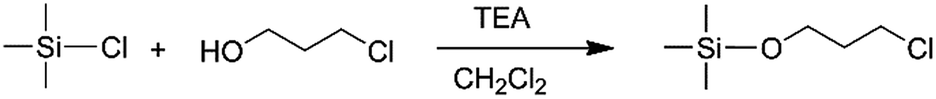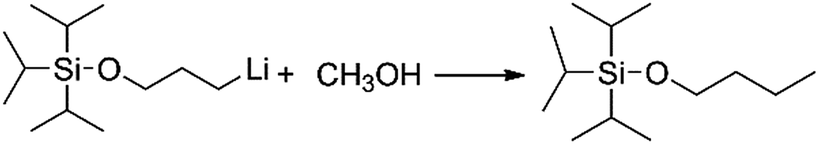DOI:
10.1039/C7RA11117G
(Paper)
RSC Adv., 2017,
7, 52712-52718
Influence of mono-lithium based initiators with different steric volumes on 1,4 unit content of hydroxyl terminated polybutadiene using anionic polymerization
Received
9th October 2017
, Accepted 2nd November 2017
First published on 15th November 2017
Abstract
In order to study the influence of steric structural factors of mono-lithium based anionic initiators on the 1,4 unit content of hydroxyl terminated polybutadiene (HTPB), a series of precursors with different steric volumes were designed and synthesized. The precursors were allowed to react with lithium in cyclohexane at 60 °C to prepare mono-lithium based initiators. The degree of lithium reaction in initiators was characterized by an indirect method in which a certain amount of highly reactive methanol was added into the mono-lithium based initiator solution, and then the highly reactive initiator was replaced by a hydrocarbon molecule to monitor the reaction process using NMR. By using the initiators prepared, HTPB was synthesized via anionic polymerization of 1,3-butadiene in cyclohexane. The structure and 1,4 unit content were characterized by SEC-MALLS and 1H NMR. It was found that the content of the 1,4 unit in HTPB was markedly increased with an increase in the initiator's steric volume. It was also found that much higher relative 1,4 unit content in HTPB was achieved when using an initiator with a higher steric volume, compared to the initiators with lower steric volume. In addition, both 1,4 unit content and molecular weight, as well as the molecular weight distribution of HTPB, can be coordinately controlled by the steric structure of the initiators during anionic polymerization.
Introduction
Hydroxyl-terminated polybutadiene (HTPB) is a telechelic type liquid polymer containing a large number of unsaturated double bonds which provide HTPB with unique physical and chemical properties. Using HTPB as one of the components to prepare polyurethane, the polymer produced presents excellent mechanical properties at a much lower temperature, and has been widely used in the automobile and aircraft industries. Additionally, its products also include tires, rubber materials, construction blocks, insulation coatings, adhesives and so on.1,2 Recently, an important development was reported of the application of HTPB in the field of solid rocket propellants.3–12
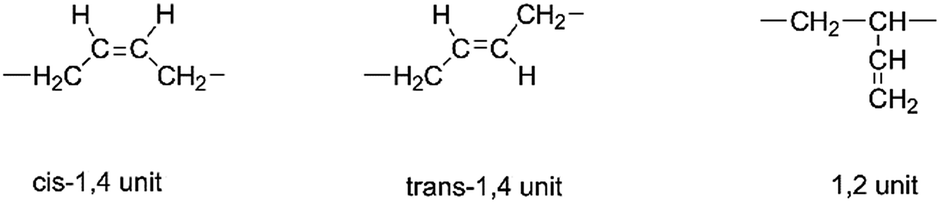
Usually, three different segment structures in the HTPB backbone are detected; they are cis-1,4 unit, trans-1,4 unit and 1,2 unit as shown in Scheme 1. Due to these structures, different physical and mechanical properties can be observed. For example, if HTPB possesses a higher 1,4 unit content, the resulting polyurethane can exhibit extremely low temperature strength.
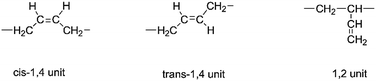 |
| | Scheme 1 Three different segment structures in the HTPB backbone. | |
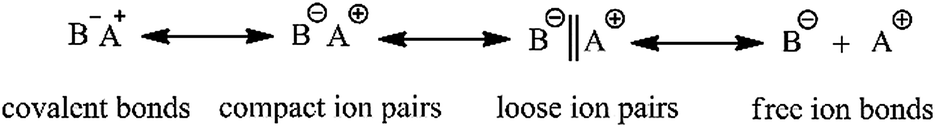
To date, HTPB has mainly been synthesized by adopting free radical polymerization. The polymer synthesized via this technique possesses poor molecular weight controllability, wide molecular weight distribution and low 1,4 unit content; the highest is around 75%. In contrast, HTPB synthesized via anionic polymerization has a narrow molecular weight distribution and higher molecular weight controllability, and in particular, contains a much higher 1,4 unit content.13–17 The advantage of this technique is because the active center for anionic polymerization needs an equilibrium among the polarized covalent bonds, compact ion pairs, loose ion pairs and free ion bonds as shown in Scheme 2. During the polymerization, the formation of ion pairs has a great effect on the final polymer products. For example, compact ions are highly selective for the spatial stereochemistry of butadiene, which facilitates the preparation of stereo-regular polymers. However, its polymerization rate is lower due to the large binding of counter ions. Counter ions of loose ion pairs are much more free and have a higher polymerization rate, but the controlling style of the monomer's addition is weak. Therefore, only when the initiator species, solvent type and polymerization parameters are suitably matched can relatively ideal polymer products be finally obtained.
