DOI:
10.1039/C7RA09343H
(Paper)
RSC Adv., 2017,
7, 52702-52711
Dual-phase spinel Li4Ti5O12/anatase TiO2 nanosheet anchored 3D reduced graphene oxide aerogel scaffolds as self-supporting electrodes for high-performance Na- and Li-ion batteries†
Received
23rd August 2017
, Accepted 7th November 2017
First published on 15th November 2017
Abstract
Self-supporting electrodes consisting of dual-phase spinel Li4Ti5O12/anatase TiO2 nanosheets and 3D reduced graphene oxide (RGO) aerogel for lithium ion batteries (LIBs) and sodium ion batteries (SIBs) were prepared via facile hetero-assembly, freeze-drying, mechanical compression and annealing. The 3D RGO aerogel acts as both conductive medium and self-supporting scaffold for anchored dual-phase nanosheets. The synergistic effect between the dual-phase nanosheets and the 3D highly conductive interconnected RGO network not only guarantees rapid reaction kinetics and strong structural stability of the electrodes during ion insertion/extraction, but also provides abundant accommodation for additional interfacial Li/Na storage. The self-supporting electrodes have desirable electrochemical performance such as a high reversible capacity (∼200/180 mA h g−1 for LIB/SIB at 1C/0.1C), good rate capability (∼141/117 mA h g−1 for LIB/SIB at 30C/10C) and superior cyclic performance (∼154/101 mA h g−1 for LIB/SIB at 10C/6C after 1000/700 cycles). Our results have great potential in constructing self-supporting RGO electrodes embedded with anode materials for LIB and SIB applications.
1. Introduction
With ever-increasing demands in automobile and stationary energy storage applications, such as electric vehicles and renewable energy integration, considerable efforts have been devoted to developing rechargeable cost-effective energy storage devices.1–6 Among various rechargeable energy storage systems, lithium ion batteries (LIBs) and sodium ion batteries (SIBs) are two typical energy storage devices due to their high energy densities, non-memory effect, environmentally friendly technology and long cycle life.7,8 In particular, SIBs have great potential in smart-grid applications owing to highly abundant natural resources and the low-cost of sodium. Nevertheless, further application extensions of current LIBs and SIBs are impeded by the facts of relatively low specific capacity, inferior rate capability and poor cycle stability. Among the challenges associated with electrolytes,9,10 separators11,12 and electrodes8,13 for these battery technologies, the anode material is one of the most important factor for both LIBs and SIBs in electrochemical properties and safety.14–18 Therefore, effective material designs and preparation of reliable anodes with excellent Li/Na storage ability are critical to solve the bottleneck faced by LIBs and SIBs for their large-scale applications.
Spinel Li4Ti5O12 (LTO) has been extensively investigated as a promising anode material for LIBs owing to its unique characteristics, such as “zero-strain” feature in the lattice on charging/discharging, flat operating plateau at a high potential of ∼1.55 V vs. Li/Li+ with a theoretical capacity of 175 mA h g−1 and excellent environmental benignity.19–22 In recent years, this anode material has also been expanded to utilization in SIBs with a work potential of ∼0.91 V vs. Na/Na+.23–25 The higher work voltages for LIBs and SIBs endow it with enough safety compared with traditional carbon anodes. However, its practical applications in high-performance LIBs and SIBs are hampered because of the unsatisfactory high-rate capability due to kinetic issues associated with poor electrical conductivity (ca. 10−13 S cm−1) and slow ion diffusion coefficient (ca. 10−9–10−13 cm2 s−1 for Li+ and 10−16 cm2 s−1 for Na+). To solve these problems, many effective strategies have been proposed and executed, such as reducing particle size,7,17,26,27 aliovalent ion doping,28–30 surface modification by secondary materials (for example, conductive carbon and carbon-free inorganic nanocrystals),31–34 and particularly their combinations.21,30,35–37 Among them, the integration of material engineering of nanostructured LTO and surface modification by TiO2 in nanoscale is an effective method to augment electrochemical properties of the LTO-based anodes. In such composites, nanostructure can shorten ion diffusion distance, while introducing TiO2 particles will result in abundant grain boundaries to provide additional accommodation for Li/Na ions to store in the interfacial area. Wang et al. reported LTO nanosheets coated with rutile TiO2 through a one-pot hydrothermal synthesis process, demonstrating higher discharge capacity and superior rate capability as LIBs anode compared to pure LTO nanosheets.7 Wu et al., reported ultrathin dual phase nanosheets consisting of alternating LTO and rutile TiO 2 lamellas through a facile and scalable hydrothermal method. The alternating nanoscaled lamella structure facilitates the transfer of both Li ions and electrons into spinel LTO, giving rise to an excellent cyclability with a capacity retention of 93.1% even after 500 cycles at 50C and fast rate performance with stable discharge capacities of 131.4 mA h g−1 at 50C.38 Li et al., prepared three-phase LTO/TiO2 (anatase)/TiO2 (rutile) nanoplates with high capacity of about 139 A h g−1 after 1000 cycles when fully charged at high current density of 4000 mA g−1.39 Nevertheless, the above-mentioned LTO/TiO2 materials require the use of electrically insulating polymeric binder, conductive additive and metallic current collector during slurry-coating process. The traditional electrode preparation process not only increases the total cost and weight of the LIBs and SIBs, but also results in adverse effect on effective gravimetric capacity. Thus, in order to spur the applications of LTO/TiO2 based anodes in LIBs and SIBs, designing a self-supporting electrode is highly desired. Due to its high surface area, lightweight property and highly interconnected electron pathways, three dimensional (3D) reduced graphene oxide (RGO) aerogel has been proposed as a suitable scaffolding material to anchor electroactive materials to prepare self-supporting electrodes.40,41 Despite recent investigation on dual-phase LTO/TiO2 materials and LTO/RGO composites, there have been few investigations on the fabrication and Li/Na storage performance of self-supporting electrode consisted of dual-phase LTO/TiO2 nanostructures and 3D RGO aerogel.
In this work, dual-phase spinel LTO/anatase TiO2 nanosheets anchored 3D RGO aerogel scaffold (designated as LTO-AT/RGO) was prepared as self-supporting LIB and SIB electrodes. The RGO aerogel paper acts as both conductive medium and self-supporting scaffold for dual-phase LTO-AT nanosheets. The synergistic effect between dual-phase LTO-AT nanosheets and 3D highly conductive interconnected RGO network not only guarantees rapid reaction kinetics and strong structural stability of the electrodes during ion insertion/extraction, but also provides abundant accommodations for additional interfacial Li/Na storage. The self-supporting LTO-AT/RGO electrodes have desirable electrochemical performance such as a high reversible capacity (∼200/180 mA h g−1 for LIB/SIB at 1C/0.1C), good rate capability (∼141/117 mA h g−1 for LIB/SIB at 30C/10C) and superior cyclic performance (∼154/101 mA h g−1 for LIB/SIB at 10C/6C after 1000/700 cycles). Our results provide an effective method of constructing self-supporting RGO electrodes embedded with anode materials for LIB and SIB application.
2. Experimental details
2.1 Synthesis of LTO nanosheets
LTO nanosheets were fabricated by a simple way as before our reported.31 1.7 ml (5 mM) of tetrabutyl titanate, 0.03 g of GdCl3·6H2O, and 0.189 g of LiOH·H2O were thoroughly mixed in 20 ml of ethanol at room temperature. The solution was mixed completely with a magnetic stirrer in a closed container for 24 h and then, 25 ml of deionized water were added to the container. After stirring for 0.5 h, the solution was transferred to a 50 ml Teflon-lined stainless autoclave and placed in an oven at 180 °C for 36 h. The white powder on the bottom of the reactor was collected, washed with ethanol 3 times, and dried at 80 °C for 6 h. Finally, the white powder was heated at 700 °C for 6 h in a horizontal tube furnace in air to obtain the ultrathin LTO nanosheets.
