
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
α-Alkylation of ketimines using visible light photoredox catalysis†
Allegra
Franchino
,
Antonia
Rinaldi
and
Darren J.
Dixon
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: darren.dixon@chem.ox.ac.uk
First published on 11th September 2017
Abstract
A novel α-alkylation of N-diphenylphosphinoyl ketimines with α-bromocarbonyl compounds has been accomplished using visible light photoredox catalysis. The reaction proceeds under remarkably mild conditions: at 35 °C, in 20 hours, under blue light irradiation (1 W), in the presence of a tertiary amine and catalytic amounts of Ru- and Ni-based complexes. Chemoselective transformation of the products provides access to 1,4-dicarbonyl compounds, protected GABA analogues and γ-lactams.
In recent years, photoredox catalysis has emerged as a powerful methodology to construct C–C bonds under mild conditions, typically involving visible light irradiation and the use of a catalytic amount of an organic dye or metal complex.1 In the continuation of our work on new reactivity of imines under photoredox catalysis,2 we were interested in studying the behaviour of ketimines with the aim of achieving C–C bond formation at the α-position (Fig. 1). The general approach for the α-alkylation of carbonyl compounds involves their conversion into carbon nucleophiles (such as enamines,3 enolates and enol silyl ethers, either pre-formed or formed in situ) followed by SN2 reaction with alkyl halides (Fig. 1, top). Photoredox catalysis has recently added a new dimension to this long-established reactivity, uncovering novel mechanistic pathways and often allowing the reactions to be carried out without the use of organometallic reagents nor strong bases. Since MacMillan's seminal work on the enantioselective alkylation of aldehydes through merging photoredox catalysis with aminocatalysis,4 alkylations of aldehydes,5 ketones,6 2-acylimidazoles7 and silyl enol ethers8 with α-halocarbonyl compounds under light irradiation have been reported. In this context we speculated that, if a similar reactivity could be extended to a new class of substrates, i.e. ketimines, a mild method for the generation of valuable γ-imino esters would be realised (Fig. 1, bottom). Synthetic manipulation of the products would provide access not only to 1,4-dicarbonyl compounds,8 but also to γ-amino esters (protected analogues of the neurotransmitter γ-aminobutyric acid, GABA),9 and to γ-lactams, which are found in both natural and synthetic molecules possessing a range of biological properties.10 However, we anticipated a series of challenges in the development of the reaction: (i) the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CO bonds of the substrates must survive the reaction conditions; (ii) overalkylation should be controlled; (iii) reductive dehalogenation of the α-bromocarbonyl compound should be avoided.
N and CO bonds of the substrates must survive the reaction conditions; (ii) overalkylation should be controlled; (iii) reductive dehalogenation of the α-bromocarbonyl compound should be avoided.
