
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(N-isopropylacrylamide-co-L-proline)-catalyzed Claisen–Schmidt and Knoevenagel condensations: unexpected enhanced catalytic activity of the polymer catalyst†
Hao Zhang‡
ab,
Mengting Han‡ab,
Tian Chena,
Lin Xuab and
Lei Yu *ab
*ab
aInstitute of Pesticide, School of Chemistry and Chemical Engineering, School of Horticulture and Plant Protection, Yangzhou University, Yangzhou, Jiangsu 225002, China. E-mail: yulei@yzu.edu.cn
bJiangsu Yangnong Chemical Group Co. Ltd., Yangzhou, Jiangsu 225002, China
First published on 13th October 2017
Abstract
The polymer catalyst is more effective than the corresponding monomer catalyst? Yes! The proline-modified polymer, poly(N-isopropylacrylamide-co-L-proline), was unexpectedly found to be more effective than the corresponding monomer L-proline catalyst in Claisen–Schmidt and Knoevenagel condensation reactions. 1H NMR, GC analysis and control reactions revealed that this abnormal phenomenon might be attributed to an enhanced concentration of the reactant on the surface of the polymer catalyst, which might be due to adsorption of the reactants to the polymer through hydrogen-bonding of the proline moiety with the reactants. This new polymer catalyst was so robust that it could be reused at least 10 times without deactivation. The polymer-catalyzed method was rather tolerant of substrates bearing sensitive groups that are usually incompatible with conventional acid- or base-catalyzed methods, reducing the protection–deprotection steps of the substrates.
Introduction
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond formation is an important subject in organic synthesis for allowing carbon chain growth in building complex large molecules.1 Among known methods, the reactions of aldehydes with active C–H, such as the Claisen–Schmidt and Knoevenagel condensations, are efficient tools to achieve this objective.2 The transformations have good potential in industrial application owing to the processes being of high atom economy, and not generating waste other than water. However, many of these reactions are catalyzed by acids or bases, which are corrosive and intolerant to sensitive functional groups. Thus, developing novel tolerant catalysts that allow the direct use of sensitive substrates free of tedious protection–deprotection steps is still desirable to improve the synthetic efficiency.
C bond formation is an important subject in organic synthesis for allowing carbon chain growth in building complex large molecules.1 Among known methods, the reactions of aldehydes with active C–H, such as the Claisen–Schmidt and Knoevenagel condensations, are efficient tools to achieve this objective.2 The transformations have good potential in industrial application owing to the processes being of high atom economy, and not generating waste other than water. However, many of these reactions are catalyzed by acids or bases, which are corrosive and intolerant to sensitive functional groups. Thus, developing novel tolerant catalysts that allow the direct use of sensitive substrates free of tedious protection–deprotection steps is still desirable to improve the synthetic efficiency.
L-Proline is a kind of organocatalyst that can work under mild conditions.3 It has been widely employed in many useful organic reactions, such as the Michael addition, Mannich reaction, Diels–Alder addition, aldol condensation and the related domino/multi-component reactions,4 providing unique and powerful synthetic tools for the accessible catalyst, metal-free conditions and high reaction selectivities. L-Proline is milder than traditional organic or inorganic acids or bases, and may be more tolerant to sensitive functional groups in reaction substrates. However, because L-proline is a cheap and abundant natural chiral molecule, much attention has been paid to its applications in asymmetric synthesis, while the design and fabrication of proline-based catalysts for simple CC bond formation is so far not well documented. We have been interested in organocatalysis for a long time.5 Recently, we investigated proline-based catalysts for CC bond formation through the Claisen–Schmidt and Knoevenagel condensations. During the research, we designed and synthesized poly(N-isopropylacrylamide-co-L-proline), which is a proline-modified polymer, and examined its catalytic activity in the reactions. Herein, we wish to report our findings.
Results and discussion
Using the Claisen–Schmidt condensation of benzaldehyde 1a with acetone 2 to produce (E)-4-phenylbut-3-en-2-one 3a as the template, a series of amino acids as catalysts and amines as additives were initially examined (eqn (1)).
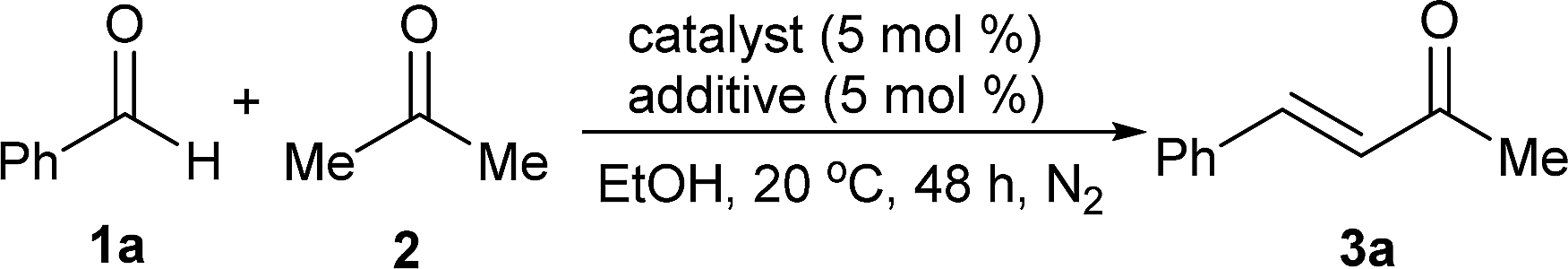 | (1) |