2.2 Fabrication of the self-supporting LTO-AT/RGO aerogel
The self-supporting LTO-AT/RGO composite were fabricated via a hetero-assemble process, freeze-drying, mechanical compression and annealing. In a typical run, 140 mg LTO nanosheets were dispersed in the solution with 0.2 M NH4HCO3 under magnetic stirring for one hour to form Solution A. 60 mg GO was dissolved in deionized water to produce Solution B. Solution A was added dropwise to Solution B under fierce stirring. Flocculent GO/LTO hydrogel with uniform color and a solid-like rheological behavior was quickly produced. Subsequent washing and freeze-drying afford the formation of LTO-AT/RGO intermediate aerogel. Lastly, after mechanical compression and annealing at 600 °C, the self-supporting LTO-AT/RGO electrodes were obtained.
2.3 Materials characterization
Powder X-ray diffraction (XRD) with a copper Kα radiation source (λ = 0.154 nm) was performed to identify the crystal structures of RGO and self-supporting LTO-AT/RGO. The scanning electron microscopy (SEM, JEOL, JSM-6490) and transmission electron microscopy (TEM, JEOL 2100) were used to characterize morphologies and microstructures of the samples. X-ray photoelectron spectroscopy (XPS) with an Al Kα source (Kratos Analytical Ltd., UK) was carried out and calibrated by using C 1s peak with the binding energy of 284.8 eV. Nitrogen adsorption–desorption isotherm measurements of the samples were conducted at 77 K (Quantachrome NOVA 4000e). Raman spectra were obtained on a Renishaw inVia system with a laser excitation source operating at λ = 532 nm. Thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed using a TGA 2050 thermogravimetric analyzer. Fourier transform infrared (FTIR) spectra were measured on a FTS-3000 Fourier transform infrared spectrophotometer.
2.4 Electrochemical characterization
The electrochemical tests were performed on two-electrode CR2032-type coin cells. The self-supporting LTO-AT/RGO composites were directly used as the working electrodes in the lithium and sodium cells. The assembly of coin cells were conducted in an glove box with filled argon using Na/Li foils as counter electrodes, 1 M NaClO4 in ethylene carbonate (EC)/propylene carbonate (PC) with volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1 M LiPF6 in a 1:1 (v/v) mixture of ethylene carbonate/dimethyl carbonate as electrolyte, the glass fibre (Whatman GF/D) and Celgard 2400 Polypropylene as separators for the sodium and lithium cells, respectively. Cyclic voltammetry (CV) was measured on a CHI660E electrochemical workstation. Galvanostatic charge–discharge (GCD) measurements were carried out at various current densities on multi-channel battery test system (NEWARE BTS-610). Electrochemical impedance spectroscopy (EIS) measurements were conducted on an electrochemical workstation (CHI660D) with the frequency range from 0.01 to 100 kHz.
:1 and 1 M LiPF6 in a 1:1 (v/v) mixture of ethylene carbonate/dimethyl carbonate as electrolyte, the glass fibre (Whatman GF/D) and Celgard 2400 Polypropylene as separators for the sodium and lithium cells, respectively. Cyclic voltammetry (CV) was measured on a CHI660E electrochemical workstation. Galvanostatic charge–discharge (GCD) measurements were carried out at various current densities on multi-channel battery test system (NEWARE BTS-610). Electrochemical impedance spectroscopy (EIS) measurements were conducted on an electrochemical workstation (CHI660D) with the frequency range from 0.01 to 100 kHz.
3. Results and discussion
3.1 Material preparation and characterization
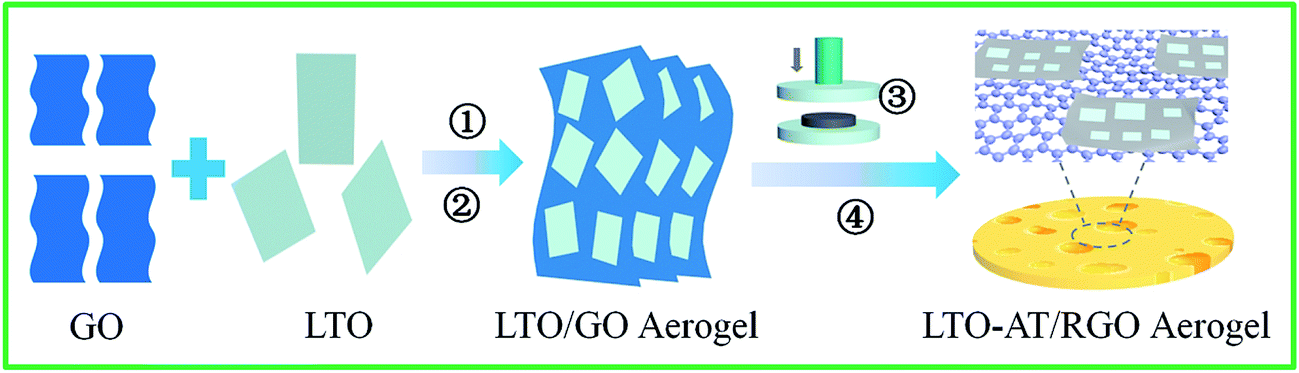
Fig. 1 illustrates the overall fabrication process of self-supporting LTO-AT/RGO electrodes, including hetero-assembly of GO and LTO nanosheets, freeze-drying, mechanical compression and annealing. In our previous studies,32 LTO nanosheets prepared by hydrothermal method followed by calcinations have abundant charged ions on the surface in the solution with high ionic strength containing 0.2 M NH4HCO3, and a fine colloid of the LTO nanosheets is readily obtained. When the GO nanosheets with negative charges and the LTO nanosheets in the solution with high ionic strength are mixed, hetero-assembly between GO and LTO nanosheets accompanying spontaneous co-precipitation occurs. As a result, flocculent GO/LTO hydrogel with uniform color and a solid-like rheological behavior was quickly produced. Subsequent washing and freeze-drying afford the formation of LTO-AT/RGO intermediate aerogel, which can be plastically deformed via mechanical compression to form self-supporting intermediate. During this process, a certain amount of Li in LTO nanosheets was washed away, depending on the time of water washing. The deficiency of Li plays a key role in the formation of dual-phase LTO-AT nanosheets in the following thermal treatment.7 After annealing at 600 °C, the self-supporting LTO-AT/RGO electrodes are obtained accompanying reduction of GO to RGO and complete decomposition of the remaining NH4HCO3. As shown in Fig. 2a and b, the self-supporting LTO-AT/RGO electrodes are mechanically robust with excellent flexibility (even sustaining 180° bending). The excellent mechanical properties suggest direct use for LIB and SIB electrodes without any binders.