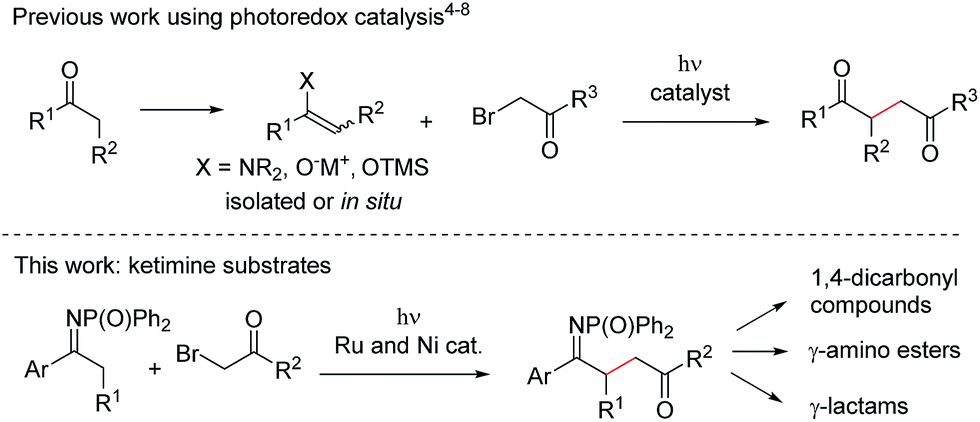 | ||
| Fig. 1 Photoredox-catalysed α-alkylation of carbonyl compounds and derivatives. | ||
Keeping these considerations in mind, we began our investigation by studying then optimising a model reaction between N-diphenylphosphinoyl acetophenone-derived ketimine 1a and ethyl bromoacetate 2, using N,N-diisoproprylamine (DIPEA) as a base, 5 mol% [Ru(bpy)3]Cl2·6H2O and 5 mol% [NiCl2(PPh3)2] (Table 1, see ESI for full data†).11 Pleasingly, the desired alkylation reaction proceeded with encouraging conversion in polar solvents that were able to solubilise all components (Table 1, entries 1–5). DMF was selected as the solvent of choice due to the good yield and the clean reaction profile (entry 5, 56% conversion, 50% yield for the desired ketimine 3a). A range of photocatalysts were then screened for performance (entries 6–9) and [Ru(bpy)3]Cl2·6H2O was confirmed to be the most effective. Among the Ni(II) salts that were tested, inexpensive nickel(II) acetate tetrahydrate was found to promote the reaction without significant detriment to the yield (45%, entry 10). Several organic and inorganic bases were trialled and their stoichiometry was varied. When using triethanolamine (TEOA) instead of DIPEA, the reaction proceeded to nearly full conversion (95%, entry 11), however the NMR yield for the alkylated ketimine 3a was a modest 28%. The poor yield observed in this case is explained by the formation of E and Z enamines 3a′ (21%) and dialkylated product (41%). Similarly, the addition of 2 further equivalents of both DIPEA and bromoacetate 2 enhanced the conversion to 72% (entry 12), but the yield for 3a remained 45%.
|
|
|||
|---|---|---|---|
| Entry | Change to conditions above | Conversiona (%) | Yielda3a + 3a′ (%) |
| a Consumption of ketimine 1a (conversion) and NMR yields for 3a and 3a′ determined by 31P and 1H NMR analysis of the reaction mixture after work-up using mesitylene as an internal standard. b 2 further equivalents of DIPEA and bromoacetate 2 were added after 7 hours. | |||
| 1 | Acetone | 29 | 21 + 3 |
| 2 | CH3CN | 34 | 34 + 0 |
| 3 | MeOH | 51 | 32 + 14 |
| 4 | DMSO | 59 | 34 + 23 |
| 5 | — | 56 | 50 + 5 |
| 6 | Eosin Y disodium form | 53 | 41 + 5 |
| 7 | [Ru(bpz)3](PF6)2 | 37 | 35 + 2 |
| 8 | [Ir{dF(CF3)ppy}2(dtbpy)]PF6 | 23 | 16 + 7 |
| 9 | [Ir(ppy)2(dtbpy)]PF6 | 31 | 29 + 0 |
| 10 | Ni(OAc)2·4H2O | 55 | 45 + 8 |
| 11 | N(CH2CH2OH)3 | 95 | 28 + 21 |
| 12 | second addition of DIPEA and 2b | 72 | 45 + 14 |
| 13 | Without [NiCl2(PPh3)2] | 38 | 28 + 10 |
| 14 | Without DIPEA | 12 | 0 |
| 15 | In the dark | 0 | 0 |
| 16 | Without [Ru(bpy)3]Cl2·6H2O | 7 | 4 + 2 |
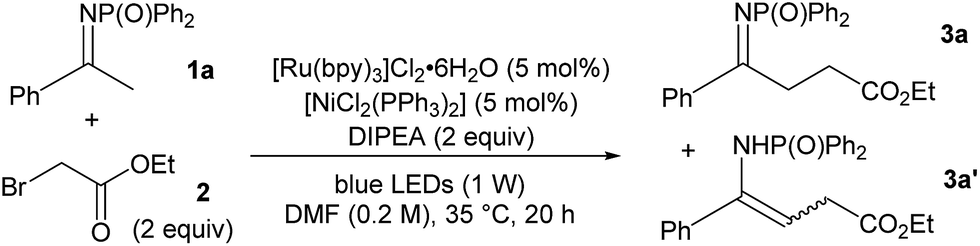
Control experiments were run to gather more information (Table 1). Omission of [NiCl2(PPh3)2] resulted in a lower yield of the product, showing that Ni(II), although not essential for reactivity, improved the conversion (entry 13). No product formation was observed in the absence of base or light, thus ruling out simple radical or SN2-type reactivity, while small amounts of 3a were formed under light irradiation without photocatalyst12 (entries 14–16).