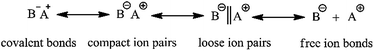 |
| | Scheme 2 The existing species of ion pairs. | |
Currently, HTPB synthesized by anionic polymerization can be applied for use in two different initiators: di-lithium and mono-lithium initiators. Di-lithium initiator is often used in the earlier synthesis process. It is composed of butyl lithium and divinylbenzene derivatives or naphthalene–lithium based initiators. These initiators usually have poor solubility in cyclohexane and other suitable solvents, so employing these kinds of initiators to achieve high 1,4 unit content in HPTB is difficult. On the other hand, mono-lithium initiator carrying protective groups consists of only one C–Li bond, which can effectively trigger 1,3-butadiene to react, at the same time, has good solubility in the hydrocarbon solvents, and has efficient stereo-selectivity. Recently, Quirk and Chen et al.15,17 successfully synthesized HTPB by using 3-tert-butyldimethylsiloxy-1-propyllithium as a mono-lithium initiator. The study found that the character of the solvent, initiator concentration and reaction temperature can equally affect the 1,4 unit content in HTPB. Most importantly, compared to di-lithium initiator, the mono-lithium initiator carries a protecting group with which these groups in the mono-lithium initiator can markedly affect the 1,4 unit content in HPTB. Unfortunately, there are only a few reports of research in this direction so far. Studies have also shown that reduction of the intermolecular aggregation via initiators can be beneficial to increase the 1,4 unit content in HTPB,18,19 while increasing the steric volume of the initiators could be another key factor to increase the 1,4 unit content in HTPB.
In order to obtain a higher 1,4 unit content and prepare highly qualified HTPB products, in this article, five different mono-lithium based initiators with different steric volumes were designed and synthesized. The molecular structures for all mono-lithium based initiators were completely confirmed via FTIR 1H NMR and SEC-MALLS. Their steric volume data were calculated via the Gaussian 09 program. Their applications in the synthesis of HTPB via anionic polymerization was also carried out. The research found that an initiator with a higher steric volume can markedly increase the 1,4 unit content in HTPB, due to its reduction effect on the intermolecular aggregation during anionic polymerization.
Experimental section
Materials
Trimethylchlorosilane, tert-butyldimethylchlorosilane, triisopropylchlorosilane, chlorotriisopropylsiane, 1-chloro-4-ethoxybutane, 3-chloro-1-propanol, 3-chloro-2,2-dimethyl-1-propanol and imidazole were all purchased from Aladdin and used as received. N,N-Dimethylformamide and cyclohexane were purified by sequential passage through activated alumina followed by Q-5 (Engelhard) to remove impurities and trace oxygen. Epoxyethane (EO) (99.5%) was purchased from Sinopharm Chemical Reagent Co., Ltd (China), and was stored in a sealed condition. 1,3-Butadiene (15% cyclohexane solution) was purchased from TCI and before use was passed under argon gas for 15 min. Triethylamine (AR), dichloromethane (AR) and methanol (AR) were used as received.
Standard Schlenk techniques were used to manipulate all moisture and air sensitive compounds under a high-purity argon atmosphere.
Synthesis of trimethylsilico propyl chloride (TMSOPrCl)
Initially 33 g 3-chloro-1-propanol (0.35 mol) was dissolved in 400 mL dichloromethane and cooled down to 0 °C under a nitrogen atmosphere. Next, 58 mL triethylamine (0.4 mol) was added to the solution. After addition, 42 g trimethylchlorosilane was put into 100 mL dichloromethane solution containing 3-chloro-1-propanol and trimethylamine. The reaction was carried out for 2 h and terminated with 200 mL diethyl ether. Finally, the salt of the triethylamine was filtrated, and the mixture solvent was removed under vacuum using a rotary flash evaporator at 38 °C. The product obtained was distilled again under reduced pressure at 70 °C under a pressure of 0–5 mmHg to obtain pure trimethylsilico propyl chloride. The yield was 63%. 1H-NMR (400 MHz, CDCl3): δ 3.72 (2H, O–CH2–), 3.65 (2H, Cl–CH2–), 1.96 (m, 2H, –CH2–), 0.13 (9H, –CH3).
Synthesis of triisopropyl silicon propyl chloride (TIPSOPrCl)
TIPSOPrCl was prepared using a modified version of the previously reported procedure. First, 0.12 mol imidazole and 0.1 mol triisopropylchlorosilane were added to 40 mL anhydrous DMF under an argon atmosphere, and stirred, until they formed a clear solution. Then, 0.12 mol 3-chloro-2,2-dimethyl-1-propanol was added to the solution. The mixture was stirred at 50 °C for 12 h. In this case, an excess mole of 3-chloro-2,2-dimethyl-1-propanol and imidazole were used to ensure complete reaction of triisopropylchlorosilane. After continuously stirring for 12 h, 100 mL cyclohexane was added. Subsequently, three successive aqueous extractions were performed to remove HCl, DMF and the excess imidazole base from the mixture solution. The cyclohexane solvent was evaporated under vacuum, and TIPSOPrCl was purified by silica gel chromatography. The yield was 96.7%. 1H NMR (400 MHz, CDCl3) δ 3.86 (2H, –O–CH2–), δ 3.71 (2H, –O–CH2–CH2–Cl), δ 2.04–1.96(2H, –O–CH2CH2–), δ 1.18–1.02 (21H, [(CH3)2CH]3–Si–).
Synthesis of tert-butyldimethylsiloxypropyl chloride (TBDMSOPrCl)
The TBDMSOPrCl was synthesized using a similar procedure to TIPSOPrCl. Except for tert-butyldimethylsilyl chloride was used instead of triisopropylchlorosilane. The yield was 95.2%. 1H NMR (400 MHz, CDCl3): δ 3.87 (2H, –O–CH2–), δ 3.75 (2H, –O–CH2–CH2–Cl), δ 2.21–2.03 (2H, –O–CH2CH2–), δ 0.92(9H, [CH3]3C–), δ 0.07(6H, [CH3]2–Si–).
Synthesis of triisopropylsiloxane-2,2-dimethyl-1-chloropropane (TBDMSODMPrCl)
The TBDMSODMPrCl was synthesized using a similar procedure to TIPSOPrCl. Except for 3-chloro-2,2-dimethyl-1-propanol was used instead of 3-chloro-1-propanol. The yield was 95.7%. 1H NMR (400 MHz, CDCl3): δ 3.54–3.46 (4H, –O–CH2–), δ 1.18–1.05 (21H, [(CH3)2CH]3–Si–), δ 1.02–0.95 (6H, –CH3).
Preparation of initiator of 4-methoxy-1-butyl lithium. A three-necked flask with a condenser was put into an oil bath with a magnetic stirrer. Under an argon atmosphere, 50 mL cyclohexane and 5 g metallic lithium particles were successively added to the reactor containing the magnetron. Stirring was started and the temperature of the oil bath was raised to 60 °C. The cyclohexane and 4-methoxy-1-butyl chloride (0.05 mol) were mixed and added dropwise within 60 min. After the addition, the reaction was carried out for 2 h and terminated. The supernatant was transferred to a closed and pre-removed receiver by internal transfer under high purity argon protection to give a solution of 4-methoxy-1-butyllithium in cyclohexane.
Preparation of initiator of 3-trimethylsiloxy-1-propyllithium (TMSOPrLi). The TMSOPrLi was synthesized using the same procedure as for 4-methoxy-1-butyl lithium. The reaction time lasted for 5 h.
Preparation of initiator of 3-tert-butyldimethylsilyloxy-1-propyllithium (tBDMSOPrLi). The tBDMSOPrLi was synthesized using the same procedure as for 4-methoxy-1-butyl lithium. The reaction time lasted for 8 h.
Preparation of initiator of 3-triisopropylsilyloxy-1-propyllithium (TIPSOPrLi). The TIPSOPrLi was synthesized using the same procedure as for 4-methoxy-1-butyl lithium. The reaction time lasted for 24 h.
Preparation of 3-triisopropylsiloxane-2,2-dimethyl-1-propyllithium (TIPSODMPrLi). The TIPSODMPrLi was synthesized using the same procedure as for 4-methoxy-1-butyl lithium. The reaction time lasted for 36 h.
Synthesis of hydroxyl-terminated polybutadiene (HTPB)
Anionic polymerization of 1,3-butadiene at 50 °C in cyclohexane was initiated by different mono-lithium based initiators. Taking TIPSOPrLi as an example, initiator (1 mL) was added to cyclohexane (10 mL) containing 15% 1,3-butadiene under an argon atmosphere thermo-state at 50 °C. After polymerization for 5 h, ethylene oxide (0.5 mL) was added to cap the living PB and allowed to react for another 2 h. The reaction mixture was terminated by the addition of degassed methanol (0.5 mL) to form PB-OH. The resultant PB-OH (1.2 g) was precipitated in methanol solution to obtain a white viscous liquid which was then dissolved in THF (5 mL). Subsequently, excess tetrabutylammonium fluoride (TBAF) was added to the solution at room temperature and allowed to react for 24 h to obtain the hydroxy groups at both ends of the polybutadiene. Finally, the polymer was precipitated in methanol and residual solvent was removed by rotary evaporation (100 mL). A pale yellow viscous liquid product was obtained at this stage.
Instruments and characterization
1H NMR spectra were recorded with a Bruker 400 MHz at room temperature using CDCl3 as the solvent and tetramethylsilane as an internal reference. The molecular weight and polydispersity index of HTPB were determined on a DAWN EOS size exclusion chromatography/multi-angle light scatter instrument (SEC-MALLS, Wyatt Technology). HPLC grade THF was used as the eluent at a flow rate of 0.5 mL min−1 at 25 °C.
The steric volume calculation
All calculations were carried out using the Gaussian 09 program. Quantum chemistry calculations were performed using the density function theory (DFT) method, B3LYP with electron basis set 6-31G* (d,p) for all atoms. The final obtained geometries were recognized as local minima energy and the volume was computed inside a contour of 0.001 electron per bohr3 density.20,21
1,4 unit content in HTPB measurements
The 1,4 unit content in HTPB was calculated based on the 1H NMR datum, following equation was used to conduct the calculation:| |