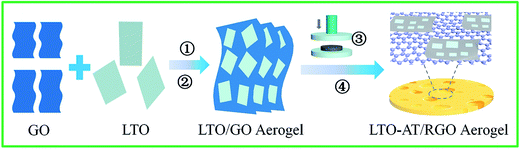 |
| | Fig. 1 Schematic of the fabrication procedures of monolithic LTO-AT/RGO aerogel. Herein, ①, ②, ③ and ④ represent hetero-assembly, washing and freeze-drying, mechanical compression and annealing, respectively. | |
 |
| | Fig. 2 (a and b) Optical image of the self-supporting LTO-AT/RGO electrodes; (c and d) cross-sectional SEM image of the self-supporting LTO-AT/RGO electrodes. | |
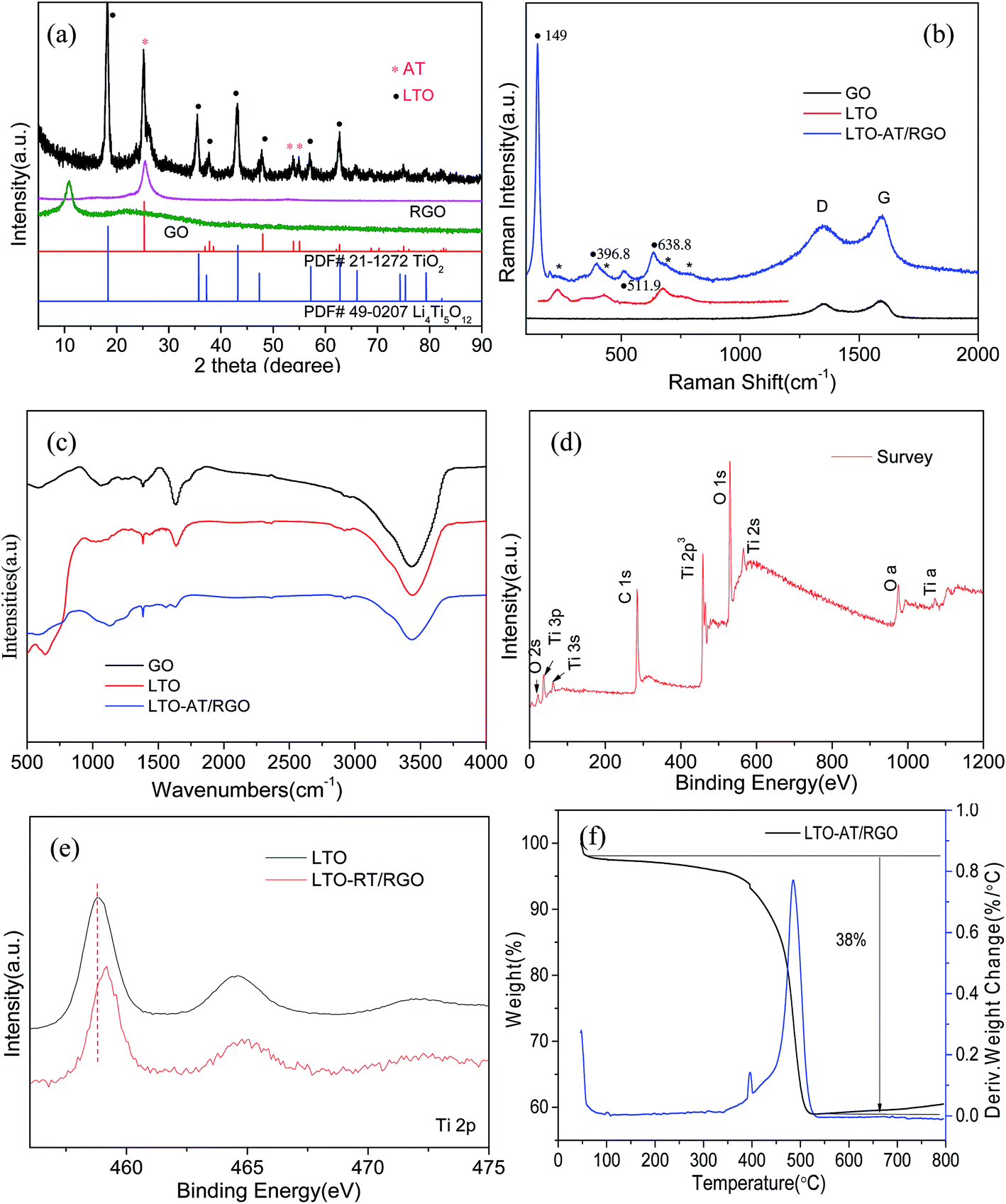
Fig. 2c depicts the cross-sectional SEM image of the self-supporting LTO-AT/RGO electrodes with a thickness of about 25 μm, disclosing a layered feature and good thickness uniformity. The high-magnification SEM image (Fig. 2d) reveals that the anchored LTO-AT nanosheets are well distributed within interconnected RGO networks. The results confirm that during hetero-assembly and spontaneous co-precipitation of LTO nanosheets and GO, the former with positive ions prefer to settle at the functional group sites on the surface and at the periphery of the latter. Fig. 3a shows the TEM image of LTO nanosheets with clear surface and large size. Meanwhile, the TEM images of LTO-AT/RGO electrode are shown in Fig. 3b and c, in which the LTO nanosheets with small nanosheets on the surface are tightly anchored on the RGO sheets. Further HRTEM observation of the marked region in Fig. 3c (see Fig. 3d) indicates that characteristic (101) lattice plane of anatase TiO2 with a lattice spacing of 0.35 nm and (111) one of spinel LTO with a spacing of 0.48 nm can be distinguished. The results reveal that these nanosheets are composed of spinel LTO and anatase TiO2. The crystal structure of the LTO-AT/RGO electrodes is further characterized by XRD. As shown in Fig. 4a, besides LTO (JCPDS no. 49-0207),42 anatase TiO2 (JCPDS card no. 21-1272)33 was also detected in the sample, in which the most intensity of the XRD peaks of LTO at 18.4 degrees in sample may be ascribed to the preferential orientation of the (111) planes. While the samples have been obtained in different time of the water washing and characterized by XRD. As shown in Fig. S1,† the ratio of the most intensity peak of the LTO (111) vs. AT (101) is 1.12, 1.33, 1.4 and 1.6 at 48 h, 24 h, 12 h and 6 h, respectively. These results indicated that the change of ratio is not obvious with the time of washing. In general, the intensity of XRD peaks from one crystalline phase in a multiphase composite depends on the weight fraction of the corresponding phase in the composite. Thus, the weight fraction of anatase TiO2 can be calculated by measuring the intensities of (101) peak of anatase TiO2 and (111) peak of LTO.38 Accordingly, the weight fraction of the anatase TiO2 in the prepared LTO-AT/RGO aerogel paper was measured to be around 0.4. No XRD peak located at 12.4° from GO is observed, suggesting that GO has been reduced to RGO via thermal treatment at 600 °C. The should peak at 26° ascribed to characteristic (002) interlayer diffraction of few-layer RGO nanosheets is weak, suggesting that the RGO sheets are effectively separated without serious self-stacking by the anchored LTO-AT nanosheets.