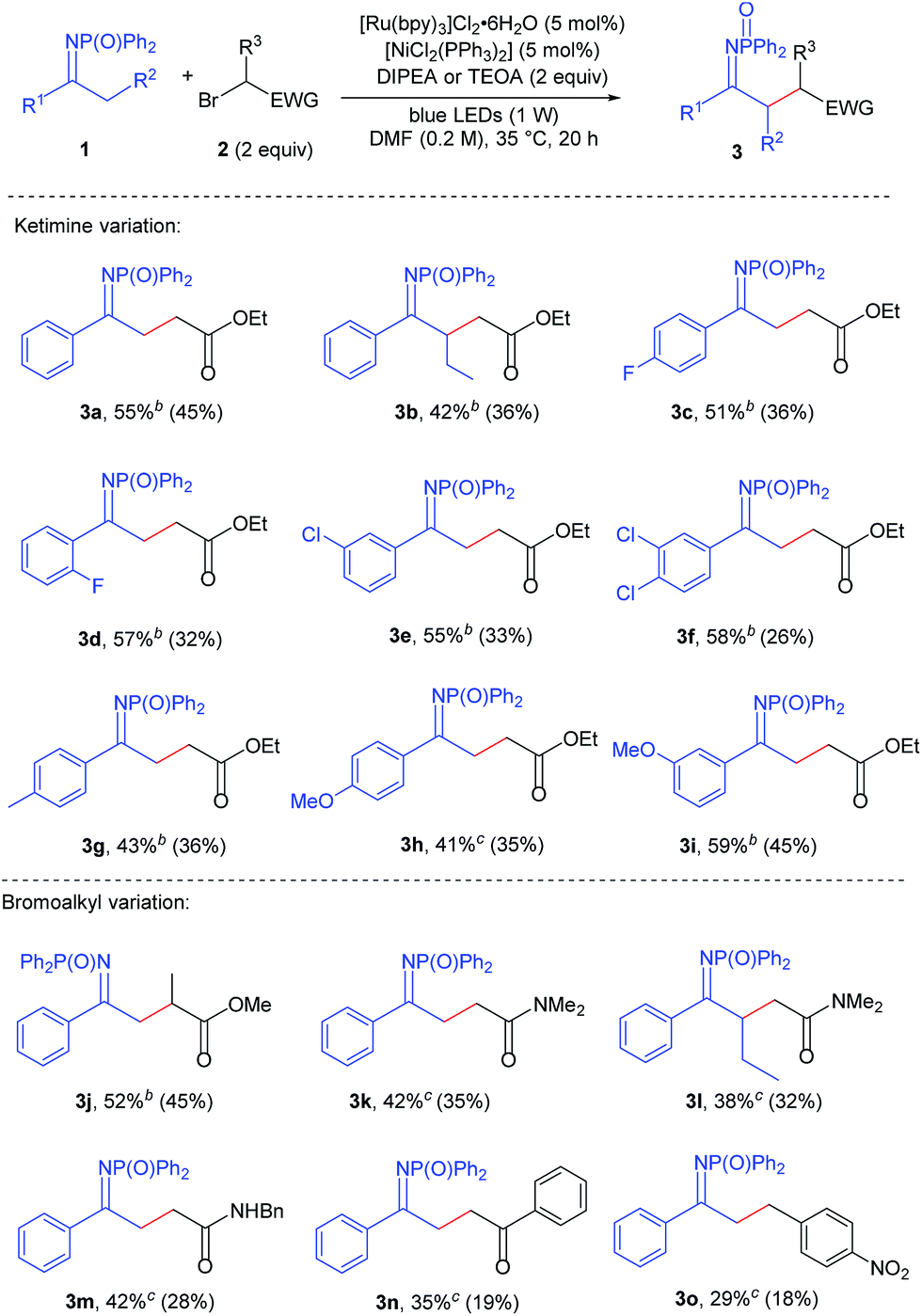
At this point the substrate scope of the reaction was assessed, using [Ru(bpy)3]Cl2·6H2O (5 mol%), [NiCl2(PPh3)2] (5 mol%) and 2 equivalents of either DIPEA or TEOA. With respect to acetophenone-derived ketimine 1a, substrates possessing electron-withdrawing groups on the aromatic ring were generally found to be more reactive. The most appropriate base to carry out their alkylation was DIPEA, that when compared to TEOA minimised the amounts of dialkylated product and enamine tautomers arising from the reaction. Aryl methyl ketimines 1c–1f, bearing F and Cl substituents in the para, meta and ortho positions of the aromatic ring were alkylated affording 51–58% NMR yields of products (ketimine plus enamine tautomers, Table 2, compounds 3c–3f). Isolated yields for the ketimine tautomers were lower (26–36%), as these compounds were prone to tautomerise to the enamine form. In line with this trend, electron-rich ketimines 1g and 1h were found to be less reactive than the model substrate.13 Alkylated ketimines with p-methyl (3g), p-methoxy (3h) and m-methoxy (3i) substituents on the aromatic ring were obtained in 35–45% yields.