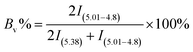 | (1) |
where I is the integral area and Bv% is the content of 1,2 unit in HTPB, and the content of 1,4 unit in HTPB is (1 − Bv) × 100%.
Results and discussion
Synthesis of different structural precursors
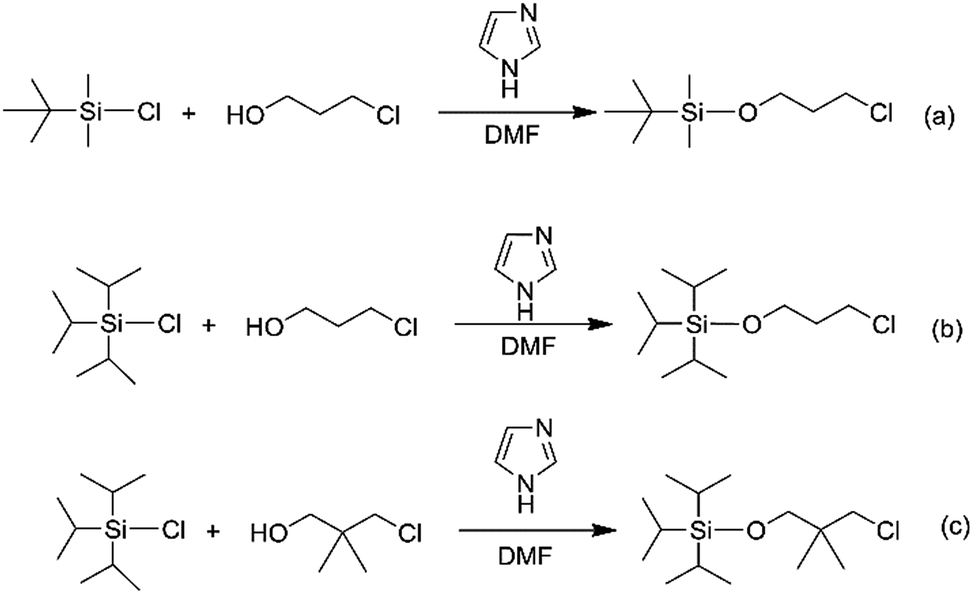
In order to obtain the different steric volume precursors, per-tert-butyldimethylchlorosilane (tBDMSCl) or triisopropylchlorosilane (TIPSCl) was reacted with 3-chloro-1-propanol, and TIPSCl was reacted with 3-chloro-2,2-dimethyl-1-propanol in DMF containing imidazole, respectively. The chemical routes are shown in Scheme 3. Then, tert-butyldimethylsiloxypropyl chloride (tBDMSOPrCl), triisopropylsiloxypropyl chloride (TIPSOPrCl) or triisopropylsiloxane-2,2-dimethyl-1-propyllithium (TIPSODMPrLi) was separated and purified by a silica gel column.22,23 As the reactivity of chlorosilane with hydroxyl is much higher, the yield of the product can reach 95%. As the reaction of 3-trimethylsiloxy-1-propylchloride (TMSOPrCl) with trimethylchlorosilane (TMSCl) as a protecting group was highly reactive, the trimethylsilyl group could not only be easily removed in saturated NaHCO3 solution, but could also be removed through a silicone column. So the reaction route of TMSCl with 3-chloro-1-propanol was adopted as another way, as shown in Scheme 4. Then, the resulting product was distilled at 70 °C under a pressure of 0–5 mmHg to obtain pure TMSOPrCl, and the yield was 63%.24
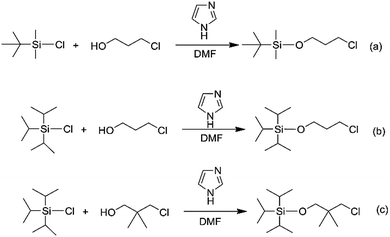 |
| | Scheme 3 Synthesis routes of TBDMSOPrCl, TIPSOPrCl and TIPSODMPrCl. | |
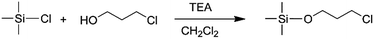 |
| | Scheme 4 Synthesis route of TMSOPrCl. | |
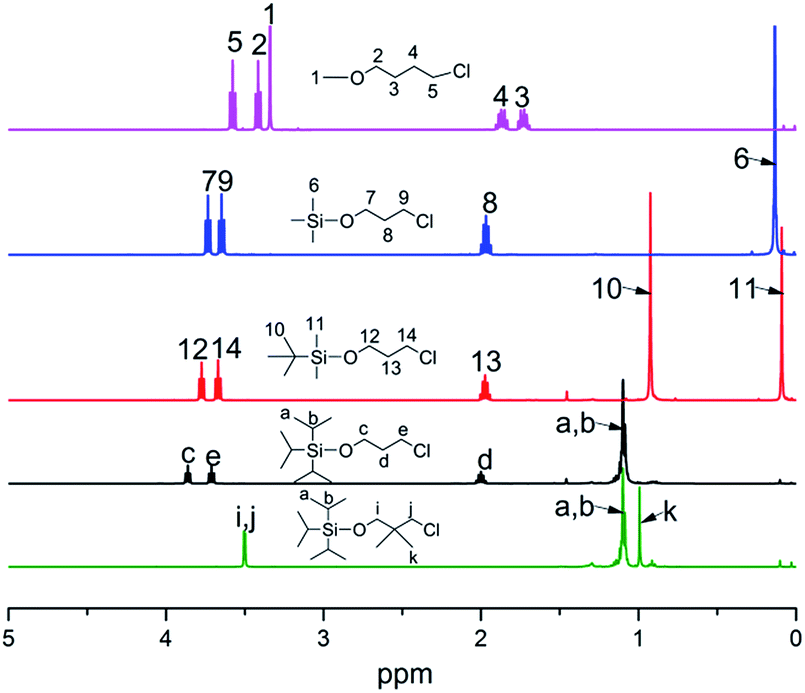
All molecular structures of TIPSODMPrCl, tBDMSOPrCl, TIPSOPrCl, TMSOPrLi and 1-chloro-4-methoxybutane were confirmed by 1H NMR spectra, as shown in Fig. 1.
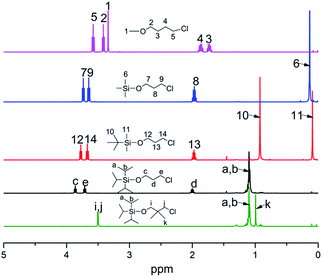 |
| | Fig. 1 1H NMR spectra of different steric volume precursors. | |
Preparation of mono-lithium based initiators
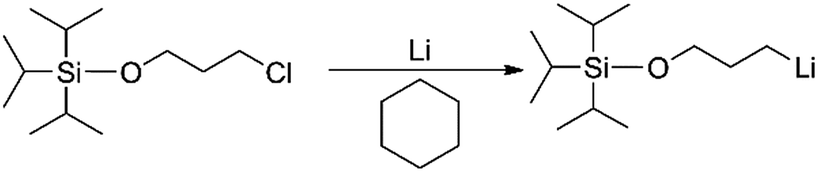
For the preparation of all mono-lithium based initiators, a similar reaction procedure was employed. Taking TIPSOPrLi as an example, as shown in Scheme 5. Conventionally, TIPSOPrCl is difficult to react with ordinary lithium; therefore in this case, lithium wires with a large specific surface area were used. During the experiment, the lithium wires were first treated to show a silver-white metallic lustre, and then, kept under argon to avoid air contact. Tong25 previously reported a similar sensitivity in the synthesis of a mono-lithium based compound. They suggested that lithium should be washed in cyclohexane repeatedly to obtain “a silvery metal surface”. Meuler26 also found that without first preparing the lithium by mechanically abrading the surface of lithium, the reaction of mono-lithium based compounds could not proceed.
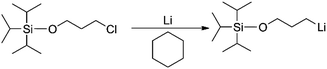 |
| | Scheme 5 The synthetic route of TIPSOPrLi. | |
During synthesis, it was found that TIPSOPrLi and other initiators exhibited extremely high activity, and were very difficult to obtain as a solid product. The best way to preserve them is in solution. Due to this reason, lithium-containing initiators could not be characterized by NMR technique directly. Previously, our determination of the lithium degree of tBDMSOPrLi was achieved using a Gilman double-titration assay, and it basically confirmed the existence of active organ-lithium.27 However, how to precisely analyze the initiator's lithium degree during reaction still needs to be further explored.