It was found that L-proline was a uniquely good catalyst for the reaction and piperazine was the favourable additive, while other amino acid catalysts or amine additives were less effective.6 Based on this experimental result, the polymer-supported proline catalyst was then designed. As shown in Scheme 1, esterification of the commercially available reagents 4 and 5 promoted by CF3SO3H/CF3CO2H first afforded the (2S,4R)-4-(acryloyloxy)pyrrolidine-2-carboxylic acid 6, which then led to poly(N-isopropylacrylamide-co-L-proline) 8 through a co-polymerization reaction with N-isopropylacrylamide (NIPA) 7.6,7 Time of flight mass spectrometer (TOF) analysis showed that the polymer 8 had an average molecular weight of ca. 4.9 × 104 and 1H NMR analysis of the material indicated that the molar ratio of the proline fragment was 12.3%.7
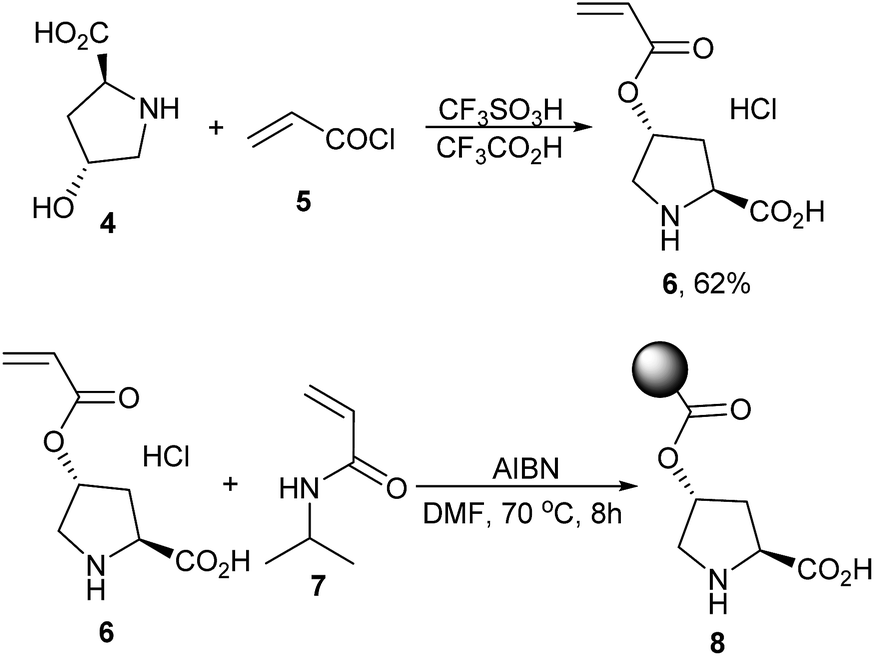 | ||
| Scheme 1 Preparation of the polymer-supported proline catalyst 8. | ||
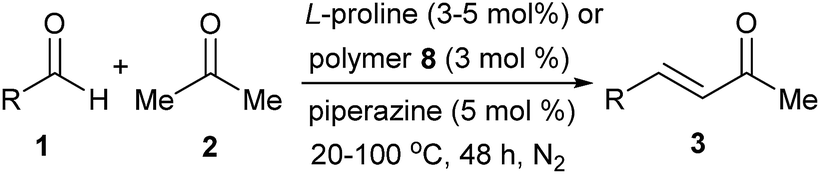
Claisen–Schmidt condensation reactions catalyzed by L-proline or polymer 8 were then performed simultaneously for comparison (Table 1). Condition optimizations6 showed that ethanol was a favourable solvent for the free L-proline-catalyzed reactions, while the polymer 8-catalyzed reaction could be performed in acetone without additional solvent. In the free L-proline-catalyzed reactions, 4-methylbenzaldehyde 1b as an electron-enriched substrate led to 3b in an even lower yield than that of the reaction of the simple substrate 1a (entry 2 vs. 1). The product yield was slightly enhanced at elevated reaction temperatures and the reaction at 80 °C afforded the highest product yield at 55% (entry 2). Similarly, the electron-enriched substrates 1c–e led to the related products 3c–e in moderate yields (entries 3–5). Elevated reaction temperatures were also required for the condensation of the electron-deficient aldehydes 1f–i with acetone, affording 3f–i in 41–51% yields (entries 6–9). The aliphatic aldehyde 1j was a favourable substrate for the reaction, giving 3j in 71% yield at room temperature (entry 10). The reactions catalyzed by polymer 8 were also examined. Although the polymer 8-catalyzed reaction of 1a with acetone in ethanol at 50 °C could afford the desired product 3a in 77% yield, for practical application considerations, we developed the method without additional solvent, in which the excess acetone could be easily recycled and reused in large-scale preparations. Catalyzed by polymer 8, the reactions of substrates 1a–j with 2 at 50 °C produced 3a–j smoothly. It was very interesting that although the proline loadings were reduced to 3 mol%, the polymer 8-catalyzed Claisen–Schmidt condensations afforded the products in yields higher than those of the free L-proline-catalyzed reactions (entries 1–10). For comparison, reactions with a reduced L-proline amount (3 mol%) were also conducted, but resulted in even lower product yields (see yields inside of the brackets).
|
|||
|---|---|---|---|
| Entry | R (1) | 3: yieldb/% | |
| L-Prolinec | Polymer 8 | ||
| a For L-proline catalyzed reactions, 1 mmol of 1, 3 mmol of acetone 2, 0.03–0.05 mmol (see note c) of L-proline and 0.05 mmol of piperazine were stirred in 1 mL of EtOH at 20–100 °C (20 °C without special instruction) for 48 h; for polymer 8-catalyzed reactions, 1 mmol of 1, 30 mg of polymer 8 and 0.05 mmol of piperazine were stirred in 1 mL of acetone 2 at 50 °C for 48 h.b Isolated yields of 3 based on 1.c Yields outside the brackets are for the reactions with 5 mol% of L-proline catalyst; yields inside the brackets are for the reactions with 3 mol% of L-proline catalyst.d Reaction at 40 °C.e Reaction at 80 °C.f Reaction at 100 °C. | |||
| 1 | Ph (1a) | 3a: 70 (53) | 75 |
| 2 | 4-MeC6H4 (1b) | 3b: 40, 52d, 55 (40)e, 53f | 70 |
| 3 | 3-MeC6H4 (1c) | 3c: 53 (42)e | 70 |
| 4 | 2-MeC6H4 (1d) | 3d: 47 (40)e | 71 |
| 5 | 4-MeOC6H4 (1e) | 3e: 51 (45)e | 61 |
| 6 | 4-ClC6H4 (1f) | 3f: 42, 51 (40)e | 60 |
| 7 | 3-ClC6H4 (1g) | 3g: 48 (36)e | 50 |
| 8 | 2-ClC6H4 (1h) | 3h: 41 (33)e | 42 |
| 9 | 4-FC6H4 (1i) | 3i: 44 (34)e | 54 |
| 10 | c-C6H11 (1j) | 3j: 71 (58) | 73 |
Notably, the polymer-supported proline was tolerant to acidic functional groups in the substrates. Catalyzed by polymer-supported proline 8, 4-hydroxybenzaldehyde 1k and 3-hydroxybenzaldehyde 1l in acetone led to 3k and 3l respectively in moderate yields; the product yields decreased with L-proline as the catalyst. The same reactions could not happen when using Ca(OH)2 as the catalyst (Scheme 2).2c
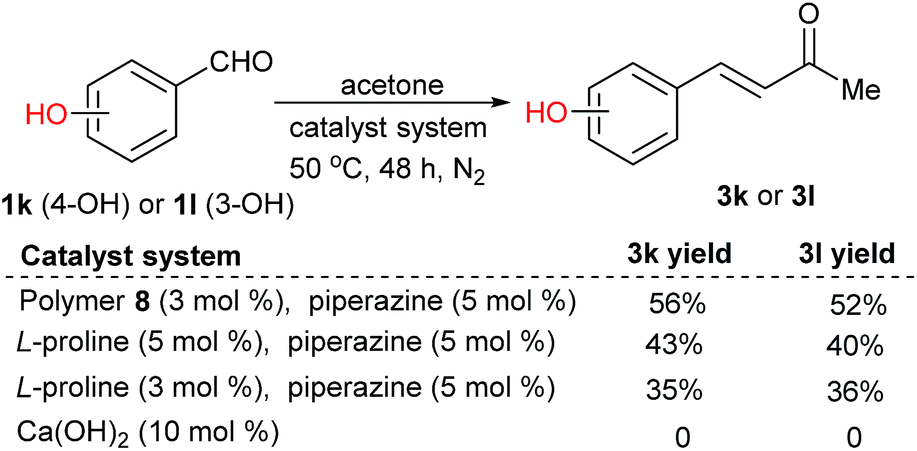 | ||
| Scheme 2 Catalyst evaluations for the reactions of a substrate with an acidic functional group. | ||
For substrate 1m, with an alkaline functional group, the polymer-supported proline-catalyzed reaction led to the desired product 3m in 50% yield, while the reaction catalyzed by free L-proline led to 3m in only 20–23% yields; the reaction could not happen with catalytic H2SO4 (Scheme 3).