| a Reactions performed on 0.2 mmol scale. Reaction yields (ketimine product plus enamine tautomers) determined by 1H NMR analysis of the reaction mixture after work-up using mesitylene as an internal standard. Isolated yields, given in parentheses, refer to spectroscopically pure product in predominantly ketimine tautomeric form as obtained after chromatographic purification. b DIPEA as a base. c TEOA as a base. |
|---|
|
Next, the substrate scope on the bromoalkyl component was investigated.14 The use of ethyl 2-bromopropionate delivered product 3j in 45% yield. Electron-withdrawing groups different from an ester could be used, although they showed lower reactivity, thus requiring the use of TEOA as a base. 2-Bromoacetamides were found to be compatible alkylating agents, delivering compounds 3k and 3m in 35% and 28% yield, respectively. 2-Bromoacetophenone effected the alkylation of the model ketimine in 19% yield (3n). Alkylating agents lacking the α-carbonyl group were not suitable reaction partners, with p-nitrobenzyl bromide affording product 3o in 18% yield.
Elongating the alkyl chain on the ketimine did not affect the reactivity significantly. Indeed ketimine 1b possessing a n-propyl chain could be alkylated with ethyl bromoacetate and 2-bromo-N,N-dimethylacetamide obtaining compounds 3b and 3l with isolated yields (32–36%) nearly comparable to those of ketimine 1a.
To demonstrate synthetic applicability, chemoselective transformations of compound 3a were performed (Scheme 1). The imine functionality was hydrolysed to afford γ-keto ester 4 in 78% yield. Alternatively, reduction of the CN bond using sodium borohydride provided γ-amino ester 5 in 82% yield. The latter compound could be deprotected and then cyclised in a one-pot procedure, affording γ-lactam 6 in 81% yield.
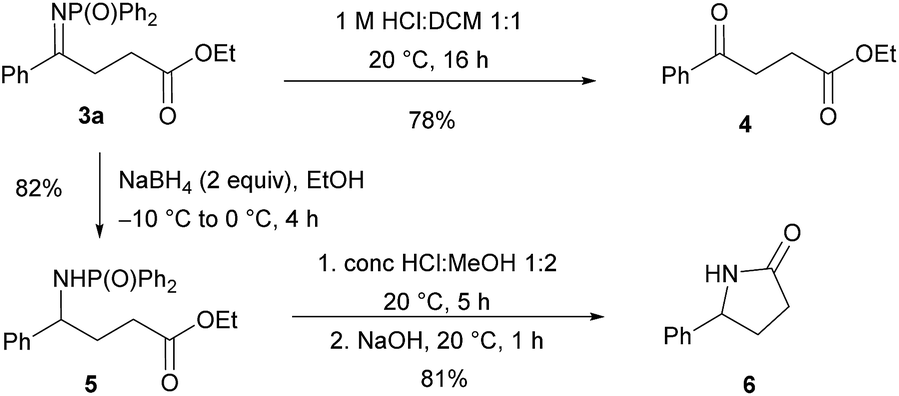 | ||
| Scheme 1 Synthetic transformations of product 3a. | ||
Finally, a series of experiments were performed to investigate the mechanism of the reaction. Addition of 1 equivalent of TEMPO inhibited product formation, indicating that radical intermediates are involved in the reaction (Scheme 2a). When N-DPP ketimine 1a was replaced by enamide 7, formation of the expected alkylated product 8 (alongside hydrolysed ketoester 4 and unreacted starting material, Scheme 2b) was observed, suggesting that the addition of an electron-deficient radical to an enamide double bond is the likely pathway, in accordance with the intramolecular mechanism postulated by Rueping for the cyclisation of α-chloroenamides.10b This mechanistic hypothesis, at first glance, appears to conflict with the observation that electron-rich ketimines are less reactive than electron-poor ones. However, this apparent discrepancy is explained by the unfavourable ketimine/enamine equilibrium of electron-rich substrates. The ketimine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :enamine ratio for 3 different substrates in the presence of DIPEA and TEOA was determined by NMR experiments (Fig. 2).15 Using either base, electron-poor ketimine 1f derived from 3′,4′-dichloroacetophenone existed in equilibrium with a higher percentage of enamine tautomer when compared to standard ketimine 1a and electron-rich substrate 1h possessing a p-methoxyphenyl group. No significant changes to the ketimine:enamine ratio were detected after 1 h or upon addition of 5 mol% [NiCl2(PPh3)2], indicating that the equilibrium position for the tautomerism is reached relatively quickly and is not influenced by [NiCl2(PPh3)2].