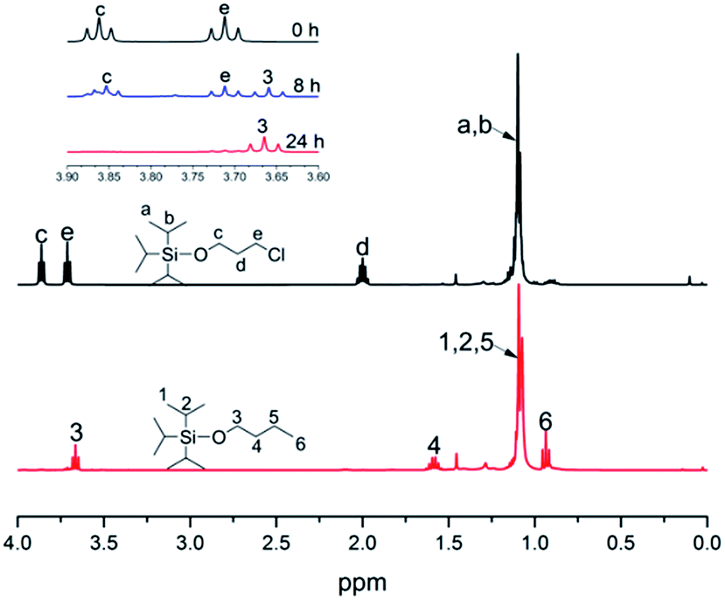
Generally speaking, the lithium degree in the initiator depends on the reactivity of TIPSOPrCl with lithium, as shown in Scheme 5. In this case, when the reaction was proceeding, the amount of TIPSOPrLi increased following the reduction of TIPSOPrCl. Therefore, by monitoring the partial content of TIPSOPrCI, the lithiation could be easily obtained. Thus, in order to conveniently control the lithium degree, an “indirect method” was used in which the high reactivity of TIPSOPrLi with methanol to produce 3-triisopropylsilyloxy-1-butane (TIPSOBu) was employed, as shown in Scheme 6. By using this method, if we took TIPSOPrCI as an analysis target at different time intervals, the data of lithiation could be measured by NMR as shown in Fig. 2. Clearly from Fig. 2, the peak intensity of TIPSOBu triplets (δ 3.67) increased, while the TIPSOPrCl triplets (δ 3.87, δ 3.73) diminished following the reaction. After 24 h, the triplets of TIPSOPrCI completely disappeared indicating the lithium degree in the initiator had reached 100%.
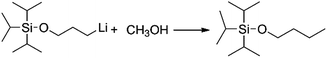 |
| | Scheme 6 The mechanism of determining lithium content in initiator via reaction of TIPSOPrLi and methanol. | |
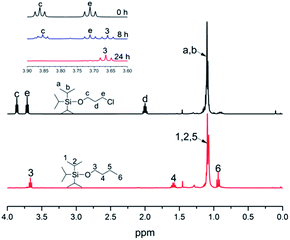 |
| | Fig. 2 1H-NMR spectra of TIPSOPrCI and TIPSOBu at different reaction intervals. | |
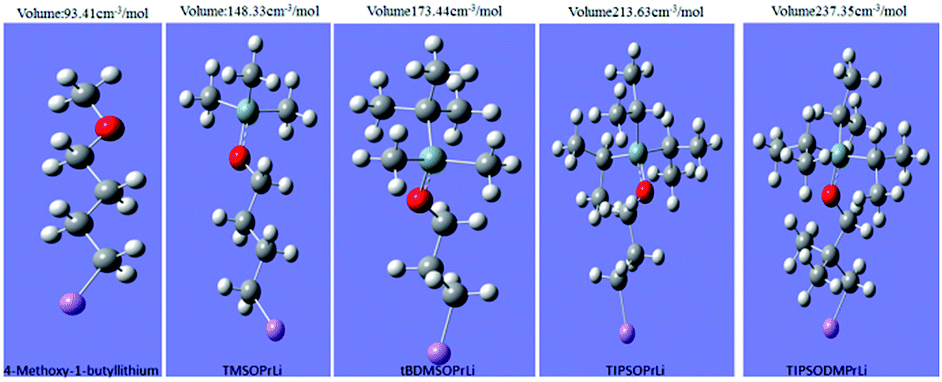
For further measuring of the magnitude of steric volume of initiators, the Gaussian 09 program was used to calculate their steric volume data. To estimate the steric volume of each mono-lithium structure, we computed the volume inside a contour of 0.001 electron per bohr3 density. The results are given in Table 1. In order to increase accuracy, importing the numeric value was adopted as Iop(6/45 = 2000).21 Both the molecular spatial configuration and the steric volume data are presented in Fig. 3. Evidently from Fig. 3, the steric volume data calculated were in good accordance with our original molecular designs. The order was satisfactorily based on the complexity of the spatial structures of these initiators, in the order TIPSODMPrLi > TIPSOPrLi > tBDMSOPrLi > TMSOPrLi > 4-methoxy-1-butyllithium.
Table 1 Molecular structural parameters of HTPB initiated via mono-lithium based initiators
| Entry |
Initiator |
Steric volumea (cm−3 mol−1) |
Mnb (g mol−1) |
Mw/Mnb |
% 1,4-unit contentc |
| Calculation by Gaussain 09 program. Measured by SEC-MALLS at 25 °C in a tetrahydrofuran mobile phase. Measured by 1H NMR and the 1,4 unit content calculated according to eqn (1).28 |
| 1 |
4-Methoxy-1-butyllithium |
93.41 |
3100 |
1.10 |
72.1 |
| 4200 |
1.06 |
74.8 |
| 4900 |
1.07 |
75.8 |
| 2 |
TMSOPrLi |
148.33 |
3080 |
1.09 |
79.1 |
| 3900 |
1.05 |
79.8 |
| 5030 |
1.04 |
81.0 |
| 3 |
tBDMSOPrLi |
173.44 |
3100 |
1.05 |
84.9 |
| 4090 |
1.09 |
85.7 |
| 5410 |
1.08 |
86.5 |
| 4 |
TIPSOPrLi |
213.63 |
3200 |
1.10 |
90.7 |
| 4300 |
1.03 |
90.8 |
| 4850 |
1.05 |
91.0 |
| 5 |
TIPSODMPrLi |
237.35 |
4100 |
1.10 |
92.1 |
| 5250 |
1.08 |
92.4 |
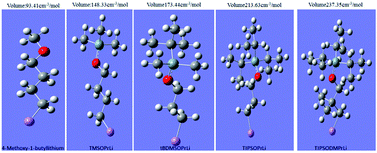 |
| | Fig. 3 Spatial configurations and their steric volume data for mono-lithium based initiators. | |
Synthesis and characterization of the hydroxyl terminated polybutadiene
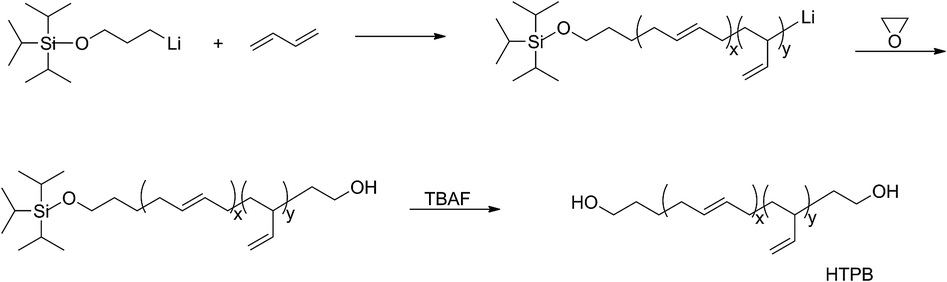
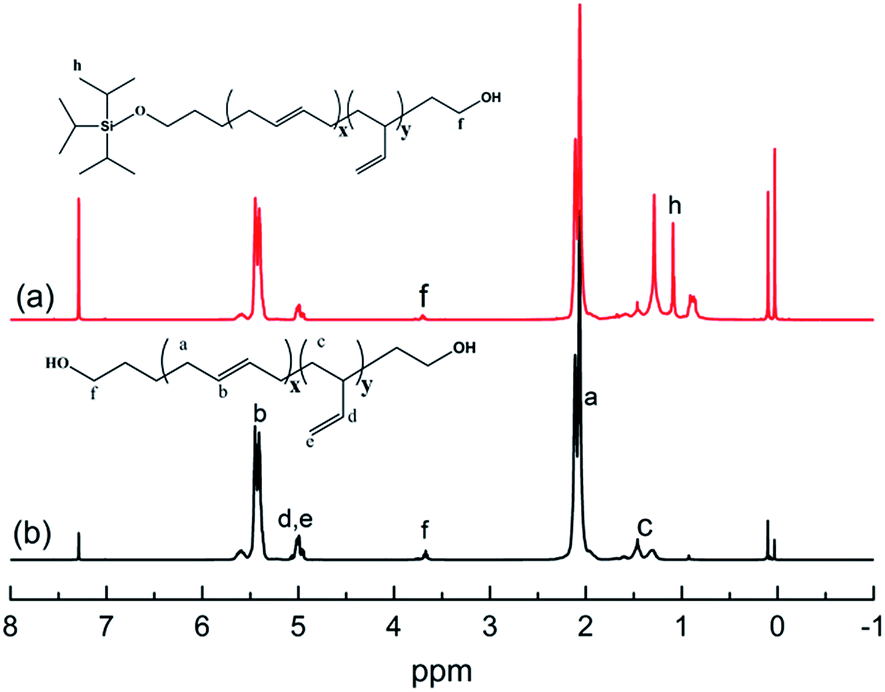
The chemical routes of anionic polymerization initiated by mono-lithium based initiators, taking TIPSOPrLi as an example, are illustrated in Scheme 7. As can be clearly seen from Scheme 7, the syntheses of hydroxyl terminated polybutadiene needed two major steps. The first step determined how many 1,4 units were achieved via monomer addition style by choosing a suitable initiator, and the second step guarantees hydroxyl groups are fixed to both ends of HTPB. During the synthesis process, we found that mono-lithium based initiators could not only efficiently control the 1,4 unit content, but also be easily subjected to hydrolysis to remove their silicon protecting groups. This function can certainly help us to make a perfect hydroxyl terminated HTPB polymer. The experimental conditions for the hydrolysis of the silicon protecting groups were using tetrahydrofuran as the solvent, plus tetrabutylammonium fluoride (TBAF) at room temperature for 24 h. The hydrolysis date of the protecting group in HTPB using TIPSOPrLi as an example is shown in Fig. 4. Clearly, after hydrolysis of triisopropoxysilane (TIPS) in TIPSOPrLi, the chemical shift at 0.95 ppm, which was assigned to the proton in the methyl of isopropyl, completely disappeared, indicating that the removal of the protecting group was fulfilled.
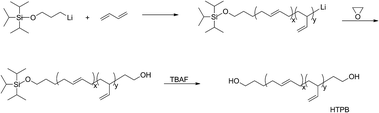 |
| | Scheme 7 The synthesis routes of HTPB via anionic polymerization initiated by TIPSOPrLi. | |
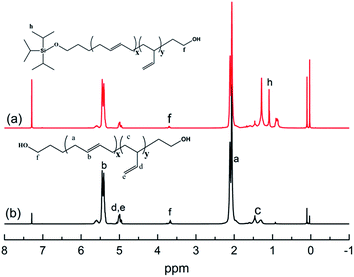 |
| | Fig. 4 1H-NMR spectra of HTPB before and after hydrolysis of the protecting group. | |
Table 1 presents the molecular structure characterization data of HTPB. As can be seen from Table 1, the molecular weight distributions for HTPBs were all around 1.1, indicating the polymers were synthesized in a mono-dispersed state. At the same time, the molecular weight distribution of HTPBs was not changed with the change of the initiator's steric volume. Meuler et al.26 reported that the magnitude of the steric volume can reduce the intermolecular aggregation and lead to an increase in the initiation efficiency, which might have an impact on the molecular weight distribution of polystyrene. However, the result showed that using different initiators with different steric volumes could not have an impact on the molecular weight distribution of HTPB.