 |
| | Fig. 3 (a) TEM image of the LTO nanosheets; (b) and (c) TEM images at low and high magnification, respectively; (d) HRTEM image acquired from the region marked by dot circle in (c). | |
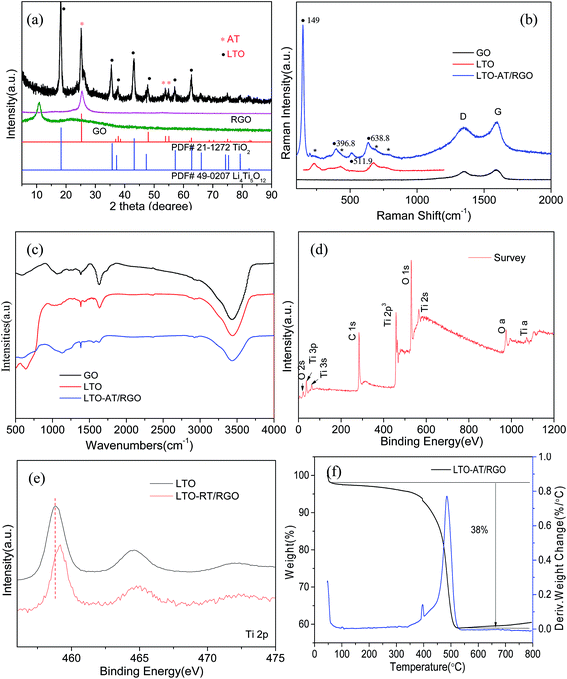 |
| | Fig. 4 (a) XRD patterns of the self-supporting LTO-AT/RGO aerogel and referenced samples; (b and c) Raman spectra and FTIR spectra of the self-supporting LTO-AT/RGO aerogel and referenced samples; (d) survey XPS spectrum of self-supporting LTO-AT/RGO aerogel; (e) high-resolution Ti XPS spectra of the self-supporting LTO-AT/RGO aerogel and referenced LTO nanosheets; (f) TGA/DSC data of the self-supporting LTO-AT/RGO aerogel. | |
Fig. 4b displays the Raman spectra of the as-prepared LTO-AT/RGO electrodes and reference samples. The Raman bands at 230, 432 and 687 cm−1 are attributed to characteristic vibration modes in cubic spinel LTO, associated with the vibration (F2g) of lithium which is octahedrally-coordinated by oxygen, stretching vibration of Li–O ionic bonds located in the LiO4 tetrahedra (Eg) and stretching vibration of Ti–O covalent bonding in TiO6 octahedra, respectively. The Raman bands located at 149, 396.8, 511.9 and 638.8 cm−1 can be assigned to the Eg, B1g, A1g and Eg modes of anatase TiO2,36 respectively. Besides these Raman modes from spinel LTO and anatase phase TiO2, two strong peaks, denoted as the disorder peak (D, centered at 1350 cm−1) and graphitic peak (G, at 1594 cm−1) are also observed from as-prepared LTO-AT/RGO electrodes, suggesting that LTO-AT nanosheets are highly dispersed into surrounding RGO matrix. Compared with peak intensity ratio for the D and G bands (ID/IG = 1.03) of the referenced RGO sample, the ratio of the LTO-AT/RGO electrodes increase to 1.09, indicating strong electronic coupling between LTO-AT nanosheets and RGO, leading to effective electron transport of the overall electrode during electrochemical processes. Fig. 4c shows FTIR spectra of the as-prepared LTO-AT/RGO electrodes and reference samples. The peaks at 450–900 cm−1 can be attributed to the Ti–O–Ti stretching vibration modes in spinel LTO and anatase TiO2. The intensity of the peak at 1626 cm−1 decreases notably and a new peak at 1564 cm−1 appears reflecting the skeletal vibration of RGO sheets, indicates effective elimination of carboxyl and epoxy functional groups and the transformation of GO to RGO.43 The Raman and FTIR spectra confirm that the LTO-AT/RGO electrodes via above-mentioned fabrication contains RGO matrix with highly dispersed LTO-AT nanosheets, which is consistent with the XRD results. The surface chemical composition and electronic states of the LTO-AT/RGO electrodes are determined by XPS. The survey XPS spectrum and high-resolution Li 1s, Ti 2p, O 1s, C 1s and N 1s spectra (see Fig. 4d) reveal the presence of Li, Ti, O, C and N. Fig. 4e shows high-resolution XPS spectrum of Ti 2p, revealing two characteristic peaks at 458.7 and 464.5 eV assigned to Ti 2p3/2 and 2p1/2, respectively, from titanium in the IV oxidation state. Compared with those from the referenced LTO nanosheets, the binding energy of Ti 2p from the LTO-AT/RGO electrode shifts toward high-energy direction, suggesting strong electron interaction between the LTO-AT nanosheets and surrounding RGO matrix. In addition, TGA/DSC results (see Fig. 4f) shows that the LTO-AT/RGO electrode is composed of ∼62 wt% LTO-AT nanosheets.
3.2 Electrochemical performance in sodium ion batteries
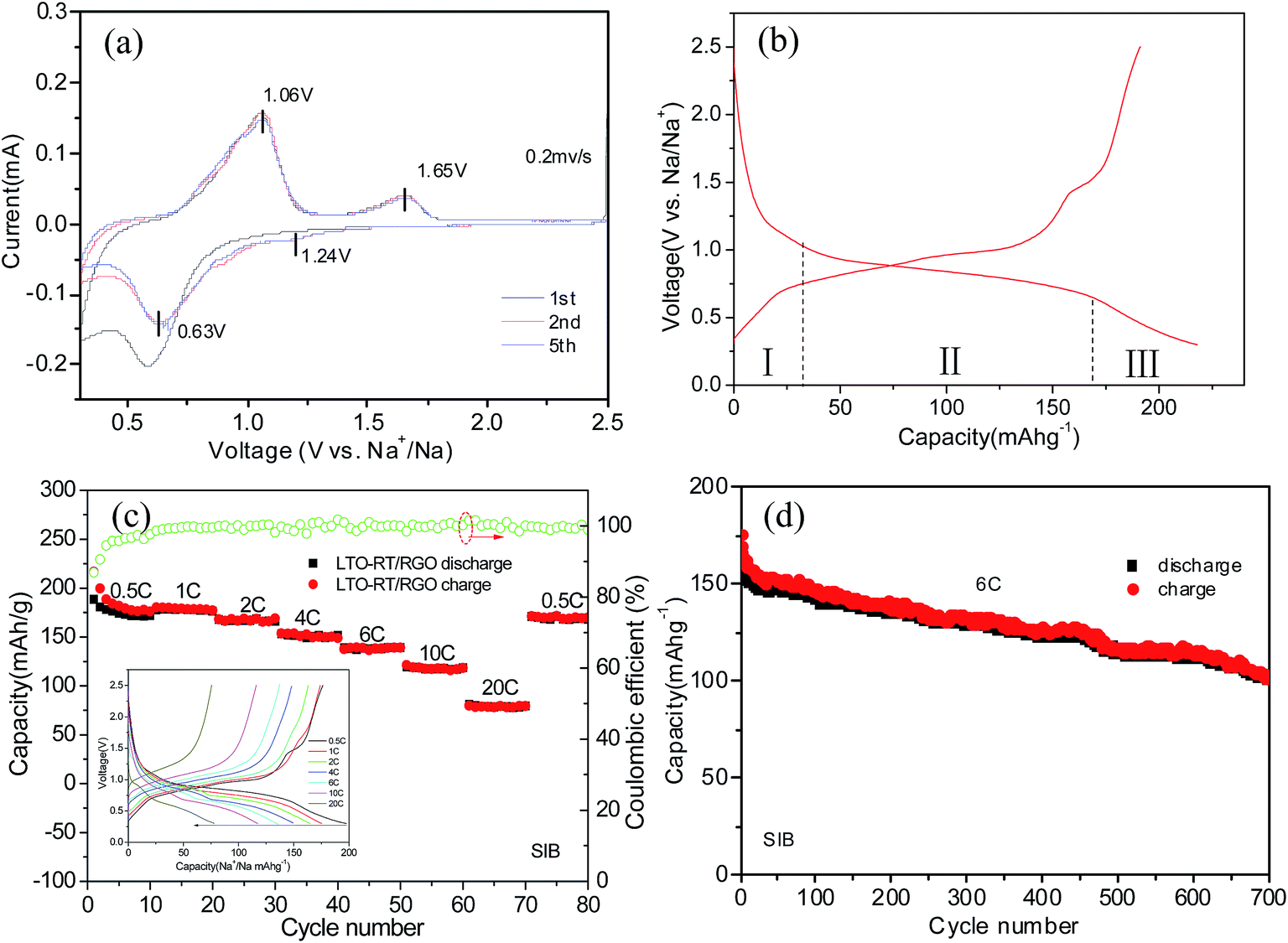
The Na storage ability of the LTO-AT/RGO electrode is firstly investigated. Fig. 5a shows the CV curve at a scanning rate of 0.2 mV s−1 in the potential range between 2.5 and 0.3 V vs. Na+/Na, demonstrating the CV trace in the first cycle is different from those of the subsequent ones, suggesting possible existence of structural deformation and reconstruction induced by electrochemical atomic rearrangements around the LTO-AT nanosheets due to the formation of SEI layers. Previous studies reveals that the redox peaks assigned to Na+ insertion/extraction in anatase TiO2 were located at about 0.76 and 0.83 V, which overlap those representing Na+ insertion/extraction in the LTO crystal lattice according to the following three-phase separation mechanism: 2Li4Ti5O12 + 6Na+ + 6e → Li7Ti5O12 + LiNa6Ti5O12.34,44 Therefore, the couple of redox peaks located at ∼0.63 V (cathodic peak) and ∼1.06 V (anodic peak) vs. Na+/Na observed in the subsequent cycles include the contribution from reversible insertion/extraction of sodium in both LTO and anatase TiO2. The redox peak positions are almost invariable during successive scans, implying superior cycling stability of the LTO-AT/RGO electrode. In addition, another couple of weak redox peaks at 1.65 V (anodic peak) and 1.24 V (cathodic peak) can be observed. Since they cannot be attributed to Na+ insertion/extraction into anatase TiO2 and LTO, this pair of redox peaks likely originates from Na+ storage at interfaces of LTO/anatase TiO2, LTO/RGO and anatase TiO2/RGO. Fig. 5b shows the typical discharging/charging profiles of the LTO-AT/RGO electrode at a current rate of 0.5C, demonstrating initial discharge and charge capacities are 217 and 190 mA h g−1, respectively. The Na+ insertion process includes three domains with a sloping region (I) from open circuit voltage to ∼1.02 V, plateau (II) about 0.75 V, and another sloping region (III) from ∼0.65 to 0.3 V. Domains I and II represents Na+ insertion into the LTO and anatase TiO2, while Domain III originates from Na+ storage at the LTO/anatase TiO2, LTO/RGO and anatase TiO2/RGO interfaces.