:enamine ratio for 3 different substrates in the presence of DIPEA and TEOA was determined by NMR experiments (Fig. 2).15 Using either base, electron-poor ketimine 1f derived from 3′,4′-dichloroacetophenone existed in equilibrium with a higher percentage of enamine tautomer when compared to standard ketimine 1a and electron-rich substrate 1h possessing a p-methoxyphenyl group. No significant changes to the ketimine:enamine ratio were detected after 1 h or upon addition of 5 mol% [NiCl2(PPh3)2], indicating that the equilibrium position for the tautomerism is reached relatively quickly and is not influenced by [NiCl2(PPh3)2].
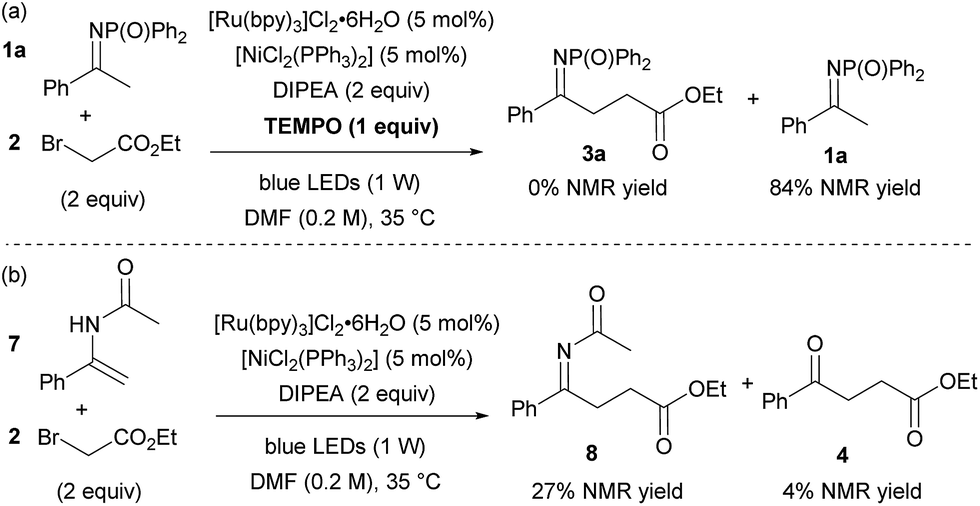 | ||
| Scheme 2 Experiments to investigate the mechanism of the reaction. | ||
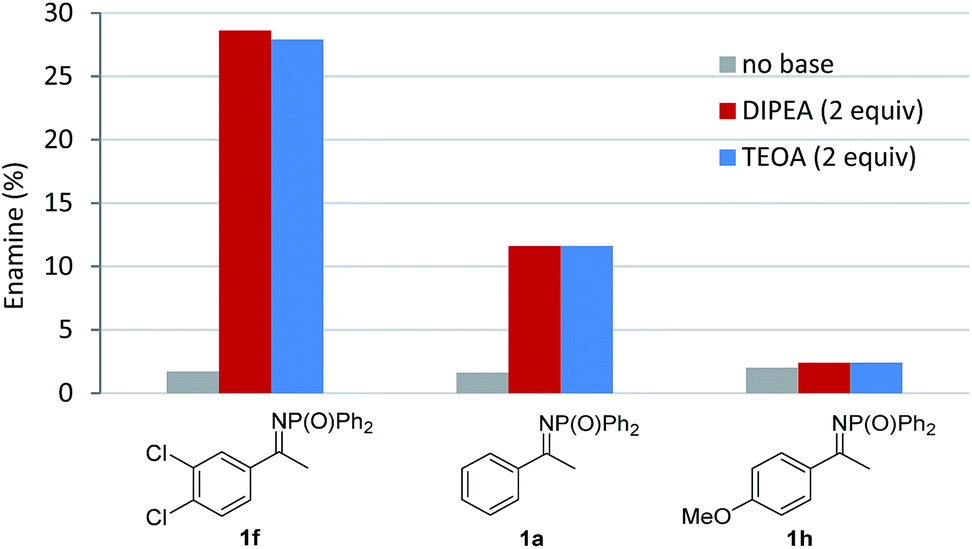 | ||
| Fig. 2 Percentage of enamine present in a 0.2 M solution of 1f, 1a and 1h in DMSO-d6 after 5 hours, as determined by 1H and 31P NMR analysis. | ||
Light/dark cycle experiments (see ESI†) confirmed that the reaction proceeds only under light irradiation. Spectroscopic studies were then performed to determine which species was responsible for the quenching of the luminescence emitted by the excited Ru2+ photocatalyst (Fig. 3, see ESI for details†).16 Whilst DMF solutions of DIPEA and of 3′,4′-dichloroacetophenone-derived ketimine 1f with 2 equivalents of TEOA (ketimine:enamine ratio 79:21 by 31P NMR) were found to quench the luminescence of *[Ru(bpy)3]2+, ketimines 1a, 1f, TEOA and ethyl bromoacetate 2 did not.