The relationship between steric volume of initiator and 1,4 unit content in HTPB
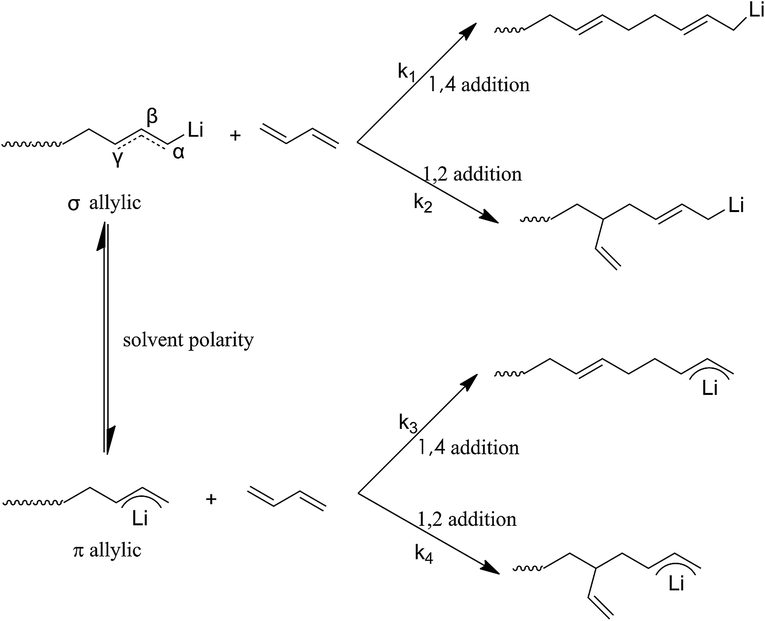
In order to further understand the mechanism at the molecular level for above result, a fundamental theory study based on the anionic polymerization was conducted. According to the previously reported literature,29,30 the active center of the chain growth for anionic polymerization relies on ion pairs, which exist in two forms, σ allylic type and the π allylic type, as shown in Scheme 8, where the σ allylic favors the formation of the 1,4-addition (1,4 unit), and π allylic favors the formation of 1,2-addition (1,2 unit).
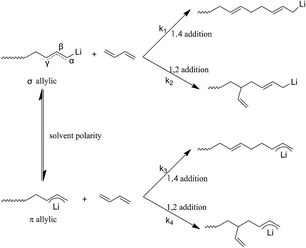 |
| | Scheme 8 Molecular mechanism of anionic polymerization of polybutadiene. | |
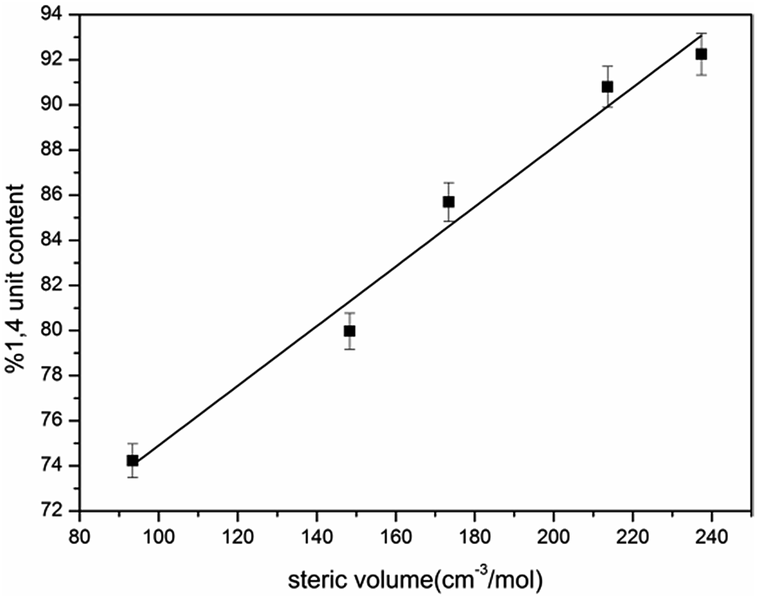
As our polymerization was conducted in cyclohexane, the active centers were mainly adopted as the σ allylic type, which can facilitate the formation of a 1,4 unit. On the other hand, due to the natural behavior of the intermolecular association of the lithium element, the associated initiators could prohibit monomers attacking the α position, and as a result, the γ position becomes the main attacking place by monomers which could easily form a 1,2 addition. As the result the 1,2 addition might directly create a 1,2 unit in HTPB, therefore, association of mono-lithium based initiators could certainly reduce the 1,4 unit content in HTPB during the polymerization period. Evidently, if an initiator possesses a large steric volume, the intermolecular aggregation among initiators might be effectively avoided leading to reduction of the 1,2 addition. As a result, the percentage of 1,4 unit content, which also included the amount of 1,2 unit content, could be tremendously increased. Here, as we can see from Table 1, the lowest 1,4 unit content in HTPB initiated by 4-methoxy-1-butyllithium (steric volume: 93.41 cm−3 mol−1) is 72.1%, and the highest 1,4 unit content in HTPB initiated by TIPSOPrLi (steric volume: 213.63 cm−3 mol−1) is 91.0%. The result clearly proved the fundamental theory that initiators with larger spatial structure can significantly prevent the intermolecular association, and as a result, increase the 1,4 unit content in HTPB. The relationship between the steric volume of different initiators with 1,4 unit contents is shown in Fig. 5. Interestingly, the 1,4 unit content in HTPBs increased linearly with the initiator's steric volume.
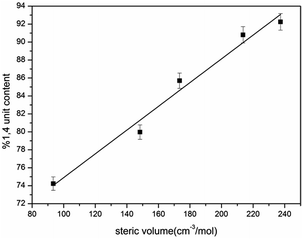 |
| | Fig. 5 1,4 unit content of HTPB initiated by mono-lithium with different steric volume. | |
Conclusions
Five mono-lithium based initiators with different steric volumes were designed and synthesized. Through the calculation of their stereo volume using the Gaussian 09 program, the order of steric volume for initiators was successfully established as being, TIPSODMPrLi > TIPSOPrLi > tBDMSOPrLi > TMSOPrLi > 4-methoxy-1-butyllithium. By using these initiators, HTPBs with different molecular weights and molecular distributions were prepared. It was found that the 1,4 unit content in HTPB linearly increased with the increase in the initiator's stereo volume. The result can be attributed to the large steric volume which played a role in preventing intermolecular aggregation among the mono-lithium based initiators, and led to the increase in the 1,4 unit content in HTPB.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by the Scientific Research Project of Ministry of Industry and Information Technology of P. R. China (no. JSJL2016140B004) and the Key Laboratory Project of the Department of Science and Technology of Shannxi Province of China (no. 2013SZS17-Z02).
Notes and references
- P. Georgopanos, G. A. Scheider, A. Dreyer, U. A. Handge, V. Filiz, A. Feld, E. D. Yilmaz, T. Krekeler, M. Ritter, H. Weller and V. Abetz, Sci. Rep., 2017, 7, 7314 CrossRef PubMed.
- Y. L. Luo, Y. Miao and F. Xu, Macromol. Res., 2011, 19, 1233 CrossRef CAS.
- C. J. Chen, I. H. Tseng, H. T. Liu, W. W. Tsai and S. L. Huang, Mater. Sci. Eng., A, 2011, 528, 4917–4923 CrossRef CAS.
- M. Amrollahi, G. Sadeghi and Y. Kashcooli, Mater. Des., 2011, 32, 3933 CrossRef CAS.
- B. N. Rao, P. J. P. Yadav, K. Malkappa, T. Jana and P. U. Sastry, Polymer, 2015, 77, 323 CrossRef CAS.
- V. Sekkar, A. S. Alex, V. Kumar and G. G. Bandyopadhyay, J. Macromol. Sci., Part A: Pure Appl.Chem., 2017, 31, 171 CrossRef.
- J. Yang, H. Lin and W. Lai, J. Membr. Sci., 2002, 208, 105 CrossRef CAS.
- K. Hailu, G. Guthausen, W. Becker, A. konig, A. Bendfeld and E. Geissier, Polym. Test., 2010, 29, 513 CrossRef CAS.
- K. Ganesh, S. Sundarrajan and K. Kishore, Macromolecules, 1999, 33, 326 CrossRef.
- W. B. Zhang, X. D. Fan, W. Tian, H. Chen, X. Z. Zhu and H. T. Zhang, RSC Adv., 2015, 5, 66073 RSC.
- L. T. Deluca, L. Galfetti, G. Colombo, L. Merotto, M. Boiocchi, C. Paravan, A. Reina, P. Tadini and L. Fanton, Acta Astronaut., 2013, 92, 150 CrossRef CAS.
- L. Poussard, F. Burel, J. P. Couvercelle, C. L. Bourhis and C. Bunel, J. Appl. Polym. Sci., 2006, 15, 3312 CrossRef.
- K. N. Ninan, V. P. Balagangadharan and K. B. Catherine, Polymer, 1991, 32, 628 CrossRef CAS.
- X. T. Zhang, Y. Li, C. Q. Zhang, Y. M. Hu, S. X. Song, H. H. Guo and Y. R. Wang, Polymer, 2009, 50, 5427 CrossRef CAS.
- J. M. Chen, Z. J. Lu, G. Q. Pan, Y. X. Yi and H. J. Bai, Chin. J. Polym. Sci., 2010, 28, 715 CrossRef CAS.
- I. Lee and F. S. Bates, Macromolecules, 2013, 46, 4529 CrossRef CAS.
- R. P. Quirk, S. H. Jang, H. M. Yang and Y. J. Lee, ACS Symp. Ser., 1998, 704, 71 CrossRef CAS.
- S. K. Jiao, Theory and practice of olefin coordination polymerization, Chemical Industry Press, Beijing, 2004, p. 269 Search PubMed.
- K. F. O'Driscoll, J. Macromol. Chem., 1966, 1, 1 CrossRef.
- D. K. Yadav, B. Maiti and M. A. Quraishi, Corros. Sci., 2010, 52, 3586 CrossRef CAS.
- Y. A. Gruzdkov, Z. A. Dreger and Y. M. Gupta, J. Phys. Chem. A, 2004, 108, 6216 CrossRef CAS.
- S. E. Leonaed, K. G. Reddie and K. S. Carroll, ACS Chem. Biol., 2009, 4, 783 CrossRef PubMed.
- C. E. Ayala, A. Villalpando, A. L. Nguyen, G. T. Mccandless and R. Kartika, Org. Lett., 2012, 14, 3676 CrossRef CAS PubMed.
- F. Costantino, R. Lenna and S. Pouri, US Pat., 7585971B2, 2009.
- J. Tong, C. Zhou, S. Ni and M. A. Winnik, Macromolecules, 2001, 34, 696 CrossRef CAS.
- A. J. Meuler, M. K. Mahanthappa, M. A. Hillmyer and F. S. Bates, Macromolecules, 2007, 40, 760 CrossRef CAS.
- T. W. Greene and P. G. M. Wuts, in ProtectiVe Groups in Organic Synthesis, Wiley-Interscience., New York, 2nd edn, 1991, p. 473 Search PubMed.
- Y. Tanaka and Y. J. Takeuchi, J. Polym. Sci., Part A-2, 1971, 9, 43 CrossRef CAS.
- G. Natta, L. Porri and A. Carbonaro, Macromol. Chem. Phys., 1964, 77, 126 CrossRef CAS.
- G. T. Jin and T. H. Lin, J. Chem. Eng., 1985, 2, 215 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Xiao-dong Fan*,
Na Zhao,
Xin Min,
Jie Liu and
Zi-chao Wang
,
Xiao-dong Fan*,
Na Zhao,
Xin Min,
Jie Liu and
Zi-chao Wang