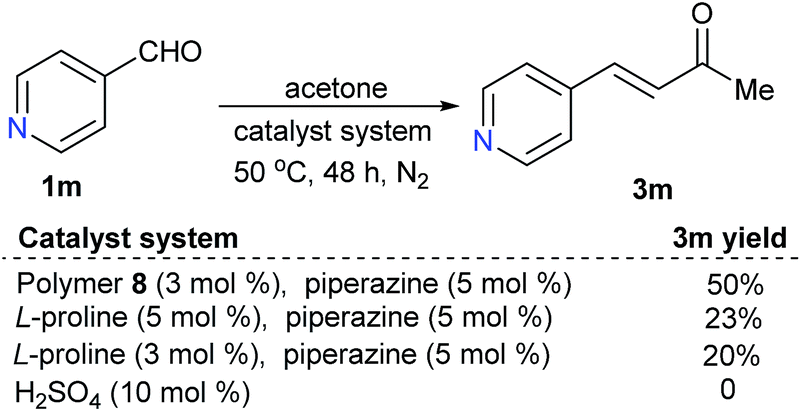 | ||
| Scheme 3 Catalyst evaluations for the reactions of a substrate with an alkaline functional group. | ||
Moreover, for substrate 1n, that contained both acidic and alkaline groups, the polymer-supported proline-catalyzed reaction led to the desired product 3n in 50% yield, which was higher than that of the reaction catalyzed by free L-proline. In contrast, neither of the acid nor base catalysts were effective for the same reaction (Scheme 4).
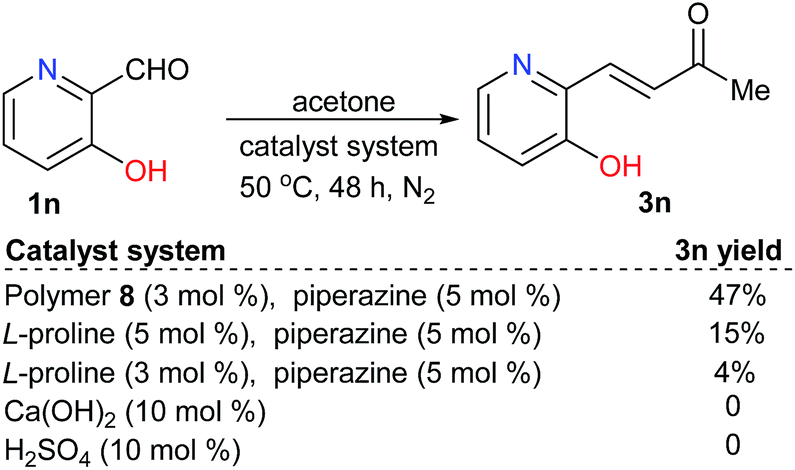 | ||
| Scheme 4 Catalyst evaluations for the reactions of a substrate with both acidic and alkaline functional groups. | ||
As a heterogeneous catalyst, polymer 8 could be easily recycled and reused in the next round of reaction. It was impressive that the catalyst was so robust that it could be reused for at least 10 times without deactivation (Fig. 1).
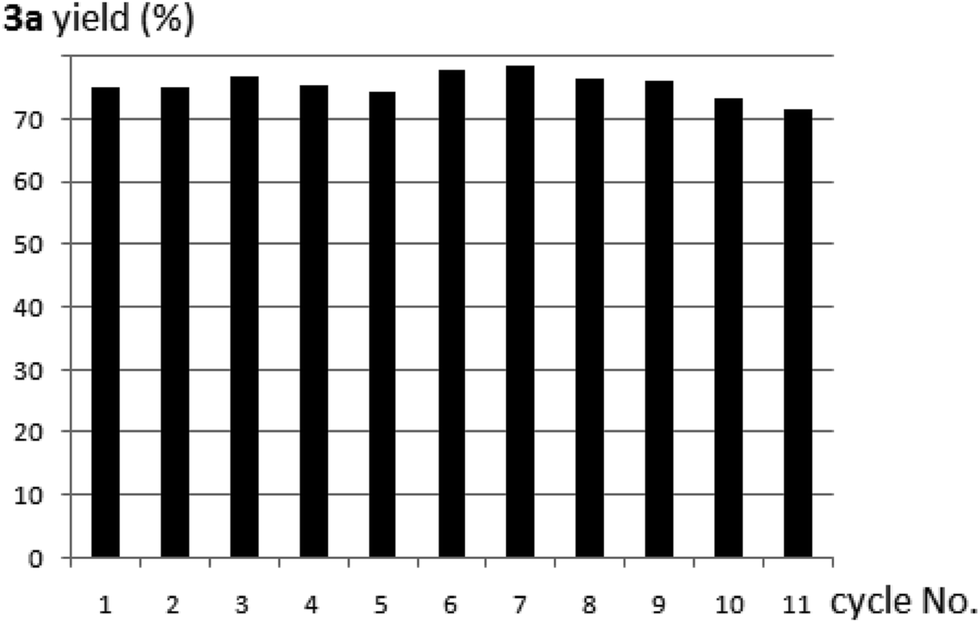 | ||
| Fig. 1 Recycle and reuse of the polymer-supported proline catalyst 8 in the reaction of 1a with acetone to produce 3a. | ||
The proline catalysts were also efficient for Knoevenagel condensations (Table 2). Reactions catalyzed by polymer 8 could even use water as the solvent, while the reactions with free L-proline as the catalyst were still performed in ethanol. Catalyzed by polymer 8, the condensation of benzaldehyde 1a with malononitrile 9 afforded 10a in 75% yield; by contrast, the same reaction catalyzed by L-proline led to 10a in only 48–50% yield (entry 1). For the electron-enriched substrate 1b, free L-proline was better than polymer 8 (entry 2). The two catalysts were similarly reactive for the electron-deficient substrate 1f, giving 10c in 68–74% and 78% yields respectively (entry 3). The heterocycle-contained substrate 1m was also favourable for the reaction to produce 10d in moderate to good yields (entry 4). Interestingly, polymer 8 was better than L-proline in the reaction of substrate 1n, which bears a bulky group (entry 5). In the reaction of the aliphatic aldehyde 1j with malononitrile, L-proline was the preferable catalyst, giving 10f in 37–50% yield (entry 6).