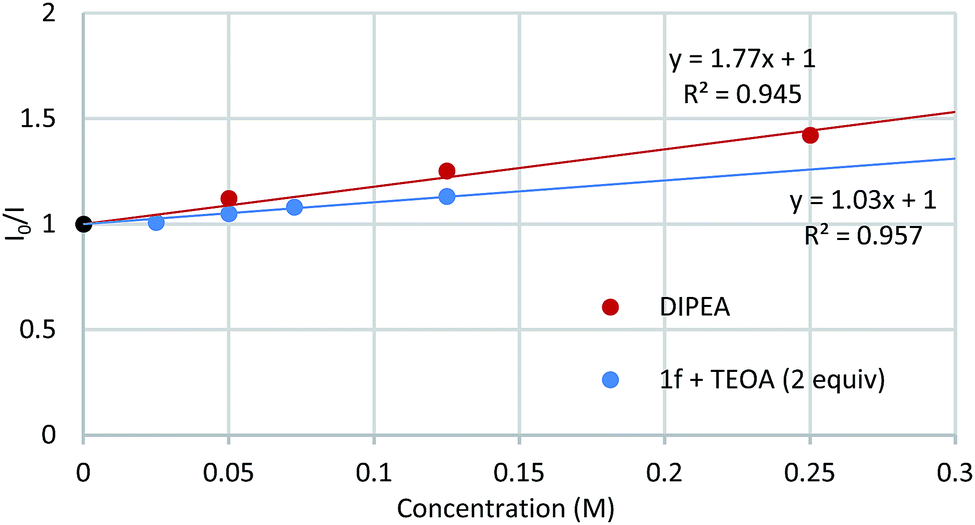 | ||
| Fig. 3 Luminescence quenching studies. [Ru(bpy)3]Cl2·6H2O 1 μM in DMF, λ(excitation) 452 nm, λ(emission) 620 nm. | ||
On the basis of these observations and of literature precedents,4 we propose the mechanism depicted in Scheme 3 for the visible-light promoted, Ru-catalysed α-alkylation of N-diphenylphosphinoyl ketimine 1a with ethyl bromoacetate 2. The photoexcited ruthenium catalyst is reductively quenched by DIPEA or the enamine form of the starting material to a Ru(I) species (EII/I1/2 = −1.33 V vs. SCE in CH3CN),17 which is then able to reduce ethyl bromoacetate (Ered1/2 = −1.08 V vs. SCE in CH3CN)18 generating the α-carbonyl radical I. This electron-deficient radical attacks the electron-rich double bond of the enamine tautomer (1a′) of the starting material, leading to the formation of α-amino radical II. The latter (Ered1/2 = ca. −1.0 V vs. SCE in CH3CN)19 can be oxidised to the protonated product III by *[Ru(bpy)3]2+ (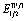 = +0.77 V vs. SCE in CH3CN),17 or by the less oxidising but more abundant bromoacetate 2, in the case a radical chain mechanism is operative.20 Finally, deprotonation of compound III affords product 3a. Depending on the nature of the ketimine and on reaction conditions, 3a may tautomerise and undergo a second alkylation.
= +0.77 V vs. SCE in CH3CN),17 or by the less oxidising but more abundant bromoacetate 2, in the case a radical chain mechanism is operative.20 Finally, deprotonation of compound III affords product 3a. Depending on the nature of the ketimine and on reaction conditions, 3a may tautomerise and undergo a second alkylation.
 | ||
| Scheme 3 Proposed mechanism for the visible light-promoted, Ru-catalysed α-alkylation of N-diphenylphosphinoyl ketimine 1a with ethyl bromoacetate 2. | ||
At present, we cannot exclude an alternative mechanism based on the coupling between enamine radical cation IV (or radical V) with the α-carbonyl radical I (Scheme 4).21
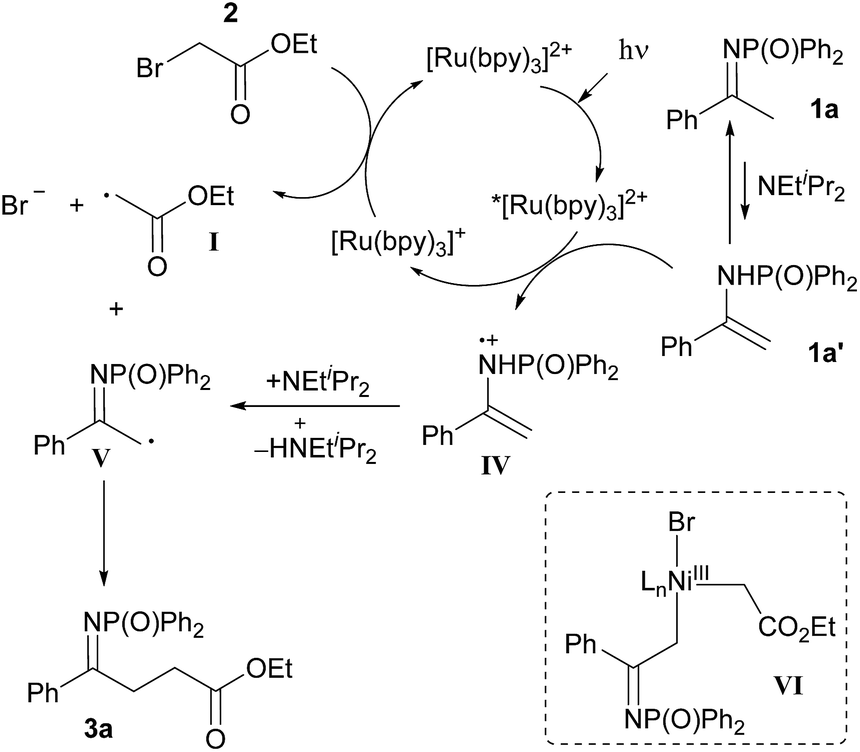 | ||
| Scheme 4 Alternative mechanism for the visible light-promoted, Ru-catalysed α-alkylation of N-diphenylphosphinoyl ketimine 1a with ethyl bromoacetate 2. | ||
The beneficial effect of [NiCl2(PPh3)2] on yield and conversion might be linked to its ability to stabilise the radical species generated during the reaction or promote a more efficient reaction pathway.22 Based on the Stern–Volmer studies,23 it is not possible to rule out electron transfer24 or energy transfer processes25,26 from the excited photocatalyst to the nickel complex. Alternatively, the intermediacy of organonickel(III) species such as VI, from which C–C bond-forming reductive elimination would take place, can be imagined (see ESI for discussion†).22 Under this hypothesis, reductive elimination from organonickel(III) intermediate VI would help to overcome the unfavourable electronic effects associated with the direct coupling of the two electron-poor radicals I and V.
In summary, a mild α-alkylation of N-diphenylphosphinoyl ketimines with α-bromocarbonyl compounds has been accomplished in moderate yields using nickel and ruthenium light-promoted catalysis. The mechanism, investigated through a combination of control, NMR and luminescence quenching experiments, likely proceeds via the attack of an α-carbonyl radical to the enamine tautomer of the starting material. The product γ-imino esters were easily transformed into 1,4-dicarbonyl compounds, GABA analogues and γ-lactams. Further studies to elucidate the role of the nickel cocatalyst are ongoing in our laboratories and the results will be disclosed in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Angel L. Fuentes de Arriba for useful discussion, the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme for funding (A. F., FP7/2007–2013, REA grant agreement no. 316955) and the EPSRC (EP/G007802/1 Leadership Fellowship to D. J. D.).Notes and references
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (c) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC; (d) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (e) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (f) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- A. L. Fuentes de Arriba, F. Urbitsch and D. J. Dixon, Chem. Commun., 2016, 52, 14434–14437 RSC.
- G. Stork, A. Brizzolara, H. Landesman, J. Szmuszkovicz and R. Terrell, J. Am. Chem. Soc., 1963, 85, 207–222 CrossRef CAS.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- (a) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed; (b) M. Silvi, E. Arceo, I. D. Jurberg, C. Cassani and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120–6123 CrossRef CAS PubMed.
- (a) E. Arceo, A. Bahamonde, G. Bergonzini and P. Melchiorre, Chem. Sci., 2014, 5, 2438–2442 RSC; (b) Y. Zhu, L. Zhang and S. Luo, J. Am. Chem. Soc., 2014, 136, 14642–14645 CrossRef CAS PubMed.
- H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100–103 CrossRef CAS PubMed.
- N. Esumi, K. Suzuki, Y. Nishimoto and M. Yasuda, Org. Lett., 2016, 18, 5704–5707 CrossRef CAS PubMed , and references therein.
- A. Bertelli, L. Donati, V. Lami, G. Primo and M. A. Rossano, Int. J. Neuropharmacol., 1968, 7, 149–154 CrossRef CAS PubMed.
- (a) J. Caruano, G. G. Muccioli and R. Robiette, Org. Biomol. Chem., 2016, 14, 10134–10156 RSC. For a photoredox-catalysed synthesis of γ-lactams, see (b) E. Fava, M. Nakajima, M. B. Tabak and M. Rueping, Green Chem., 2016, 18, 4531–4535 RSC.
- [NiCl2(PPh3)2] was chosen based on reports for Reformatsky reactions of bromoacetates, where it has been identified as a highly effective catalyst: J. C. Adrian Jr and M. L. Snapper, J. Org. Chem., 2003, 68, 2143–2150 CrossRef PubMed.
- This might be due to the direct, uncatalysed photoexcitation of 1a′, the enamine tautomer of the starting ketimine (ref. 5b and 20) or of a donor–acceptor complex between enamine 1a′ and ethyl bromoacetate (ref. 5a).
- N-DPP ketimines derived from tert-butyl methyl ketone, cyclohexyl methyl ketone and 2-acetylthiophene gave the α-alkylated products using TEOA in 15%, 22% and 22% NMR yields respectively (see Table S1 in the ESI†).
- When using ethyl iodoacetate and chloroacetate, 3a was obtained in 36% and 12% NMR yield respectively. Benzyl bromide, ethyl bromodifluoroacetate and diethyl bromomalonate were not suitable reaction partners (see Table S1 in the ESI†).
- Given the similar reaction profile, DMSO-d6 was chosen as a cheaper alternative to DMF-d7 to avoid peak overlap.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- C. R. Bock, J. A. Connor, A. R. Gutierrez, T. J. Meyer, D. G. Whitten, B. P. Sullivan and J. K. Nagle, J. Am. Chem. Soc., 1979, 101, 4815–4823 CrossRef CAS.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- D. D. M. Wayner, J. J. Dannenberg and D. Griller, Chem. Phys. Lett., 1986, 131, 189–191 CrossRef CAS.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- (a) K. Narasaka, T. Okauchi, K. Tanaka and M. Murakami, Chem. Lett., 1992, 2099–2102 CrossRef CAS; (b) T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582–585 CrossRef CAS PubMed; (c) Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2012, 48, 5355–5357 RSC.
- (a) J. J. Douglas, M. J. Sevrin and C. R. J. Stephenson, Org. Process Res. Dev., 2016, 20, 1134–1147 CrossRef CAS; (b) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed; (c) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Chem., 2017, 1, 1–18 CrossRef.
- Luminescence quenching studies with [NiCl2(PPh3)2] were undertaken (see ESI†), but due to inner filter effects they did not provide any conclusive evidence of quenching.
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed.
- D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715–12718 CrossRef CAS PubMed.
- E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Science, 2017, 355, 380–385 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimisation tables, experimental procedures, characterisation data, copies of 1H and 13C NMR spectra, NMR and luminescence quenching studies. See DOI: 10.1039/c7ra09248b |
| This journal is © The Royal Society of Chemistry 2017 |