J = 8.2 Hz, 1H), 7.43 (td, J1 = 7.8 Hz, J2 = 0.8 Hz, 1H), 7.54 (d, J = 9.2 Hz, 1H), 7.58 (td, J1 = 7.8 Hz, J2 = 1.2 Hz, 1H), 7.83 (d, J = 8.4 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 9.66 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 67.5, 117.4, 118.7, 119.0, 123.0, 124.3, 124.7, 127.0, 128.7, 130.0, 140.6, 166.0; IR v (cm−1): 3189.9, 2961.3, 2877.5, 1681.8, 1458.9; HRMS calcd for C12H9NO2 (M + Na)+ 222.0531, found 222.0527.8-Amino-2H-benzo[b][1,4]oxazin-3(4H)-one 2s20
Known compound, 85% yield; 1H NMR (400 MHz, CDCl3): δ ppm 4.73 (s, 2H), 6.73 (dd, J1 = 8.0 Hz, J2 = 1.2 Hz, 1H), 6.90 (t, J = 8.0 Hz, 1H), 7.06 (dd, J1 = 8.0 Hz, J2 = 1.2 Hz, 1H), 8.61 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 67.4, 114.3, 122.2, 122.9, 125.1, 127.1, 139.8, 165.4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Natural Science Foundation of the Jiangsu Higher Education (Grant No. 15KJB150029), the Project of Science and Technology of Xuzhou Government (No. KC16SG250) and the Natural Science Foundation of Jiangsu Province (Grants No. BK 20171175). The work is also sponsored by Qing Lan Project of Jiangsu Province and Zhen Xing Project of XZMC.Notes and references
- (a) H. Otsuka, Y. Hirani, T. Nagao and K. Yamasaki, J. Nat. Prod., 1988, 51, 74 CrossRef CAS PubMed; (b) A. S. Bourlot, I. Sanchez, G. Dureng, G. Guillaumet, R. Massingham, A. Monteil, E. Winslow, M. D. Pujol and J. Y. Merour, J. Med. Chem., 1998, 41, 3142 CrossRef CAS PubMed; (c) D. A. Dudley, A. M. Bunker, L. Chi, W. L. Cody, D. R. Holland, D. P. Ignasiak, N. Janiczek-Dolphin, T. B. McClanahan, T. E. Mertz, L. S. Narasimhan, S. T. Rapundalo, J. A. Trautschold, C. A. Van Huis and J. J. Edmunds, J. Med. Chem., 2000, 43, 4063 CrossRef CAS PubMed; (d) R. Fringuelli, D. Pietrella, F. Schiaffella, A. Guarraci, S. Perito, F. Bistoni and A. Vecchiarelli, Bioorg. Med. Chem., 2002, 10, 1681 CrossRef CAS PubMed.
- T. B. Lanni Jr, K. L. Greene, C. N. Kolz, K. S. Para, M. Visnick, J. L. Mobley, D. T. Dudley, T. J. Baginski and M. B. Liimatta, Bioorg. Med. Chem. Lett., 2007, 17, 756 CrossRef PubMed.
- C. Su, C. Tseng, C. Ramesh, H. Liu, C. Huang and C. Yao, Eur. J. Med. Chem., 2017, 132, 90 CrossRef CAS PubMed.
- H. Chan, Synthesis, 1984, 10, 851 Search PubMed.
- (a) T. Miki, M. Kori, H. Mabuchi, H. Banno, R. Tozawa, M. Nakamura, S. Itokawa, Y. Sugiyama and H. Yukimasa, Bioorg. Med. Chem., 2002, 10, 401 CrossRef CAS PubMed; (b) E. Feng, H. Huang, Y. Zhou, D. Ye, H. Jiang and H. J. Liu, J. Org. Chem., 2009, 74, 2846 CrossRef CAS PubMed; (c) A. Sharifi, M. Barazandeh, A. Saeed and M. Mirzaei, Tetrahedron Lett., 2010, 51, 1852 CrossRef CAS; (d) D. Chen, Z. Wang and W. Bao, J. Org. Chem., 2010, 75, 5768 CrossRef CAS PubMed.
- (a) D. Chen, G. Shen and W. Bao, Org. Biomol. Chem., 2009, 7, 4067 RSC; (b) A. Sharifi, M. Ansari, H. Darabi and M. Abaee, Tetrahedron Lett., 2016, 57, 529 CrossRef CAS.
- (a) J. F. Hartwig, Acc. Chem. Res., 1998, 31, 852 CrossRef CAS; (b) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534 CrossRef CAS PubMed.
- (a) J. P. Wolfe, S. Wagaw, J. F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805 CrossRef CAS; (b) A. R. Muci and S. L. Buchwald, Top. Curr. Chem., 2002, 219, 131 CrossRef CAS.
- (a) B. P. Fors, P. Krattiger, E. Strieter and S. L. Buchwald, Org. Lett., 2008, 10, 16 CrossRef PubMed; (b) D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27 RSC.
- B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1998, 120, 7369 CrossRef CAS.
- N. C. Bruno, N. Niljianskul and S. L. Buchwald, J. Org. Chem., 2014, 79, 4161 CrossRef CAS PubMed.
- N. A. Isley, S. Dobarco and B. H. Lipshutz, Green Chem., 2014, 16, 1480 RSC.
- J. K. Laha, P. U. Shah and K. P. Jethava, Chem. Commun., 2013, 49, 7623 RSC.
- L. Zhang, L. Sonaglia, J. Stacey and M. Lautens, Org. Lett., 2013, 15, 2128 CrossRef CAS PubMed.
- (a) Z. Xu, H. Chen, Z. Wang, A. Ying and L. Zhang, J. Am. Chem. Soc., 2016, 138, 5515 CrossRef CAS PubMed; (b) Z. Xu, R. L. Zhai, T. Liang and L. Zhang, Chem.–Eur. J., 2017, 23, 14133 CrossRef CAS PubMed.
- P. Stefanic, K. Turnsek and D. Kikelj, Tetrahedron, 2003, 59, 7123 CrossRef CAS.
- (a) S. Ceylan, L. Coutable, J. Wegner and A. Kirschning, Chem.–Eur. J., 2011, 17, 1884 CrossRef CAS PubMed; (b) C. Rajitha, P. K. Dubey, V. Sunku, F. J. Piedrafita, V. R. Veeramaneni and M. Pal, Eur. J. Med. Chem., 2011, 46, 4887 CrossRef CAS PubMed.
- W. S. Huang, R. Xu, R. Dodd and W. C. Shakespeare, Tetrahedron Lett., 2013, 54, 5214 CrossRef CAS.
- F. Babudri, S. Florio, G. Indelicati and G. Trapani, J. Org. Chem., 1983, 48, 4082 CrossRef CAS.
- P. Smid, H. Coolen, H. G. Keizer, R. Hes, J. Moes, A. P. Hartog, B. Stork, R. H. Plekkenpol, L. C. Niemann, C. Stroomer, M. Tulp, H. Stuivenberg, A. McCreary, M. Hesselink, H. J. Arnoud and C. G. Kruse, J. Med. Chem., 2005, 48, 6855 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09160e |
| This journal is © The Royal Society of Chemistry 2017 |