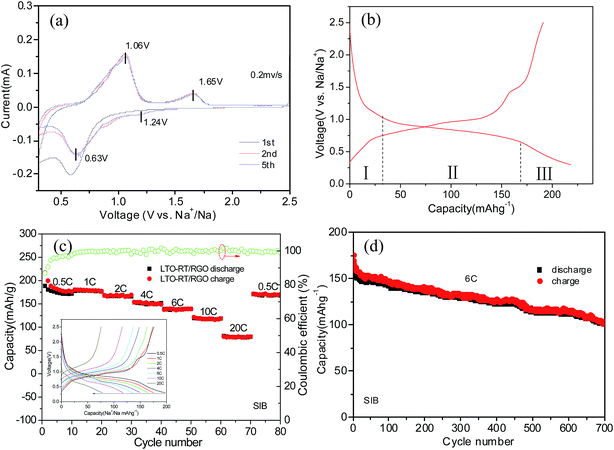 |
| | Fig. 5 (a) Typical CVs of the self-supporting LTO-AT/RGO electrode for SIBs at scan rate 0.2 mV s−1; (b) typical charge–discharge profiles of the self-supporting LTO-AT/RGO electrode at 0.5C; (c) rate performance, coulombic efficient and charge–discharge profiles (see inset) of the self-supporting LTO-AT/RGO electrode; (d) cycling performance of the self-supporting LTO-AT/RGO electrode for SIBs at 6C. | |
The inset in Fig. 5c is the charging–discharging profiles at different current densities, demonstrating apparent decrease in the voltage plateau region while no obvious change in the sloping region with increasing current density. The results indicate that at high rates, Na+intercalation into the LTO and anatase TiO2 becomes difficult, whereas interfacial Na storage is still sufficient. The LTO-AT/RGO electrodes deliver reversible capacities of 184, 178, 167, 150, 139 and 117 mA h g−1 at rates of 0.5, 1, 2, 4, 6 and 10C respectively. Even at a high rate of 20C, a reversible capacity of 77 mA h g−1 is maintained. When the current rate is recovered to 0.5C after cycling at various rates, the capacity can recover to 171 mA h g−1. These results demonstrate superior rate capability of the LTO-AT/RGO electrode. Furthermore, the cycling durability is tested at a large current density of 6C. As shown in Fig. 4d, after a quick decay in the initial 10 cycles, the LTO-AT/RGO electrode delivers a high capacity of 158 mA h g−1 with a coulombic efficiency approaching 100%. A capacity of 101 mA h g−1 is maintained after 700 cycles with a capacity loss of 0.067 per cycle, confirming excellent long-term cycling sustainability of the LTO-AT/RGO electrode.
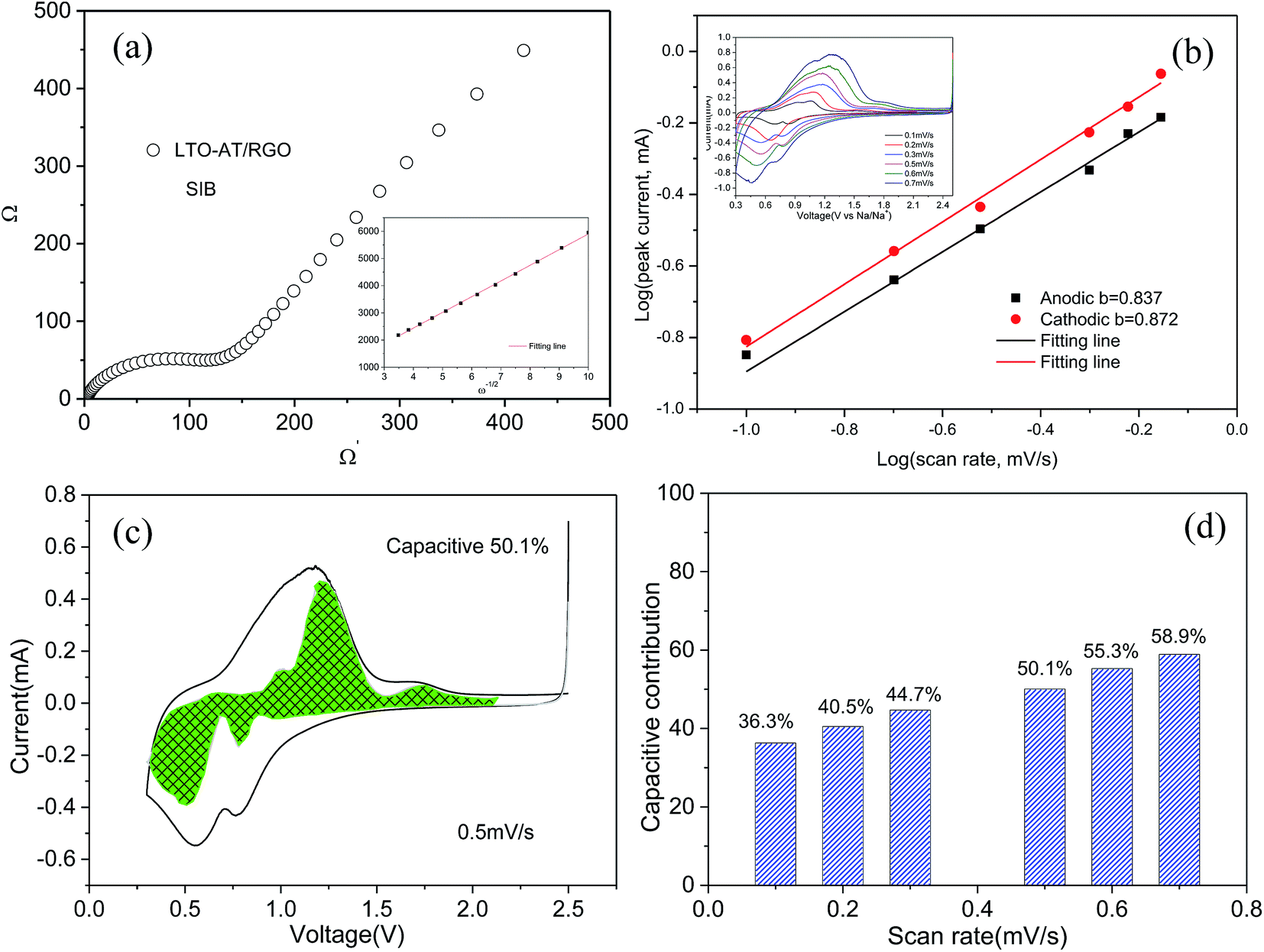
To gain insight into the superior electrochemical performance of the LTO-AT/RGO electrode, EIS measurements were performed after the rate capability test. The obtained Nyquist plots (see Fig. 6a) include mainly a semicircle related to charge-transfer impedance (Rct) on electrode–electrolyte interface in high frequency, and an incline straight line attributed to Warburg impedance (Zw) in low frequency region. Using the inset equivalent circuit model, the calculated Rct value for the LTO-AT/RGO electrode is 88.35 (see Table 1). The Na+ chemical diffusion coefficient (DNa) is also calculated according to the following equations:
| |
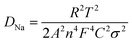 | (1) |
| | |
Z = Rs + Rct + σω−1/2
| (2) |
where
R,
T,
A,
n,
F,
C and
σ are the gas constant, absolute temperature, surface area of the electrode, number of electrons transferred in the half reaction of the redox couple, Faraday's constant, Na
+ concentration, and Warburg factor, respectively. The value of
σ can be obtained (
Table 1) from the slope of the lines between
Z′ and
ω−1/2 (see the inset of
Fig. 6a). The calculated DNa value of the LTO-AT/RGO electrode is 7.5 × 10
−15 (see
Table 1). The small Rct and large DNa in the LTO-AT/RGO electrode indicate effective charge transfer process at the electrode/electrolyte interface and rapid electrochemical kinetics due to increased grain boundary density which had a lower energy barrier for Na diffusion and in fact facilitated Na ion diffusion. Furthermore, we performed kinetic analysis based on CV results that has been proven as a powerful technique for studying electrochemical kinetics of electrode materials toward Na
+. The inset of
Fig. 6b shows the CV curves of the LTO-AT/RGO electrode at different scan rates. In general, the currents (
i) responding to the sweeping rates (
v) in the CV curves follow the equation of
i =
avb, where
a and
b are adjustable values.