|
|||
|---|---|---|---|
| Entry | R (1) | 10: yieldb/% | |
| L-Prolinec | Polymer 8 | ||
| a For L-proline catalyzed reactions, 1 mmol of 1, 1 mmol of 9, 0.03–0.05 mmol of L-proline (see note c) and 0.05 mmol of piperazine were stirred in 1 mL of EtOH at 30 °C for 48 h; for polymer 8-catalyzed reactions, 1 mmol of 1, 1 mmol of 9, 30 mg of polymer 8 and 0.05 mmol of piperazine were stirred in 1 mL of water at 50 °C for 48 h.b Isolated yields of 10 based on 1.c Yields outside of the brackets are for the reactions with 5 mol% of L-proline catalyst; yields inside of the brackets are for the reactions with 3 mol% of L-proline catalyst. | |||
| 1 | Ph (1a) | 10a: 50 (48) | 75 |
| 2 | 4-MeC6H4 (1b) | 10b: 82 (71) | 70 |
| 3 | 4-ClC6H4 (1f) | 10c: 74 (68) | 78 |
| 4 | 2-C4H3S (1m) | 10d: 88 (79) | 75 |
| 5 | 2-C10H7 (1n) | 10e: 68 (60) | 83 |
| 6 | c-C6H11 (1j) | 10f: 50 (37) | 19 |
Plausible mechanisms were then proposed on the basis of the above experimental results as well as references (Scheme 5).8,9 In the Claisen–Schmidt condensations, the proline catalysts, whether small molecule or polymer, first condense with acetone to produce 11, which leads to the resonant intermediates 12a–b after dehydration.8 Furthermore, condensation of the piperazine additive with aldehyde 1 furnishes the intermediate 13,9 which reacts with 12a–b to produce the ion pair 14.8a The chemical structure of the amino acid catalyst is an important factor that facilitates the contact of the reactants.6,8b Rearrangement in 14 releases piperazine and leads to 15, which provides 16 after hydration.8a The intermediate 16 could decompose to 17 and regenerate the proline catalyst. Dehydration of 17 affords α,β-unsaturated ketones 3 as the final products.
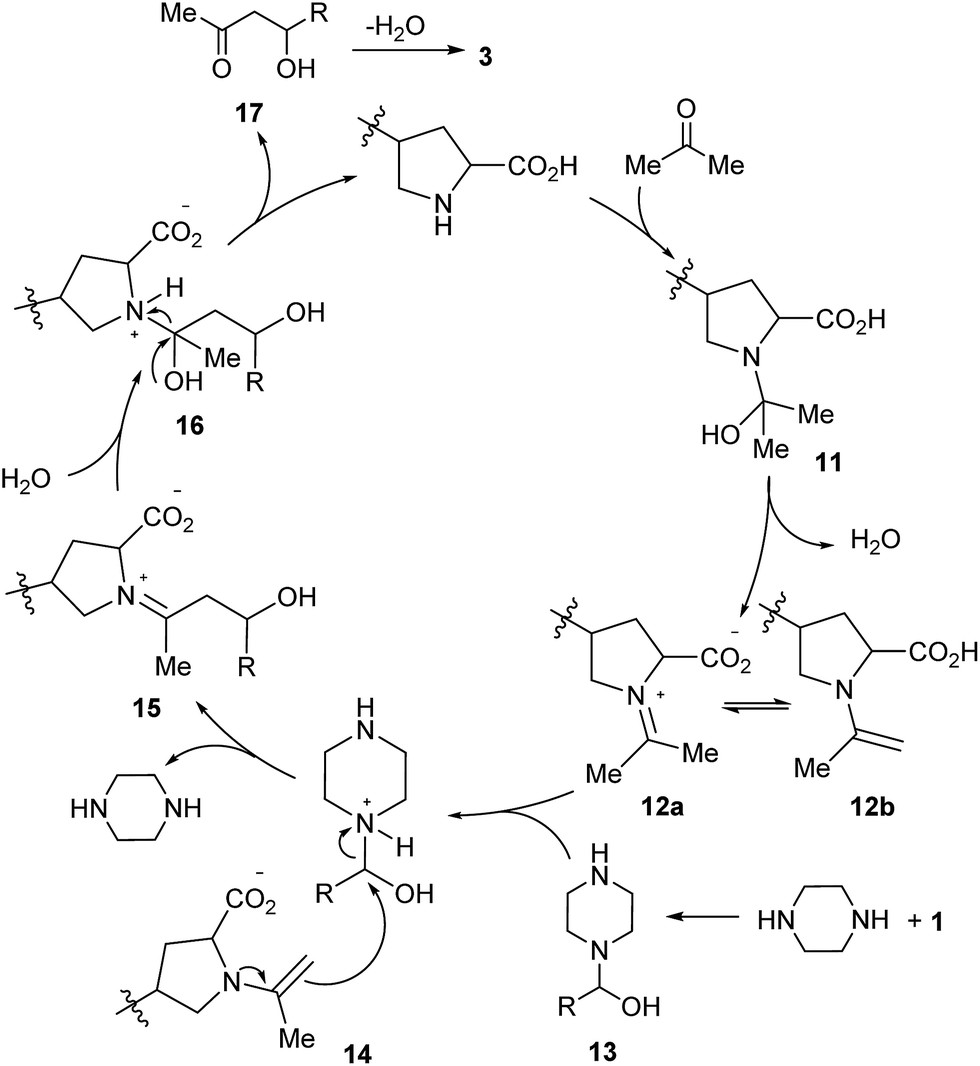 | ||
| Scheme 5 A possible mechanism for proline-catalyzed Claisen–Schmidt condensations. | ||
In 1H NMR tests, in both CDCl3 and CD3OD solvents, the chemical shifts of the aldehyde-H moved to the low field region after adding L-proline into the benzaldehyde sample, indicating interaction of the proline moieties with the reactant (Fig. 2).6,10 GC analysis experiments showed that the polymer 8 could enrich the reactants around its reaction sites (see Experimental vide infra).6
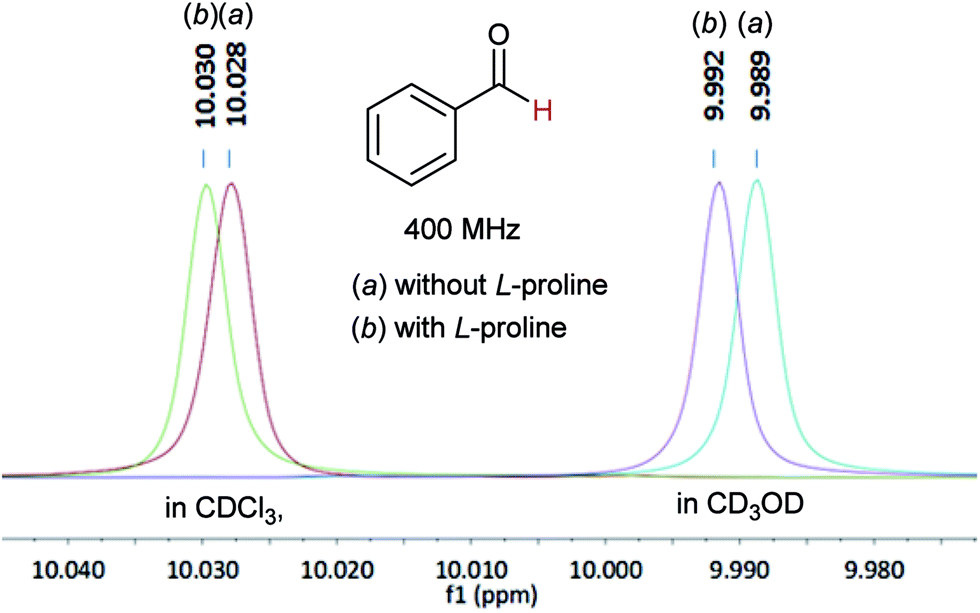 | ||
| Fig. 2 Chemical shifts of the aldehyde-H signal before and after adding L-proline in CDCl3 or CD3OD. | ||
Moreover, the control reaction of 1b with 2 at an elevated reactant concentration produced 3b in increased yield (Scheme 6). On the basis of the above experimental results, it was proposed that the reactants were enriched around the catalyst reaction sites from the solution through hydrogen bonds with the proline moieties in polymer 8 and this might have resulted in an elevated local reactant concentration, which facilitated the reaction to produce 3 in yields higher than those of the reactions catalyzed by free L-proline.
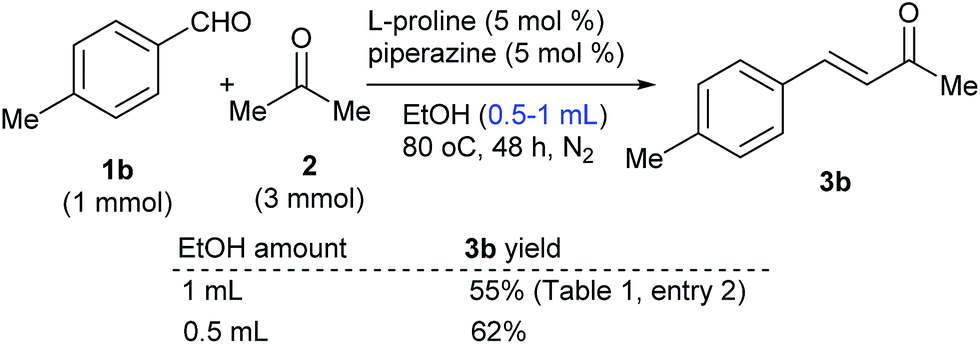 | ||
| Scheme 6 Control reactions. | ||
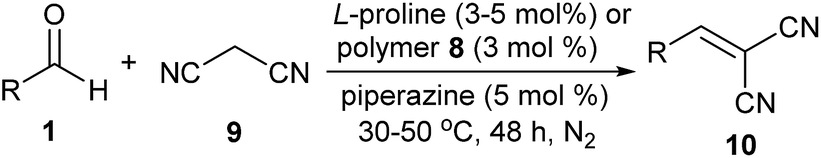
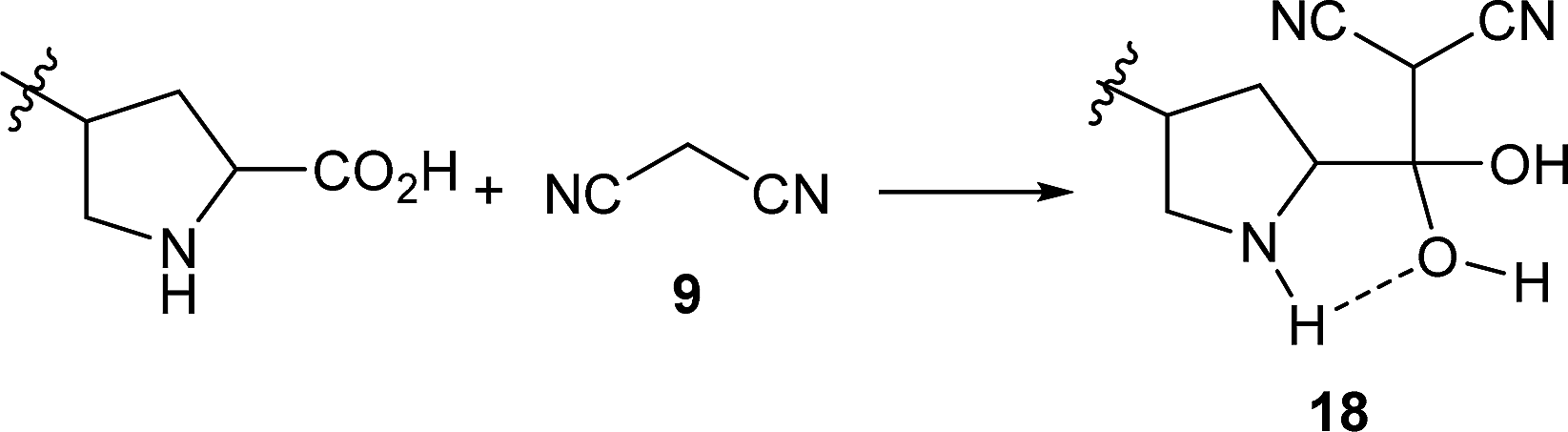
In the Knoevenagel condensations, the prolines first react with malononitrile 9 to produce the intermediate 18, which could be well stabilized by an intramolecular hydrogen bond with the adjacent pyrrolidine NH in the proline catalysts (eqn (2)).11 The reaction of 18 with the intermediate 13 leads to 19 and the regeneration of the proline catalyst as well as the piperazine additive (eqn (3)).11 Dehydration of 19 generates the final product 10 (eqn (4)). Although the mechanisms remain to be fully clarified and alternative processes may also exist due to the complex catalyst system, Scheme 5 and eqn (2)–(4) should be the most likely mechanisms based on the above experimental results as well as the related references.