45,46 If
b = 1, it suggests that the total charge (
Q) originates from the capacitive process (interfacial Na storage process) and if
b = 0.5, it is totally diffusion-controlled (Na
+ insertion/extraction process).
Fig. 5b shows the log(
v)–log(
i) plots for the LTO-AT/RGO electrode and the fitted
b values for the cathodic and anodic peak currents are 0.872 and 0.837, respectively, implying that that the charge (thus the capacity) comes from two contributions. According to the equation of
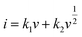
,
47 the capacitive (interfacial) contribution can be separated, where
k1v and
k2v1/2 represent the contributions from the capacitive (interfacial Na storage process) charge and diffusion-controlled (Na
+ insertion/extraction process) one, respectively. For example, as shown in
Fig. 5c, the interfacial capacitive contribution at 0.5 mV s
−1 is determined to be 50.1% in the LTO-AT/RGO electrode. With the scanning rates being increased from 0.1 to 0.7 mV s
−1 (see
Fig. 6d), the interfacial capacitive contributions increase from 36.3% to 58.9%. The results indicate that the kinetics of Na
+ intercalation process becomes limited at high rates but the kinetics of interfacial Na storage is still sufficient in the LTO-AT/RGO electrode, which is consistent with the aforementioned sodium storage performance observed at various current densities. The CV kinetic analysis disclose that the intercalation-based and interfacial Na storage behaviors take effect simultaneously, and particularly the latter keeps highly efficient at high rates in the LTO-AT/RGO electrode, which endows it with excellent Na storage performance.
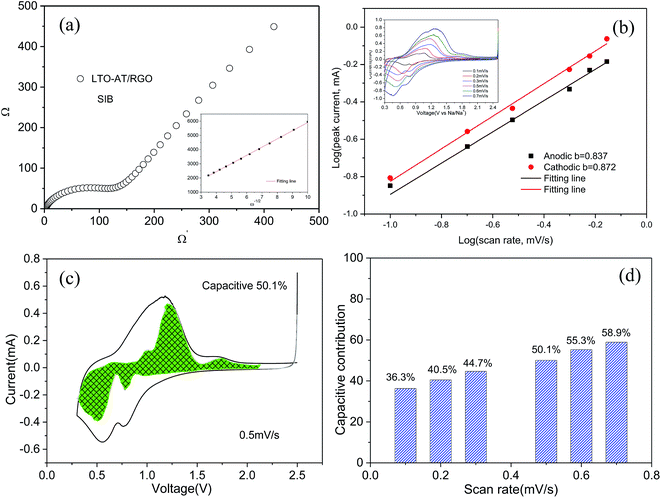 |
| | Fig. 6 (a) EIS spectra of self-supporting LTO-AT/RGO electrode and referenced one for SIBs; (b) CV curves at various scan rates of self-supporting LTO-AT/RGO electrode for SIBs (see inset) and calculating the b-value according to log(i)–log(v) plots; (c) typical CV curve with the contribution from capacitive effect shown by the highlighted region at 0.5 mV s−1; (d) comparison of the capacitive charge contributions at different scan rates. | |
Table 1 The calculated Rct and Rs values for the LTO-AT/RGO electrodes according to EIS results in Fig. 5 and 6
| Compound |
Half-cells |
Rs (Ω) |
Rct (Ω) |
D (cm2 s−1) |
| LTO-AT/RGO |
Lithium-ion battery |
3.21 |
75.85 |
2.1 × 10−12 |
| Sodium-ion battery |
6.51 |
88.35 |
7.5 × 10−15 |
3.3 Electrochemical performance in lithium ion batteries
Considering great advantage and application of LTO-based anodes in high-performance LIBs, we also examine Li storage performance of the LTO-AT/RGO electrode. Fig. 7a shows the CV curves of initial five cycles for as-prepared electrodes between 1 and 2.5 V (vs. Li/Li+) with a scan rate of 0.2 mV s−1.
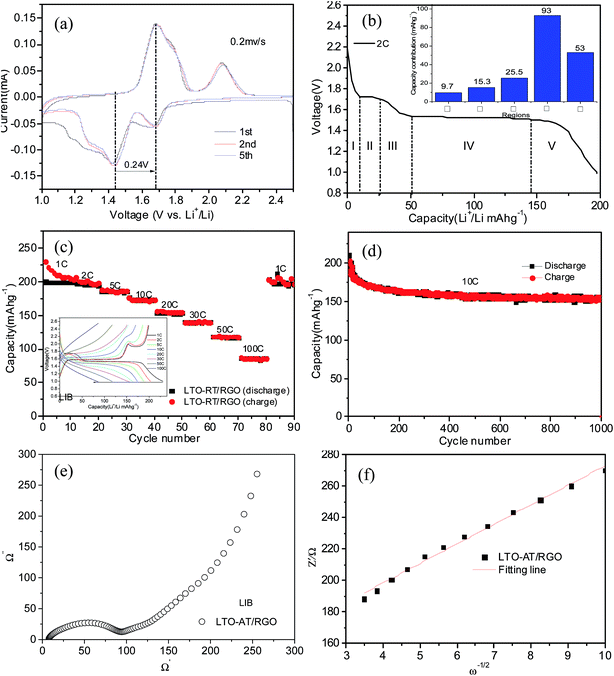 |
| | Fig. 7 (a) CVs of the self-supporting LTO-AT/RGO electrode for LIBs at scan rate of 0.2 mV s−1; (b) typical discharge profiles of the self-supporting LTO-AT/RGO electrode at 2C; (c) rate capacity and charge–discharge curves (see inset) of the LTO-AT/RGO electrode for LIBs; (d) cycling performance of the self-supporting LTO-AT/RGO electrode at 10C; (e) Nyquist plots of the self-supporting LTO-AT/RGO electrode for LIBs; (f) corresponding Z′ plots against ω−1/2 plots against at the low frequency region (10–0.1 Hz). | |
Two pairs of oxidation and reduction peaks at 2.08 V/1.68 V and 1.68 V/1.44 V, which are ascribed to anatase TiO2 and spinel LTO, respectively, indicating the feature of a dual phase composite material. The redox peak positions are unchanged during successive scans demonstrating excellent reversibility of the LTO-AT/RGO electrode. The initial discharge profiles for the LTO-AT/RGO electrode with a current rate of 2C are shown in Fig. 7b. The flat discharge plateau at about 1.55 V due to Li ions being extracted from the spinel LTO and the voltage one at around 1.7 V verified the lithium ion insertion of anatase TiO2 can be observed, which are in agreement with the CV results. Similarly, the Li+ insertion process can be divided into five domains (see inset of Fig. 7b) with a sloping region (I) from open circuit voltage to ∼1.7 V, plateau (II) about 1.7 V, a sloping region (III) from ∼1.7 to 1.55 V, plateau (IV) about 1.55 V and another sloping region (V) from 1.55 to 1 V. Domains I, III and IV originate from Li insertion into anatase TiO2 and spinel LTO, while domain II and V associates with Na storage at the interface of LTO/anatase TiO2, LTO/RGO and anatase TiO2/RGO. The charge–discharge profiles acquired at different current densities from 1 to 100C (see inset of Fig. 7c) of LTO-AT/RGO electrode show similar feature that the voltage plateaus decrease apparently while the sloping region is almost unchanged at high current densities, revealing that interfacial Li storage is more sufficient than that associated with Li+ intercalation into LTO and anatase TiO2. The corresponding rate performance (see Fig. 7c) shows high and stable capacity. At rates of 1, 2, 5, 10, 20, 30 and 50C, the LTO-AT/RGO electrode delivers discharge capacities of 208, 197, 186, 173, 154, 138 and 115 mA h g−1, respectively. Even at 100C, a reversible capacity of 86 mA h g−1 is maintained implying rapid charging and discharging within a few minutes for high energy and power densities. When the rate is returned to 1C after high-rate cycling, a high capacity of 202 mA h g−1 in subsequent cycling can be recovered, suggesting good structural integrity of the prepared electrode. Fig. 7d displays the long-term cycling performance of the LTO-AT/RGO electrode at a high current density of 10C, demonstrating a stable capacity of 173 mA h g−1 with the coulombic efficiency approaching 100% after 50 cycles. A capacity of 154 mA h g−1 is still retained after 1000 cycles with a capacity loss of only 19 mA h g−1. The Fig. S3a and b† show morphology of the as-cycled LTO-AT/RGO electrode with no apparent difference comparing the before-cycled SEM images and RGO sheets are in good contact with LTO-AT nanosheets. The crystalline structure is also tested via Raman scattering. As shown in Fig. S4,† besides the strong Raman peaks from RGO, the characteristic peaks at 230, 432 and 687 cm−1 are ascribed to characteristic vibration modes in cubic spinel LTO and 149, 396.8. 