 | (2) |
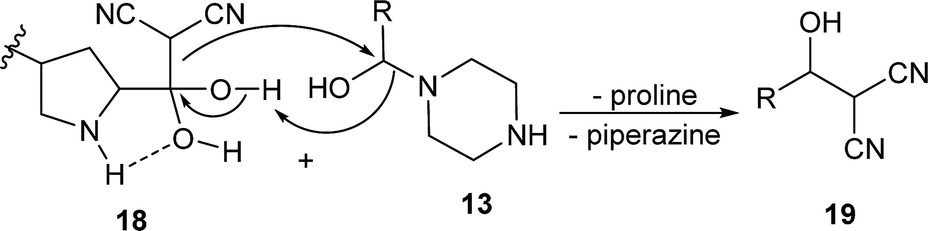 | (3) |
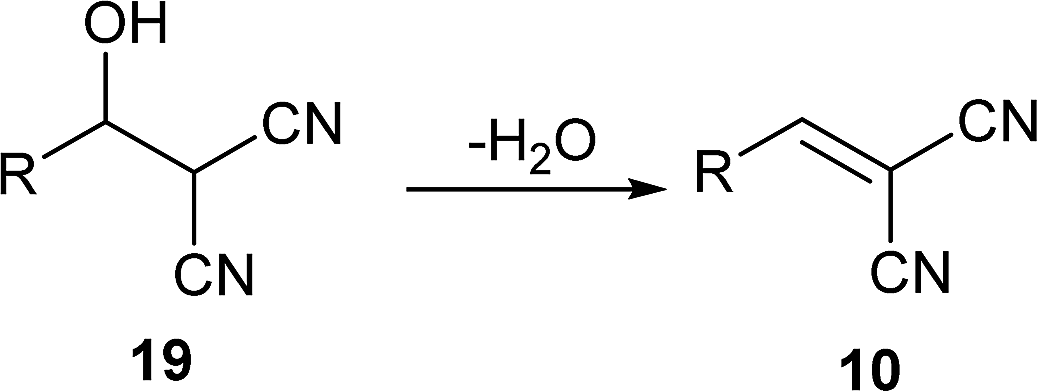 | (4) |
Conclusions
In conclusion, we reported the Claisen–Schmidt and Knoevenagel condensations catalyzed by proline catalysts. It is interesting that the polymer-supported proline catalyst is even more active than free L-proline in many cases. 1H NMR and GC analysis as well as the control experiments indicated that the proline-modified polymer might enrich the reactant around its reaction sites through hydrogen bonds and enhance the local reactant concentration to facilitate the reactions. The proline-modified polymer was impressively robust and could be recycled and reused many times without deactivation. It is also tolerant to sensitive functional groups, allowing the direct use of related substrates free of protection–deprotection steps and largely improving the synthetic efficiency.Experimental
General methods
Chemicals were purchased from reagent merchants with purities more than 98% and were directly used as received. Solvents were analytically pure (AR) and directly used without any special treatment. Melting points were measured using a WRS-2A digital instrument. IR spectra were measured on a Bruker Tensor 27 Infrared spectrometer. 1H and 13C NMR spectra were recorded on a Bruker Avance 600/400 instrument (600 or 400 MHz for 1H and 150 or 100 MHz for 13C NMR spectroscopy). Chemical shifts in the 1H NMR spectroscopy were referred to internal Me4Si (0 ppm) and J-values are shown in Hz. GC analyses were performed on an Agilent 7890B gas chromatograph with an FID detector using a J & W DB-1 column (10 m, 0.1 mm I.D.). Mass spectra were measured on a Shimadzu GCMS-QP2010 Ultra spectrometer (EI).Detailed procedure for the screening of the catalysts and additives (Table S1 in ESI†)
To a 15 mL reaction tube, 0.05 mmol of amino acid catalyst and 0.05 mmol of amine additive were added. The tube was then charged with N2 and a solution of 1 mmol of benzaldehyde 1a and 3 mmol of acetone 2 in 1 mL of ethanol was injected using a syringe. The mixture was stirred at 20 °C for 48 h. The solvent was removed by distillation under reduced pressure. The product 3a could be isolated by preparative thin layer chromatography (TLC, eluent: petroleum ether/EtOAc 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The reaction condition screenings were performed in a similar way (Table S2 in ESI†).
:1). The reaction condition screenings were performed in a similar way (Table S2 in ESI†).
Detailed procedure for the preparation of the monomer 6 (Scheme 1)
To a 250 mL three-necked flask equipped with a magnetic bar, 60 mL of CF3CO2H and 16.42 g of (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid 4 (125 mmol) were added. The mixture was stirred at 0 °C and 2 mL of CF3SO3H was added (22.9 mmol). After 5 min, 20.31 mL of acryloyl chloride 5 was injected. The mixture was stirred at room temperature for 2 h and 40 min and then cooled by ice water. 180 mL of ether was slowly added to precipitate the product 6, which was isolated by filtration and washed by ether and dried under vacuum for 12 h (yield 62%).Detailed procedure for the preparation of the polymer poly(N-isopropylacrylamide-co-L-proline) 8 (Scheme 1)
To a 50 mL Schlenk tube, 2.06 g of N-isopropylacrylamide 7 (18.2 mmol), 0.68 g of 6 (3.1 mmol), 15 mL of DMF and 52.8 mg of AIBN (0.32 mmol) were added. The mixture was heated at 70 °C under N2 protection for 8 h and cooled to room temperature and then slowly dropped into a quickly stirred 150 mL of ether. The precipitated polymer 8 was isolated by centrifuge and dissolved in water. The liquid pH was adjusted using Et3N to 6–7 and re-precipitated using 150 mL of ether. The polymer 8 was washed using methanol and ether and dried under vacuum at 50 °C overnight.Typical procedure for the Claisen–Schmidt condensations catalyzed by L-proline (Table 1)
To a 15 mL reaction tube, 5.8 mg of L-proline (0.05 mmol) and 4.3 mg of piperazine (0.05 mmol) were added. The tube was then charged with N2 and a solution of 1 mmol of aldehyde 1 and 3 mmol of acetone in 1 mL of ethanol was injected using a syringe. The mixture was stirred at the related temperature (see Table 2 in text) for 48 h. The solvent was removed by distillation under reduced pressure. The residue could be isolated by preparative TLC (eluent: petroleum ether/EtOAc 10:1).
Typical procedure for the Claisen–Schmidt condensations catalyzed by polymer 8 (Table 1)
To a 15 mL reaction tube, 30 mg of polymer 8 (containing 0.03 mmol of proline moiety) and 4.3 mg of piperazine (0.05 mmol) were added. The tube was then charged with N2 and a solution of 1 mmol of aldehyde 1 in 1 mL of acetone was injected using a syringe. The mixture was stirred at 50 °C for 48 h. The solvent was removed by distillation under reduced pressure. The residue could be isolated by preparative TLC (eluent: petroleum ether/EtOAc 10:1).
Detailed procedure for the catalyst recycle and reuse in the Claisen–Schmidt condensation of 1a with acetone catalyzed by polymer 8 (Fig. 1)
To a 35 mL reaction tube, 0.15 g of polymer 8 (containing 0.15 mmol of proline moiety) and 21.5 mg of piperazine (0.25 mmol) were added. The tube was then charged with N2 and a solution of 5 mmol of 1a in 3 mL of acetone was injected using a syringe. The mixture was stirred at 50 °C for 48 h and cooled to room temperature. Petroleum ether was added to precipitate the polymer 8. After standing for 2 h, the liquid was removed and the polymer 8 was washed with petroleum ether. The solvent of the combined organic solutions was removed by distillation under reduced pressure and the residue was subjected to preparative TLC to produce 3a (eluent: petroleum ether/EtOAc 10:1). The recycled polymer 8 was dried under vacuum at 50 °C for 24 h and could be reused as a catalyst in the next round of reaction.
Typical procedure for the Knoevenagel condensations catalyzed by L-proline (Table 2)
To a 15 mL reaction tube, 5.8 mg of L-proline (0.05 mmol) and 4.3 mg of piperazine (0.05 mmol) were added. The tube was then charged with N2 and a solution of 1 mmol of aldehyde 1 and 1 mmol of malononitrile 9 in 1 mL of ethanol was injected using a syringe. The mixture was stirred at 30 °C for 48 h. The solvent was removed by distillation under reduced pressure. The residue could be purified by preparative TLC (eluent: petroleum ether/EtOAc 10:1).
Typical procedure for the Knoevenagel condensations catalyzed by polymer 8 (Table 2)
To a 15 mL reaction tube, 30 mg of polymer 8 (containing 0.03 mmol of proline moiety) and 4.3 mg of piperazine (0.05 mmol) were added. The tube was then charged with N2 and a suspension of 1 mmol of aldehyde 1 and 1 mmol of malononitrile 9 in 1 mL of water was injected using a syringe. The mixture was stirred at 50 °C for 48 h. The products could be extracted using ether (3 × 1 mL) and purified by preparative TLC (eluent: petroleum ether/EtOAc 10:1).
Detailed procedures of the polymer absorption tests for the mechanism studies
1 mmol of freshly distilled benzaldehyde was dissolved in 5 mL of absolute ethanol in a tube. The supernatant liquid sample was sent for GC analysis three times. The values of the benzaldehyde peak area divided by the ethanol peak area were calculated to be 0.05009, 0.05009 and 0.05008 (average value: 0.05009). 10 mg of the polymer-supported proline 8 was then added and the mixture was left overnight under N2 protection. The polymer precipitated at the bottom of the tube. The supernatant liquid was sent for GC analysis again. The analysis results indicated that only benzaldehyde and ethanol were detected while no new compound was generated. The values of the benzaldehyde peak area divided by the ethanol peak area were calculated to be 0.04539, 0.04470 and 0.04479 for the three parallel experiments respectively (average value: 0.04496). The experimental results indicated that after adding the polymer 8, the concentration of the reactant had fallen by 10.2%. Since GC-analysis showed that no chemical reaction occurred during the process, the loss of reactant might be attributed to adsorption around the catalyst reaction sites of the polymer 8. The detailed GC data and spectra are given in the ESI.†Characterization of the products
C–H + 2Ar-H), 7.36 (d, J = 8.4 Hz, 2H), 6.68 (d, J = 16.2 Hz, 1H), 2.37 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 198.0, 141.9, 136.4, 133.0, 129.4, 129.3, 127.5, 27.7; MS (EI, 70 eV): m/z (%) 181 (8) [M+ + 1], 180 (27) [M+], 165 (100), 102 (53), 137 (50); known compound.2cConflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NNSFC (21202141), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions, the High Level Talent Support Project of Yangzhou University, the Open Project Program of Jiangsu Key Laboratory of Zoonosis (R1509) and the testing centre of Yangzhou University.Notes and references
- (a) M. Paraja and C. Valdes, Org. Lett., 2017, 19, 2034 CrossRef CAS PubMed; (b) B. Huang, Y. Shen, Z. Mao, Y. Liu and S. Cui, Org. Lett., 2016, 18, 4888 CrossRef CAS PubMed; (c) Y. Gao, W. Xiong, H. Chen, W. Wu, J. Peng, Y. Gao and H. Jiang, J. Org. Chem., 2015, 80, 7456 CrossRef CAS PubMed; (d) T. Nishikata, K. Nakamura, K. Itonaga and S. Ishikawa, Org. Lett., 2014, 16, 5816 CrossRef CAS PubMed.
- (a) H. Wang, C. Wang, Y. Yang, M. Zhao and Y. Wang, Catal. Sci. Technol., 2017, 7, 405 RSC; (b) C. Winter, J. N. Caetano, A. B. C. Araújo, A. R. Chaves, I. C. Ostroski, B. G. Vaz and C. N. Pérez, Chem. Eng. J., 2016, 303, 604 CrossRef CAS; (c) L. Yu, M. Han, J. Luan, L. Xu, Y. Ding and Q. Xu, Sci. Rep., 2016, 6, 30432 CrossRef PubMed; (d) L. Zhang, H. Wang, W. Shen, Z. Qin, J. Wang and W. Fan, J. Catal., 2016, 344, 293 CrossRef CAS.