511.9 and 638.8 cm−1 can be assigned to the Eg, B1g, A1g and Eg modes of anatase TiO2. The results show that the LTO-AT/RGO electrode is robust and can sustain long-term cycling. In a word, the high-rate and long-term cycling performance of the LTO-AT/RGO electrode are excellent, compared to the results reported in LTO-based LIB anodes using traditional slurry-coating process.21,23,25,26,48 In addition, EIS measurements (see Fig. 6e and f) with the simulated results (see Table 1) reveal that the LTO-AT/RGO electrode has low charge-transfer resistance and large Li ion diffusion coefficient. The CV curves of the LTO-AT/RGO electrode material as lithium-ion battery at different scan rates have been also studied (Fig. S2†). Although, compared LTO-AT/RGO electrode in sodium ion battery, the capacitive contribution and b value are smaller in lithium ion battery, which may be attributed to the anatase TiO2 with understanding diffusion-controlled processes as electrode in lithium ion batteries and larger interfacial contribution in sodium ion batteries.49 These results indicate that the kinetics for the Li+ intercalation process is limited at high rates, whereas the kinetics for the interfacial Li storage process is still sufficient. In brief, the results demonstrate that the LTO-AT/RGO anode is suitable for high-performance LIBs.
4. Conclusion
Self-supporting LTO-AT/RGO electrode was successfully prepared via a facile hetero-assembly, freeze-drying, mechanical compression and annealing. Such composite was directly utilized as anodes for LIBs and SIBs without any additives. The synergistic effect between dual-phase LTO-AT nanosheets and 3D highly conductive interconnected RGO network not only guarantee rapid reaction kinetics and strong structural stability of the electrodes during ion insertion/extraction, but also provide abundant accommodation sites for efficient interfacial Li/Na storage. For SIBs, the self-supporting LTO-AT/RGO anode have desirable electrochemical performance such as a high capacity (∼208 mA h g−1 at 0.1C), good rate capability (∼∼141/117 at 30C/10C) and superior cyclic performance (∼101 at 6C after 700 cycles). As anodes for LIBs, it demonstrates high-rate performance with a capacity of 86 mA h g−1 at 100C and long-term cycling performance with a capacity of 154 mA h g−1 after 1000 cycles at 10C. Our results are important for future research in additive-free electrode fabrication as the fabrication technique is facile and can be extended for anchoring other materials on 3D RGO scaffold material to constructing self-supporting anode for LIB and SIB applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Grants from National Natural Science Foundation of China (No. 11474242 and 51472209), the Research Foundation of Education Bureau of Hunan Province, China (Grant No. 15B237) and the Hunan Provincial Innovation Foundation for Graduate (No. CX2016B254).
References
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2012, 46, 1135–1143 CrossRef PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- X. Wang, X. Lu, B. Liu, D. Chen, Y. Tong and G. Shen, Adv. Mater., 2014, 26, 4763–4782 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2009, 22, 587–603 CrossRef.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- Y.-Q. Wang, L. Gu, Y.-G. Guo, H. Li, X.-Q. He, S. Tsukimoto, Y. Ikuhara and L.-J. Wan, J. Am. Chem. Soc., 2012, 134, 7874–7879 CrossRef CAS PubMed.
- D. Xie, X. Xia, Y. Zhong, Y. Wang, D. Wang, X. Wang and J. Tu, Adv. Energy Mater., 2017, 7, 1601804 CrossRef.
- Z. Zhang, L. Hu, H. Wu, W. Weng, M. Koh, P. C. Redfern, L. A. Curtiss and K. Amine, Energy Environ. Sci., 2013, 6, 1806–1810 CAS.
- J. Feng, Z. Zhang, L. Li, J. Yang, S. Xiong and Y. Qian, J. Power Sources, 2015, 284, 222–226 CrossRef CAS.
- S. Wei, S. Choudhury, J. Xu, P. Nath, Z. Tu and L. A. Archer, Adv. Mater., 2017, 29, 1605512 CrossRef PubMed.
- Y.-E. Miao, G.-N. Zhu, H. Hou, Y.-Y. Xia and T. Liu, J. Power Sources, 2013, 226, 82–86 CrossRef CAS.
- M. Moradi, Z. Li, J. Qi, W. Xing, K. Xiang, Y.-M. Chiang and A. M. Belcher, Nano Lett., 2015, 15, 2917–2921 CrossRef CAS PubMed.
- B. Qu, C. Ma, G. Ji, C. Xu, J. Xu, Y. S. Meng, T. Wang and J. Y. Lee, Adv. Mater., 2014, 26, 3854–3859 CrossRef CAS PubMed.
- J. Chang, X. Huang, G. Zhou, S. Cui, P. B. Hallac, J. Jiang, P. T. Hurley and J. Chen, Adv. Mater., 2014, 26, 758–764 CrossRef CAS PubMed.
- L. Gao, L. Wang, S. Dai, M. Cao, Z. Zhong, Y. Shen and M. Wang, J. Power Sources, 2017, 344, 223–232 CrossRef CAS.
- X. Jia, Y. Lu and F. Wei, Nano Res., 2016, 9, 230–239 CrossRef CAS.
- G. Hu, P. Chen, Z. Liu, Y. Cao, Z. Zhang, Z. Peng and D. Ke, J. Alloys Compd., 2017, 696, 177–184 CrossRef CAS.
- S. Chen, Y. Xin, Y. Zhou, Y. Ma, H. Zhou and L. Qi, Energy Environ. Sci., 2014, 7, 1924–1930 CAS.
- A. K. Haridas, C. S. Sharma and T. N. Rao, Small, 2015, 11, 290–294 CrossRef CAS PubMed.
- E. F. Rodriguez, F. Xia, D. Chen, A. F. Hollenkamp and R. A. Caruso, J. Mater. Chem. A, 2016, 4, 7772–7780 CAS.
- K. Qian, L. Tang, M. Wagemaker, Y. B. He, D. Liu, H. Li, R. Shi, B. Li and F. Kang, Adv. Sci., 2017, 1700205 CrossRef.
- L. Y. Yang, H. Z. Li, J. Liu, S. S. Tang, Y. K. Lu, S. Te Li, J. Min, N. Yan and M. Lei, J. Mater. Chem. A, 2015, 3, 24446–24452 CAS.
- C. Chen, H. Xu, T. Zhou, Z. Guo, L. Chen, M. Yan, L. Mai, P. Hu, S. Cheng and Y. Huang, Adv. Energy Mater., 2016, 6, 1600322 CrossRef.
- X. Feng, H. Zou, H. Xiang, X. Guo, T. Zhou, Y. Wu, W. Xu, P. Yan, C. Wang and J.-G. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 16718–16726 CAS.
- W. Liu, D. Shao, G. Luo, Q. Gao, G. Yan, J. He, D. Chen, X. Yu and Y. Fang, Electrochim. Acta, 2014, 133, 578–582 CrossRef CAS.
- H. Park, T. Song, H. Han and U. Paik, J. Power Sources, 2013, 244, 726–730 CrossRef CAS.
- J. G. Kim, M. S. Park, S. M. Hwang, Y. U. Heo, T. Liao, Z. Sun, J. H. Park, K. J. Kim, G. Jeong and Y. J. Kim, ChemSusChem, 2014, 7, 1451–1457 CrossRef CAS PubMed.
- T.-F. Yi, Y. Xie, L.-J. Jiang, J. Shu, C.-B. Yue, A.-N. Zhou and M.-F. Ye, RSC Adv., 2012, 2, 3541–3547 RSC.
- Y. Ma, B. Ding, G. Ji and J. Y. Lee, ACS Nano, 2013, 7, 10870–10878 CrossRef CAS PubMed.
- G. Xu, W. Li, L. Yang, X. Wei, J. Ding, J. Zhong and P. K. Chu, J. Power Sources, 2015, 276, 247–254 CrossRef CAS.
- Z. Yang, Q. Meng, Z. Guo, X. Yu, T. Guo and R. Zeng, Energy, 2013, 55, 925–932 CrossRef CAS.
- L. Gao, D. Huang, Y. Shen and M. Wang, J. Mater. Chem. A, 2015, 3, 23570–23576 CAS.
- L. Gao, L. Wang, S. Dai, M. Cao, Z. Zhong, Y. Shen and M. Wang, J. Power Sources, 2017, 344, 223–232 CrossRef CAS.
- B. Zhang, Y. Yu, Y. Liu, Z.-D. Huang, Y.-B. He and J.-K. Kim, Nanoscale, 2013, 5, 2100–2106 RSC.
- J.-Y. Liao, V. Chabot, M. Gu, C. Wang, X. Xiao and Z. Chen, Nano Energy, 2014, 9, 383–391 CrossRef CAS.
- K.-T. Kim, C.-Y. Yu, C. S. Yoon, S.-J. Kim, Y.-K. Sun and S.-T. Myung, Nano Energy, 2015, 12, 725–734 CrossRef CAS.
- L. Wu, X. Leng, Y. Liu, S. Wei, C. Li, G. Wang, J. Lian, Q. Jiang, A. Nie and T. Y. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 4649–4657 CAS.
- S. Wang, Y. Yang, W. Quan, Y. Hong, Z. Zhang, Z. Tang and J. Li, Nano Energy, 2017, 32, 294–301 CrossRef CAS.
- Y. Jiao, H. Zhang, T. Dong, P. Shen, Y. Cai, H. Zhang and S. Zhang, J. Mater. Sci., 2017, 52, 3233–3243 CrossRef CAS.
- M. Xu, L. Wang, X. Zhao, J. Song, H. Xie, Y. Lu and J. B. Goodenough, Phys. Chem. Chem. Phys., 2013, 15, 13032–13037 RSC.
- C. Chen, Y. Huang, C. An, H. Zhang, Y. Wang, L. Jiao and H. Yuan, ChemSusChem, 2015, 8, 114–122 CrossRef CAS PubMed.
- S. Yu, L. Yang, Y. Tian, P. Yang, F. Jiang, S. Hu, X. Wei and J. Zhong, J. Mater. Chem. A, 2013, 1, 12750–12758 CAS.
- G. Hasegawa, K. Kanamori, T. Kiyomura, H. Kurata, K. Nakanishi and T. Abe, Adv. Energy Mater., 2015, 5, 1400730 CrossRef.
- B. Wang, X. Li, B. Luo, L. Hao, M. Zhou, X. Zhang, Z. Fan and L. Zhi, Adv. Mater., 2015, 27, 1526–1532 CrossRef CAS PubMed.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- G. A. Muller, J. B. Cook, H.-S. Kim, S. H. Tolbert and B. Dunn, Nano Lett., 2015, 15, 1911–1917 CrossRef CAS PubMed.
- C. Chen, Y. Huang, C. An, H. Zhang, Y. Wang, L. Jiao and H. Yuan, ChemSusChem, 2015, 8, 114–122 CrossRef CAS PubMed.
- X. Liu, G. Xu, H. Xiao, X. Wei and L. Yang, Electrochim. Acta, 2017, 236, 33–42 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09343h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*


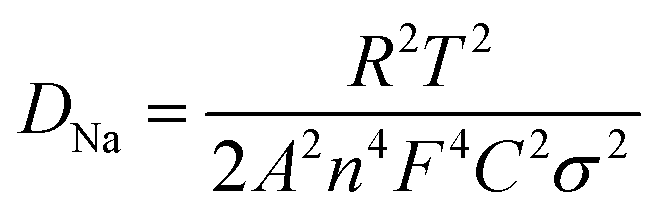
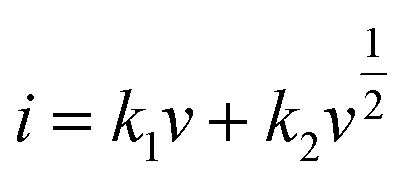 ,47 the capacitive (interfacial) contribution can be separated, where k1v and k2v1/2 represent the contributions from the capacitive (interfacial Na storage process) charge and diffusion-controlled (Na+ insertion/extraction process) one, respectively. For example, as shown in Fig. 5c, the interfacial capacitive contribution at 0.5 mV s−1 is determined to be 50.1% in the LTO-AT/RGO electrode. With the scanning rates being increased from 0.1 to 0.7 mV s−1 (see Fig. 6d), the interfacial capacitive contributions increase from 36.3% to 58.9%. The results indicate that the kinetics of Na+ intercalation process becomes limited at high rates but the kinetics of interfacial Na storage is still sufficient in the LTO-AT/RGO electrode, which is consistent with the aforementioned sodium storage performance observed at various current densities. The CV kinetic analysis disclose that the intercalation-based and interfacial Na storage behaviors take effect simultaneously, and particularly the latter keeps highly efficient at high rates in the LTO-AT/RGO electrode, which endows it with excellent Na storage performance.
,47 the capacitive (interfacial) contribution can be separated, where k1v and k2v1/2 represent the contributions from the capacitive (interfacial Na storage process) charge and diffusion-controlled (Na+ insertion/extraction process) one, respectively. For example, as shown in Fig. 5c, the interfacial capacitive contribution at 0.5 mV s−1 is determined to be 50.1% in the LTO-AT/RGO electrode. With the scanning rates being increased from 0.1 to 0.7 mV s−1 (see Fig. 6d), the interfacial capacitive contributions increase from 36.3% to 58.9%. The results indicate that the kinetics of Na+ intercalation process becomes limited at high rates but the kinetics of interfacial Na storage is still sufficient in the LTO-AT/RGO electrode, which is consistent with the aforementioned sodium storage performance observed at various current densities. The CV kinetic analysis disclose that the intercalation-based and interfacial Na storage behaviors take effect simultaneously, and particularly the latter keeps highly efficient at high rates in the LTO-AT/RGO electrode, which endows it with excellent Na storage performance.