- (a) J. Liu and L. Wang, Synthesis, 2017, 49, 960 CAS; (b) P. Kumar and N. Dwivedi, Acc. Chem. Res., 2013, 46, 289 CrossRef CAS PubMed; (c) M. Gruttadauria, F. Giacalone and R. Noto, Chem. Soc. Rev., 2008, 37, 1666 RSC.
- (a) A. Obregón-Zúñiga, M. Milán and E. Juaristi, Org. Lett., 2017, 19, 1108 CrossRef PubMed; (b) M. Bhati, S. Upadhyay and S. Easwar, Eur. J. Org. Chem., 2017, 1788 CrossRef CAS; (c) H. Jin, S. T. Kim, G.-S. Hwang and D. H. Ryu, J. Org. Chem., 2016, 81, 3263 CrossRef CAS PubMed; (d) L. Li and B. G. Janesko, J. Org. Chem., 2016, 81, 10802 CrossRef CAS PubMed; (e) S. Lai, W. Mao, H. Song, L. Xia and H. Xie, New J. Chem., 2016, 40, 8194 RSC; (f) J. Safaei-Ghomi and S. Zahedi, Appl. Organomet. Chem., 2015, 29, 566 CrossRef CAS; (g) T. E. Kristensen, K. Vestli, K. A. Fredriksen, F. K. Hansen and T. Hansen, Org. Lett., 2009, 11, 2968 CrossRef CAS PubMed.
- For selected articles, please see: (a) T. Wang, X. Jing, C. Chen and L. Yu, J. Org. Chem., 2017, 82, 9342 CrossRef CAS PubMed; (b) X. Jing, D. Yuan and L. Yu, Adv. Synth. Catal., 2017, 359, 1194 CrossRef CAS; (c) Y. Wang, L. Yu, B. Zhu and L. Yu, J. Mater. Chem. A, 2016, 4, 10828 RSC; (d) L. Yu, F. Chen and Y. Ding, ChemCatChem, 2016, 8, 1033 CrossRef CAS; (e) L. Yu, Z. Bai, X. Zhang, X. Zhang, Y. Ding and Q. Xu, Catal. Sci. Technol., 2016, 6, 1804 RSC; (f) X. Zhang, J. Sun, Y. Ding and L. Yu, Org. Lett., 2015, 17, 5840 CrossRef CAS PubMed; (g) X. Zhang, J. Ye, L. Yu, X. Shi, M. Zhang, Q. Xu and M. Lautens, Adv. Synth. Catal., 2015, 357, 955 CrossRef CAS; (h) L. Yu, J. Ye, X. Zhang, Y. Ding and Q. Xu, Catal. Sci. Technol., 2015, 5, 4830 RSC; (i) L. Yu, H. Li, X. Zhang, J. Ye, J. Liu, Q. Xu and M. Lautens, Org. Lett., 2014, 16, 1346 CrossRef CAS PubMed; (j) L. Yu, Y. Wu, H. Cao, X. Zhang, X. Shi, J. Luan, T. Chen, Y. Pan and Q. Xu, Green Chem., 2014, 16, 287 RSC.
- For details, please see ESI.†.
- L. Xu, J. Huang, M. Zhang, L. Yu and Y. Fan, ChemistrySelect, 2016, 1, 1933 CrossRef CAS.
- (a) L. Xu, J. Huang, Y. Liu, Y. Wang, B. Xu, K. Ding, Y. Ding, Q. Xu, L. Yu and Y. Fan, RSC Adv., 2015, 5, 42178 RSC; (b) L. Xu, F. Wang, J. Huang, C. Yang, L. Yu and Y. Fan, Tetrahedron, 2016, 72, 4076 CrossRef CAS and references cited therein.
- I.-H. Bhat and S. Tabassum, Spectrochim. Acta, Part A, 2009, 72, 1026 CrossRef PubMed.
- X. Han, Y. Wang, X. Gai and X. Zeng, Synlett, 2015, 26, 2858 CrossRef CAS.
- T.-X. Metro, J. Bonnamour, T. Reidon, A. Duprez, J. Sarpoulet, J. Martinez and F. Lamaty, Chem.– Eur. J., 2015, 21, 12787 CrossRef CAS PubMed , In this article, the reaction of malononitrile with carboxylic acid generated 2-hydroxymethylene malononitrile derivative 20, which was the dehydration product of 2-dihydroxymethyl malononitrile 18׳
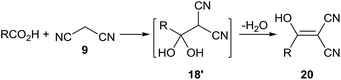 . Considering the adjacent pyrrolidine NH in the proline moiety that could lead to a hydrogen bond to form a stable ring, the intermediate 18 might exist and react with 13 before dehydration.
. Considering the adjacent pyrrolidine NH in the proline moiety that could lead to a hydrogen bond to form a stable ring, the intermediate 18 might exist and react with 13 before dehydration. - (a) I. M. Heilbron and R. Hill, J. Chem. Soc., 1928, 2863 RSC; (b) A. K. Chakraborti, L. Sharma and M. K. Nayak, J. Org. Chem., 2002, 67, 6406 CrossRef CAS PubMed; (c) D. Fattori, C. Rossi, C. I. Fincham, M. Berettoni, F. Calvani, F. Catrambone, P. Felicetti, M. Gensini, R. Terracciano, M. Altamura, A. Bressan, S. Giuliani, C. A. Maggi, S. Meini, C. Valenti and L. Quartara, J. Med. Chem., 2006, 49, 3602 CrossRef CAS PubMed; (d) M. Winter, F. Gautschi, I. Flament and M. Stoll, US Pat., 4,018,910, 1974; (e) J. Boyle, A. E. Fenwick, D. M. Gethin and C. F. Mccusker, US Pat., 20,080,267,942, 2008; (f) See webpage of the SDBS: http://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi; (g) M. Hosseini-Sarvari, H. Sharghi and S. Etemad, Chin. J. Chem., 2007, 25, 1563 CrossRef CAS; (h) J.-Y. Liu, Y.-J. Jang, W.-W. Lin, J.-T. Liu and C.-F. Yao, J. Org. Chem., 2003, 68, 4030 CrossRef CAS PubMed; (i) C. Wiles, P. Watts and S. J. Haswell, Lab Chip, 2007, 7, 322 RSC; (j) K. Yamashita, T. Tanaka and M. Hayashi, Tetrahedron, 2005, 61, 7981 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Condition optimization table, mechanism study details, product characterization, and 1H and 13C NMR spectra of the products. See DOI: 10.1039/c7ra09412d |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |