
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient and straightforward click synthesis of structurally related dendritic triazoles†
M. I. Mangione *,
R. A. Spanevello and
M. B. Anzardi
*,
R. A. Spanevello and
M. B. Anzardi
Instituto de Química Rosario, Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario – CONICET, Suipacha 531, S2002RLK Rosario, Argentina. E-mail: mangione@iquir-conicet.gov.ar
First published on 11th October 2017
Abstract
A simple, rapid and efficient copper-catalyzed 1,3-dipolar cycloaddition reaction is described for the synthesis of a novel family of twelve triazolic dendrimers structurally related. The products were the result of the click reaction of three cores and four different azides in tetrahydrofuran applying a homogeneous copper catalysis. The reaction intermediates and products were obtained in very good to excellent yields using straightforward and simple work-up procedures. This new family of compounds contain electroactive moieties such as carbazole and triphenylamine which may turn them into excellent candidates for the development of optoelectronic organic materials.
Introduction
“The act of simplifying” is probably the idea which is the essence of the “Click chemistry” paradigmatic concept, introduced by Sharpless and co-workers more than a decade ago.1 The main criterion that identified this click philosophy is the application of modular and orthogonal processes that allows to obtain in a simple and efficient manner the desired compound. In order to be consider a click-type reaction, the procedure should be wide in scope, high yielding, easy to purify, stereospecific, simple to perform and can be done in benign and/or easily removable solvents.2 This type of chemistry rapidly found a widespread application in several research areas such as dendrimers, drug discovery, biochemistry and chemical modifications of surfaces and nanostructures.3 Probably, materials and polymers sciences are areas in which this concept has found a broad implementation holding the promise to develop unprecedented functional materials for organic optoelectronic applications.Among the click reactions that are implemented in material synthesis and functionalization, the copper-catalyzed azide–alkyne [3 + 2] cycloadditions (CuAAC) became the prevalent click procedure, often being referred to as the “click reaction”.
In 2002, Meldal and Sharpless' groups independently reported a copper(I) catalyzed 1,3-dipolar cycloaddition of azides and terminal alkynes (CuAAC) that were conducted at low temperatures with high rates, efficiency and regiospecificity.4 This significant event, not only improved the understanding of 1,3-dipolar cycloaddition studied by Huisgen in the 1960's,5 but also generated an explosive growth of click chemistry as a valuable concept in materials science. The replacement of the original method for one that uses simple and robust chemistry, avoiding harsh conditions, rapidly attracted the interest of the community of scientists belonging to the field of macromolecular and material science. After the first implementation in 2004 of the copper(I) catalyzed reaction in dendrimer synthesis,6 CuAAC has been widely used to design and prepare polymeric structures with macromolecular architectures which proved to be very difficult or almost impossible to get before. Dendrimer synthesis often requires excess of reactants, high cost catalysts and tiresome chromatographic separations with the generation of considerable waste. The possibility of high efficiency and easy purification without excess of reactants make CuAAC very attractive for the synthesis of dendrimers and after 2010 several review articles describe the application of CuAAC in dendrimers synthesis or functionalization.7
Triazole unit is a non-natural heterocycle and its origin is from chemical synthesis and the copper-catalyzed 1,3-dipolar cycloaddition is an orthogonal coupling reaction which is widely tolerated for several functional groups that allowed to obtain this type of ring.8 This scaffold has been widely used in many areas such as biology, drug design, supramolecular chemistry, nano-optoelectronics, macromolecular and materials sciences. Particularly in the last two applications, the triazole ring acts simply as linkage.9 In organometallics chemistry, N(2) and N(3) of the triazole can coordinate with different metals when the macromolecules contain some spatially closed triazole rings.10 Many new materials have been successfully obtained by CuAAC, but the most intriguingly aspect is the nature and the function of the 1,2,3-triazole structure within the resulting material properties. This aromatic ring often affects the properties of the molecules to which it belongs. For example, triazole π-system provides aromatic character for monomers, conjugated polymers and fluorescent probes.11
On the other hand, the triarylamine core has been widely used in the development of electro-optical materials such as organic light emitting diodes (OLEDs), organic field-effect transistors (OFETs), non-linear materials and xerography, due to its good electron donating and transporting capabilities. Furthermore when it is used as core gives rise to star-shape molecular architectures.12
Hence, the electronic characteristics of triarylamines combined with the interesting properties that the triazole moiety can confer to polymers and dendrimers, such as solubility, swelling, metal adhesion, macromolecular architecture, among others, makes the CuAAC reaction a convenient tool for the development of novel dendritic organic materials.
Herein, we describe the design and synthesis of structurally related families of triazole derived dendrimers (Fig. 1). These families of compounds were efficiently synthesized in high yields, with no need for chromatographic separations. The new set of macromolecules with electroactive moieties such as triphenylamine and carbazole are promising targets for optoelectronic applications.
 | ||
| Fig. 1 Cores and dendrons of triazolic dendrimers. | ||
Experimental
General considerations
Melting points were taken on a Leitz Wetzlar Microscope Heating Stage, Model 350 apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on Bruker Avance-300 spectrometer with Me4Si as the internal standard and dimethylsulfoxide-d6 or chloroform-d as solvents. Abbreviations: s = singlet, d = doublet, t = triplet, and m = multiplet expected but not resolved. Mass spectra of dendrons were recorded in a Shimadzu QP2010 Plus instrument, ion source temperature = 300 °C, and detector voltage = 70 kV. High resolution mass spectra of the dendrimers were analyzed by electrospray ionization (ESI) using a Bruker micrOTOF-Q II instrument, positive ion polarity or by ultraviolet matrix assisted laser desorption-ionization mass spectrometry (UV-MALDI MS) and by ultraviolet laser desorption-ionization mass spectrometry (UV-LDI MS) performed on the Bruker Ultraflex Daltonics TOF/TOF mass spectrometer. Mass spectra were acquired in linear positive and negative ion modes. Stock solutions of samples were prepared in chloroform. External mass calibration was made using β-cyclodextrin (MW 1134) with nHo as matrix in positive and negative ion mode. Sample solutions were spotted on a MTP 384 target plate polished steel from Bruker Daltonics (Leipzig, Germany). For UV-MALDI MS matrix solution was prepared by dissolving GA (gentisic acid, 1 mg mL−1) in water and dry droplet sample preparation was used according to Nonami et al.13 loading successively 0.5 μL of matrix solution, analyte solution and matrix solution after drying each layer at normal atmosphere and room temperature. For UV-LDI MS experiments two portions of analyte solution (0.5 μL × 2) were loaded on the probe and dried successively (two dry layers). Desorption/ionization was obtained by using the frequency-tripled Nd:YAG laser (355 nm). The laser power was adjusted to obtain high signal-to-noise ratio (S/N) while ensuring minimal fragmentation of the parent ions and each mass spectrum was generated by averaging 100 lasers pulses per spot. Spectra were obtained and analyzed with the programs FlexControl and FlexAnalysis, respectively. Reactions were monitored by TLC on 0.25 mm E. Merck Silica Gel Plates (60F254), using UV light (254 nm) and phosphomolybdic acid as developing agent. Flash column chromatographies using E. Merck silica gel 60H were performed by gradient elution of mixture of n-hexane and increasing volumes of dichloromethane or ethyl acetate. Reactions were run under an argon atmosphere with freshly anhydrous distilled solvents, unless otherwise noted. Yields refer to chromatographically and spectroscopically homogeneous materials, unless otherwise stated.General procedure for the synthesis of the triazolic dendrimers are described, the synthetic protocols for the preparation of cores 4, 5 and 6 and dendrons 7, 8, 9 and 10 are fully described in the ESI.†
General procedure for the synthesis of the dendrimers 1a–d, 2a–d and 3a–d
Core (0.12 mmol) was dissolved in anhydrous tetrahydrofuran (4 mM) under Ar atmosphere. Azide (3.6 equiv.), Cu(PPh3)3Br (21% mole) and N,N-di-isopropylethylamine (3 equiv.) were added and the reaction was refluxed. The completion of the reaction was controlled by TLC until consumption of the cores. Solvent was evaporated and the resulting crude product suspended in ethanol. The solid was filtered, washed with ethanol and suspended in methyl-tert-butyl ether. Solid was filtered again and dried affording pure dendrimer as coloured solid. Numbering of each structure is arbitrary and is described in the ESI.†![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1507 (CC), 1234, 1043, 756 (Ph), 685 (Ph) cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 7.05 (6H, dd, J1 = J2 = 7.8 Hz, 7-H), 7.53 (3H, dd, J = 7.4 Hz, 8-H), 8.02 6H, (6H, d, J = 8.2 Hz, 6-H), 8.57 (3H, s, 2-H), 9.49 (3H, s, 4-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 120.0 (C-6), 120.3 (C-4), 122.0 (C-2), 128.8 (C-8), 130.0 (C-7), 131.8 (C-1), 136.6 (C-5), 146.9 (C-3). MALDI-TOF (m/z): calcd for C30H21N9Na+ 530.2 (M + Na+): obsd, 530.1.N), 1450 (CC), 1227, 1036, 748 (Ph), 721 (Ph) cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 7.09 (12H, d, J = 8.3 Hz, 10-H), 7.12 (6H, d, J = 7.4 Hz, 12-H), 7.14 (6H, d, J = 9.0 Hz, 6-H), 7.35 (12H, dd, J = 8.3, J = 7.4 Hz, 11-H), 7.87 (6H, d, J = 8.9 Hz, 7-H), 8.51 (3H, s, 2-H), 9.36 (3H, s, 4-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 119.9 (C-4), 121.4 (C-7), 121.8 (C-2), 122.9 (C-6), 123.8 (C-12), 124.6 (C-10), 129.8 (C-11), 130.7 (C-5), 131.8 (C-1), 146.6 (C-3), 146.7 (C-9), 147.7 (C-8). MALDI-TOF (m/z): calcd for C66H42N12H+ 1009.4 (M + H+): obsd, 1009.9.N), 1514 (CC), 1487, 1271, 1036, 752 (Ph), 649 (Ph) cm−1. H-NMR (300 MHz, DMSO-d6): δ = 7.33 (6H, ddd, J = 7.7, J = 2.1 Hz, 12-H), 7.46–7.50 (12H, m, 10-H, 11-H), 7.95 (6H, d, J = 8.7 Hz, 6-H), 8.28 (6H, d, J = 9.7 Hz, 13-H), 8.35 (6H, d, J = 8.6 Hz, 7-H), 8.68 (3H, s, 2-H), 9.70 (3H, s, 4-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 109.7 (C-10), 120.4 (C-12), 120.4 (C-4), 120.6 (C-13), 121.7 (C-7), 122.1 (C-2), 122.9 (C-14), 126.4 (C-11), 128.2 (C-6), 131.8 (C-1), 135.4 (C-8), 137.0 (C-5), 140.0 (C-9), 147.0 (C-3). MALDI-TOF (m/z): calcd for C66H42N12Na+ 1025.4 (M + Na+): obsd, 1025.9.N), 1489 (CC), 1364, 1261, 1036, 810 (Ph) cm−1. H-NMR (300 MHz, CDCl3): δ = 1.47 (54H, s, CH3), 7.41 (6H, d, J = 8.5 Hz, 10-H), 7.48 (6H, dd, J = 8.7, J = 1.9 Hz, 11-H), 7.79 (6H, d, J = 8.8 Hz, 6-H), 8.06 (6H, d, J = 8.8 Hz, 7-H), 8.16 (6H, d, J = 1.4 Hz, 13-H), 8.56 (3H, s, 4-H vinylic), 8.58 (3H, s, 2-H). 13C-NMR (75 MHz, CDCl3): δ = 32.0 (CH3), 34.8 (
N), 1507 (CC), 1234, 1043, 756 (Ph), 685 (Ph) cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 7.05 (6H, dd, J1 = J2 = 7.8 Hz, 7-H), 7.53 (3H, dd, J = 7.4 Hz, 8-H), 8.02 6H, (6H, d, J = 8.2 Hz, 6-H), 8.57 (3H, s, 2-H), 9.49 (3H, s, 4-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 120.0 (C-6), 120.3 (C-4), 122.0 (C-2), 128.8 (C-8), 130.0 (C-7), 131.8 (C-1), 136.6 (C-5), 146.9 (C-3). MALDI-TOF (m/z): calcd for C30H21N9Na+ 530.2 (M + Na+): obsd, 530.1.N), 1450 (CC), 1227, 1036, 748 (Ph), 721 (Ph) cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 7.09 (12H, d, J = 8.3 Hz, 10-H), 7.12 (6H, d, J = 7.4 Hz, 12-H), 7.14 (6H, d, J = 9.0 Hz, 6-H), 7.35 (12H, dd, J = 8.3, J = 7.4 Hz, 11-H), 7.87 (6H, d, J = 8.9 Hz, 7-H), 8.51 (3H, s, 2-H), 9.36 (3H, s, 4-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 119.9 (C-4), 121.4 (C-7), 121.8 (C-2), 122.9 (C-6), 123.8 (C-12), 124.6 (C-10), 129.8 (C-11), 130.7 (C-5), 131.8 (C-1), 146.6 (C-3), 146.7 (C-9), 147.7 (C-8). MALDI-TOF (m/z): calcd for C66H42N12H+ 1009.4 (M + H+): obsd, 1009.9.N), 1514 (CC), 1487, 1271, 1036, 752 (Ph), 649 (Ph) cm−1. H-NMR (300 MHz, DMSO-d6): δ = 7.33 (6H, ddd, J = 7.7, J = 2.1 Hz, 12-H), 7.46–7.50 (12H, m, 10-H, 11-H), 7.95 (6H, d, J = 8.7 Hz, 6-H), 8.28 (6H, d, J = 9.7 Hz, 13-H), 8.35 (6H, d, J = 8.6 Hz, 7-H), 8.68 (3H, s, 2-H), 9.70 (3H, s, 4-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 109.7 (C-10), 120.4 (C-12), 120.4 (C-4), 120.6 (C-13), 121.7 (C-7), 122.1 (C-2), 122.9 (C-14), 126.4 (C-11), 128.2 (C-6), 131.8 (C-1), 135.4 (C-8), 137.0 (C-5), 140.0 (C-9), 147.0 (C-3). MALDI-TOF (m/z): calcd for C66H42N12Na+ 1025.4 (M + Na+): obsd, 1025.9.N), 1489 (CC), 1364, 1261, 1036, 810 (Ph) cm−1. H-NMR (300 MHz, CDCl3): δ = 1.47 (54H, s, CH3), 7.41 (6H, d, J = 8.5 Hz, 10-H), 7.48 (6H, dd, J = 8.7, J = 1.9 Hz, 11-H), 7.79 (6H, d, J = 8.8 Hz, 6-H), 8.06 (6H, d, J = 8.8 Hz, 7-H), 8.16 (6H, d, J = 1.4 Hz, 13-H), 8.56 (3H, s, 4-H vinylic), 8.58 (3H, s, 2-H). 13C-NMR (75 MHz, CDCl3): δ = 32.0 (CH3), 34.8 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) Me3), 109.0 (C-10), 116.4 (C-13), 118.4 (C-4), 121.8 (C-7), 122.8 (C-2), 123.7 (C-14), 123.9 (C-11), 127.7 (C-6), 131.6 (C-1), 135.0 (C-8), 138.8 (C-9), 138.9 (C-5), 143.5 (C-12), 147.8 (C-3). MALDI-TOF (m/z): calcd for C90H90N12+ 1338.7 (M+): obsd, 1338.4.N, CC), 1040, 758 (Ph) cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 7.22 (6H, d, J = 8.6 Hz, 2-H), 7.50 (3H, dd, J = 7.4 Hz, 10-H), 7.63 (6H, dd, J = 8.3, J = 7.5 Hz, 9-H), 7.91 (6H, d, J = 8.6 Hz, 3-H), 7.95 (6H, d, J = 8.0 Hz, 8-H), 9.25 (3H, s, 6-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 119.0 (C-6), 119.9 (C-8), 124.2 (C-2), 125.2 (C-4), 126.7 (C-3), 128.7 (C-10), 129.9 (C-9), 136.6 (C-7), 146.7 (C-1), 147.1 (C-5). MALDI-TOF (m/z): calcd for C42H30N10+ 674.3 (M+): obsd, 674.1.N), 1491 (CC), 1227, 1032, 835 (Ph), 748 (Ph) cm−1. H-NMR (300 MHz, DMSO-d6): δ = 7.05–7.09 (18H, m, 12-H, 14-H), 7.11 (6H, d, J = 9.0 Hz, 9-H), 7.18 (6H, d, J = 8.1 Hz, 2-H), 7.34 (12H, dd, J = 7.8 Hz, J = 7.4 Hz, 13-H), 7.81 (6H, d, J = 8.6 Hz, 8-H), 7.88 (6H, d, J = 8.2 Hz, 3-H), 9.13 (3H, s, 6-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 118.8 (C-6), 121.3 (C-8), 123.0 (C-14), 123.8 (C-9), 124.5 (C-2) 124.5 (C-12), 125.2 (C-4), 126.7 (C-3), 129.7 (C-13), 130.8 (C-7), 146.6 (C-1), 146.7 (C-5), 146.7 (C-11), 147.6 (C-10). ESI (m/z): calcd for C78H58N13H+ 1176.4933 (M + H+): obsd, 1176.4925.N), 1489 (CC), 1281, 1032, 835 (Ph), 752 (Ph), 694 (Ph) cm−1. H-NMR (300 MHz, DMSO-d6): δ = 7.25–7.35 (12H, m, 2-H, 13-H), 7.46 (12H, bd, J = 3.6 Hz, 8-H, 12-H), 7.91 (6H, d, J = 8.6 Hz, 15-H), 7.97 (6H, d, J = 8.5 Hz, 3-H), 8.26 (12H, bd, J = 8.1 Hz, 9-H, 14-H), 9.42 (3H, s, 6-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 109.7 (C-12), 119.2 (C-6), 120.4 (C-7), 120.6 (C-13), 121.5 (C-14), 122.9 (C-8), 124.3 (C-2), 125.1 (C-4), 126.4 (C-9), 126.8 (C-3), 128.2 (C-15), 135.4 (C-11), 136.9 (C-10), 140.0 (C-16), 146.7 (C-1), 147.3 (C-5). ESI (m/z): calcd for C78H52N13+ 1170.4463 (M + H+): obsd, 1170.4460.N), 1489 (CC), 1364, 1261, 1032, 810 (Ph) cm−1. 1H-NMR (300 MHz, CDCl3): δ = 1.48 (54H, s, CH3), 7.32 (6H, d, 2-H), 7.40 (6H, d, J = 8.6 Hz, 12-H), 7.50 (6H, dd, J = 8.7, J = 1.9 Hz, 13-H), 7.77 (6H, d, J = 8.8 Hz, 8-H), 7.90 (6H, d, J = 8.6 Hz, 3-H), 8.04 (6H, d, J = 8.8 Hz, 9-H), 8.16 (6H, d, J = 1.6 Hz, 15-H), 8.27 (3H, s, 6-H vinylic). 13C-NMR (75 MHz, CDCl3): δ = 32.0 (CH3), 34.8 (Me3), 109.0 (C-12), 116.5 (C-15), 117.1 (C-6), 121.9 (C-9), 123.7 (C-16), 123.8 (C-13), 124.7 (C-2), 125.1 (C-4), 127.1 (C-3), 127.8 (C-8), 135.3 (C-10), 138.7 (C-7), 138.9 (C-11), 143.5 (C-14), 147.5 (C-1), 148.3 (C-5). ESI (m/z): calcd for C102H100N13+ 1506.8219 (M + H+): obsd, 1506.8189.N), 1502 (CC), 1034, 833 (Ph), 756 (Ph), 687 (Ph) cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 7.07 (6H, d, J = 8.3 Hz, 2-H), 7.20 (3H, d, J = 17.0 Hz, 6-H), 7.32 (3H, d, J = 16.0 Hz, 5-H), 7.52 (3H, d, J = 8.0 Hz, 16-H), 7.59 (6H, d, J = 8.5 Hz, 3-H), 7.64 (6H, d, J = 7.7 Hz, 8-H), 7.71 (6H, d, J = 8.6 Hz, 14-H), 7.95 (12H, bd, J = 7.8 Hz, 9-H, 15-H), 9.31 (3H, s, 12-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 119.5 (C-12), 120.0 (C-9), 123.9 (C-2), 125.6 (C-14), 126.7 (C-6), 126.9 (C-15), 127.8 (C-3), 128.1 (C-5), 128.7 (C-16), 129.1 (C-13), 129.9 (C-8), 132.0 (C-4), 136.6 (C-10), 137.2 (C-7), 146.1 (C-1), 147.1 (C-11). MALDI-TOF (m/z): calcd for C66H49N10+ 981.4 (M + H+): obsd, 981.3.N), 1506 (CC), 1315, 1227, 841 (Ph), 748 (Ph) cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 7.03–7.13 (30H, m, 2-H, 15-H, 18-H, 20-H), 7.18 (3H, d, J = 16.9 Hz, 5-H vinylic), 7.29 (3H, d, J = 16.5 Hz, 6-H vinylic), 7.35 (12H, dd, J = 7.9 Hz, 19-H), 7.57 (6H, d, J = 8.2 Hz, 3-H), 7.68 (6H, d, J = 8.3 Hz, 8-H), 7.80 (6H, d, J = 8.8 Hz, 14-H), 7.92 (6H, d, J = 8.2 Hz, 9-H), 9.18 (3H, s, 12-H triazole). 13C-NMR (75 MHz, DMSO-d6): δ = 119.5 (C-12), 121.4 (C-14), 123.0 (C-15), 123.8 (C-2), 123.8 (C-20), 125.5 (C-18), 126.9 (C-6 vinylic), 126.9 (C-8), 127.8 (C-3), 128.1 (C-5 vinylic), 129.2 (C-10), 129.8 (C-19), 130.8 (C-13), 132.0 (C-4), 137.1 (C-7), 146.1 (C-1), 146.7 (C-17), 146.9 (C-11), 147.6 (C-16). MALDI-TOF (m/z): calcd for C102H76N13+ 1482.8 (M + H+): obsd, 1482.3.N), 1505 (CC), 1273, 1031, 833 (Ph), 752 (Ph), 696 (Ph) cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 7.08 (6H, d, J = 7.6 Hz, 2-H), 7.24–7.34 (12H, m, 5-H, 6-H vinylics, 20-H), 7.47 (12H, bd, J = 3.4 Hz, 18-H, 19-H), 7.61 (6H, d, J = 8.1 Hz, 3-H), 7.75 (6H, d, J = 8.0 Hz, 9-H), 7.92 (6H, d, J = 8.6 Hz, 15-H), 7.99 (6H, d, J = 7.8 Hz, 8-H), 8.26 (6H, d, J = 8.5 Hz, 14-H), 8.27 (6H, d, J = 7.4 Hz, 21-H), 9.46 (3H, s, 12-H triazole). 13C-NMR (75 MHz, DMSO-d6): δ = 110.2 (C-18), 120.2 (C-12), 120.9 (C-20), 121.1 (C-21), 123.4 (C-22), 124.4 (C-2), 126.2 (C-8), 126.9 (C-19), 127.5 (C-5 or C-6 vinylics), 127.5 (C-9), 128.3 (C-3), 128.7 (C-5/6 vinylics), 128.7 (C-15), 129.5 (C-7), 132.5 (C-4), 135.9 (C-13), 137.4 (C-16), 137.8 (C-10), 140.5 (C-17), 146.7 (C-1), 147.8 (C-11). MALDI-TOF (m/z): calcd for C102H70N13+ 1476.6 (M + H+): obsd, 1476.3.N), 1506 (CC), 1261, 1032, 808 (Ph) cm−1. 1H-NMR (300 MHz, CDCl3): δ = 1.47 (54H, s, CH3), 7.08 (3H, d, J = 16.2 Hz, 5-H), 7.14 (6H, d, J = 8.6 Hz, 2-H), 7.17 (3H, d, J = 16.2 Hz, 6-H), 7.38 (6H, d, J = 8.6 Hz, 18-H), 7.47 (6H, d, J = 8.6 Hz, 3-H), 7.49 (6H, dd, J = 8.7, J = 1.9 Hz, 19-H), 7.62 (6H, d, J = 8.5 Hz, 8-H), 7.75 (6H, d, J = 8.8 Hz, 14-H), 7.94 (6H, d, J = 8.3 Hz, 9-H), 8.02 (6H, d, J = 8.8 Hz, 15-H), 8.16 (6H, d, J = 1.5 Hz, 21-H), 8.29 (3H, s, 12-H triazole). 13C-NMR (75 MHz, CDCl3): δ = 32.0 (CH3), 34.8 (Me3), 109.0 (C-18), 116.4 (C-21), 117.4 (C-12), 121.9 (C-15), 123.7 (C-22), 123.9 (C-19), 124.3 (C-2), 126.2 (C-10), 126.9 (C-5 o C-6 vinylics), 127.0 (C-8), 127.6 (C-3), 127.7 (C-14), 128.5 (C-5/6 vinylics), 129.0 (C-10), 132.1 (C-4), 135.1 (C-16), 137.8 (C-7), 138.7 (C-13), 138.9 (C-17), 143.5 (C-20), 146.8 (C-1), 148.4 (C-11). MALDI-TOF (m/z): calcd for C127H122N13O+ 1730.8 (M + MeOH2+ tBu group loss): obsd, 1730.4.
Me3), 109.0 (C-10), 116.4 (C-13), 118.4 (C-4), 121.8 (C-7), 122.8 (C-2), 123.7 (C-14), 123.9 (C-11), 127.7 (C-6), 131.6 (C-1), 135.0 (C-8), 138.8 (C-9), 138.9 (C-5), 143.5 (C-12), 147.8 (C-3). MALDI-TOF (m/z): calcd for C90H90N12+ 1338.7 (M+): obsd, 1338.4.N, CC), 1040, 758 (Ph) cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 7.22 (6H, d, J = 8.6 Hz, 2-H), 7.50 (3H, dd, J = 7.4 Hz, 10-H), 7.63 (6H, dd, J = 8.3, J = 7.5 Hz, 9-H), 7.91 (6H, d, J = 8.6 Hz, 3-H), 7.95 (6H, d, J = 8.0 Hz, 8-H), 9.25 (3H, s, 6-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 119.0 (C-6), 119.9 (C-8), 124.2 (C-2), 125.2 (C-4), 126.7 (C-3), 128.7 (C-10), 129.9 (C-9), 136.6 (C-7), 146.7 (C-1), 147.1 (C-5). MALDI-TOF (m/z): calcd for C42H30N10+ 674.3 (M+): obsd, 674.1.N), 1491 (CC), 1227, 1032, 835 (Ph), 748 (Ph) cm−1. H-NMR (300 MHz, DMSO-d6): δ = 7.05–7.09 (18H, m, 12-H, 14-H), 7.11 (6H, d, J = 9.0 Hz, 9-H), 7.18 (6H, d, J = 8.1 Hz, 2-H), 7.34 (12H, dd, J = 7.8 Hz, J = 7.4 Hz, 13-H), 7.81 (6H, d, J = 8.6 Hz, 8-H), 7.88 (6H, d, J = 8.2 Hz, 3-H), 9.13 (3H, s, 6-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 118.8 (C-6), 121.3 (C-8), 123.0 (C-14), 123.8 (C-9), 124.5 (C-2) 124.5 (C-12), 125.2 (C-4), 126.7 (C-3), 129.7 (C-13), 130.8 (C-7), 146.6 (C-1), 146.7 (C-5), 146.7 (C-11), 147.6 (C-10). ESI (m/z): calcd for C78H58N13H+ 1176.4933 (M + H+): obsd, 1176.4925.N), 1489 (CC), 1281, 1032, 835 (Ph), 752 (Ph), 694 (Ph) cm−1. H-NMR (300 MHz, DMSO-d6): δ = 7.25–7.35 (12H, m, 2-H, 13-H), 7.46 (12H, bd, J = 3.6 Hz, 8-H, 12-H), 7.91 (6H, d, J = 8.6 Hz, 15-H), 7.97 (6H, d, J = 8.5 Hz, 3-H), 8.26 (12H, bd, J = 8.1 Hz, 9-H, 14-H), 9.42 (3H, s, 6-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 109.7 (C-12), 119.2 (C-6), 120.4 (C-7), 120.6 (C-13), 121.5 (C-14), 122.9 (C-8), 124.3 (C-2), 125.1 (C-4), 126.4 (C-9), 126.8 (C-3), 128.2 (C-15), 135.4 (C-11), 136.9 (C-10), 140.0 (C-16), 146.7 (C-1), 147.3 (C-5). ESI (m/z): calcd for C78H52N13+ 1170.4463 (M + H+): obsd, 1170.4460.N), 1489 (CC), 1364, 1261, 1032, 810 (Ph) cm−1. 1H-NMR (300 MHz, CDCl3): δ = 1.48 (54H, s, CH3), 7.32 (6H, d, 2-H), 7.40 (6H, d, J = 8.6 Hz, 12-H), 7.50 (6H, dd, J = 8.7, J = 1.9 Hz, 13-H), 7.77 (6H, d, J = 8.8 Hz, 8-H), 7.90 (6H, d, J = 8.6 Hz, 3-H), 8.04 (6H, d, J = 8.8 Hz, 9-H), 8.16 (6H, d, J = 1.6 Hz, 15-H), 8.27 (3H, s, 6-H vinylic). 13C-NMR (75 MHz, CDCl3): δ = 32.0 (CH3), 34.8 (Me3), 109.0 (C-12), 116.5 (C-15), 117.1 (C-6), 121.9 (C-9), 123.7 (C-16), 123.8 (C-13), 124.7 (C-2), 125.1 (C-4), 127.1 (C-3), 127.8 (C-8), 135.3 (C-10), 138.7 (C-7), 138.9 (C-11), 143.5 (C-14), 147.5 (C-1), 148.3 (C-5). ESI (m/z): calcd for C102H100N13+ 1506.8219 (M + H+): obsd, 1506.8189.N), 1502 (CC), 1034, 833 (Ph), 756 (Ph), 687 (Ph) cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 7.07 (6H, d, J = 8.3 Hz, 2-H), 7.20 (3H, d, J = 17.0 Hz, 6-H), 7.32 (3H, d, J = 16.0 Hz, 5-H), 7.52 (3H, d, J = 8.0 Hz, 16-H), 7.59 (6H, d, J = 8.5 Hz, 3-H), 7.64 (6H, d, J = 7.7 Hz, 8-H), 7.71 (6H, d, J = 8.6 Hz, 14-H), 7.95 (12H, bd, J = 7.8 Hz, 9-H, 15-H), 9.31 (3H, s, 12-H vinylic). 13C-NMR (75 MHz, DMSO-d6): δ = 119.5 (C-12), 120.0 (C-9), 123.9 (C-2), 125.6 (C-14), 126.7 (C-6), 126.9 (C-15), 127.8 (C-3), 128.1 (C-5), 128.7 (C-16), 129.1 (C-13), 129.9 (C-8), 132.0 (C-4), 136.6 (C-10), 137.2 (C-7), 146.1 (C-1), 147.1 (C-11). MALDI-TOF (m/z): calcd for C66H49N10+ 981.4 (M + H+): obsd, 981.3.N), 1506 (CC), 1315, 1227, 841 (Ph), 748 (Ph) cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 7.03–7.13 (30H, m, 2-H, 15-H, 18-H, 20-H), 7.18 (3H, d, J = 16.9 Hz, 5-H vinylic), 7.29 (3H, d, J = 16.5 Hz, 6-H vinylic), 7.35 (12H, dd, J = 7.9 Hz, 19-H), 7.57 (6H, d, J = 8.2 Hz, 3-H), 7.68 (6H, d, J = 8.3 Hz, 8-H), 7.80 (6H, d, J = 8.8 Hz, 14-H), 7.92 (6H, d, J = 8.2 Hz, 9-H), 9.18 (3H, s, 12-H triazole). 13C-NMR (75 MHz, DMSO-d6): δ = 119.5 (C-12), 121.4 (C-14), 123.0 (C-15), 123.8 (C-2), 123.8 (C-20), 125.5 (C-18), 126.9 (C-6 vinylic), 126.9 (C-8), 127.8 (C-3), 128.1 (C-5 vinylic), 129.2 (C-10), 129.8 (C-19), 130.8 (C-13), 132.0 (C-4), 137.1 (C-7), 146.1 (C-1), 146.7 (C-17), 146.9 (C-11), 147.6 (C-16). MALDI-TOF (m/z): calcd for C102H76N13+ 1482.8 (M + H+): obsd, 1482.3.N), 1505 (CC), 1273, 1031, 833 (Ph), 752 (Ph), 696 (Ph) cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 7.08 (6H, d, J = 7.6 Hz, 2-H), 7.24–7.34 (12H, m, 5-H, 6-H vinylics, 20-H), 7.47 (12H, bd, J = 3.4 Hz, 18-H, 19-H), 7.61 (6H, d, J = 8.1 Hz, 3-H), 7.75 (6H, d, J = 8.0 Hz, 9-H), 7.92 (6H, d, J = 8.6 Hz, 15-H), 7.99 (6H, d, J = 7.8 Hz, 8-H), 8.26 (6H, d, J = 8.5 Hz, 14-H), 8.27 (6H, d, J = 7.4 Hz, 21-H), 9.46 (3H, s, 12-H triazole). 13C-NMR (75 MHz, DMSO-d6): δ = 110.2 (C-18), 120.2 (C-12), 120.9 (C-20), 121.1 (C-21), 123.4 (C-22), 124.4 (C-2), 126.2 (C-8), 126.9 (C-19), 127.5 (C-5 or C-6 vinylics), 127.5 (C-9), 128.3 (C-3), 128.7 (C-5/6 vinylics), 128.7 (C-15), 129.5 (C-7), 132.5 (C-4), 135.9 (C-13), 137.4 (C-16), 137.8 (C-10), 140.5 (C-17), 146.7 (C-1), 147.8 (C-11). MALDI-TOF (m/z): calcd for C102H70N13+ 1476.6 (M + H+): obsd, 1476.3.N), 1506 (CC), 1261, 1032, 808 (Ph) cm−1. 1H-NMR (300 MHz, CDCl3): δ = 1.47 (54H, s, CH3), 7.08 (3H, d, J = 16.2 Hz, 5-H), 7.14 (6H, d, J = 8.6 Hz, 2-H), 7.17 (3H, d, J = 16.2 Hz, 6-H), 7.38 (6H, d, J = 8.6 Hz, 18-H), 7.47 (6H, d, J = 8.6 Hz, 3-H), 7.49 (6H, dd, J = 8.7, J = 1.9 Hz, 19-H), 7.62 (6H, d, J = 8.5 Hz, 8-H), 7.75 (6H, d, J = 8.8 Hz, 14-H), 7.94 (6H, d, J = 8.3 Hz, 9-H), 8.02 (6H, d, J = 8.8 Hz, 15-H), 8.16 (6H, d, J = 1.5 Hz, 21-H), 8.29 (3H, s, 12-H triazole). 13C-NMR (75 MHz, CDCl3): δ = 32.0 (CH3), 34.8 (Me3), 109.0 (C-18), 116.4 (C-21), 117.4 (C-12), 121.9 (C-15), 123.7 (C-22), 123.9 (C-19), 124.3 (C-2), 126.2 (C-10), 126.9 (C-5 o C-6 vinylics), 127.0 (C-8), 127.6 (C-3), 127.7 (C-14), 128.5 (C-5/6 vinylics), 129.0 (C-10), 132.1 (C-4), 135.1 (C-16), 137.8 (C-7), 138.7 (C-13), 138.9 (C-17), 143.5 (C-20), 146.8 (C-1), 148.4 (C-11). MALDI-TOF (m/z): calcd for C127H122N13O+ 1730.8 (M + MeOH2+ tBu group loss): obsd, 1730.4.Results and discussion
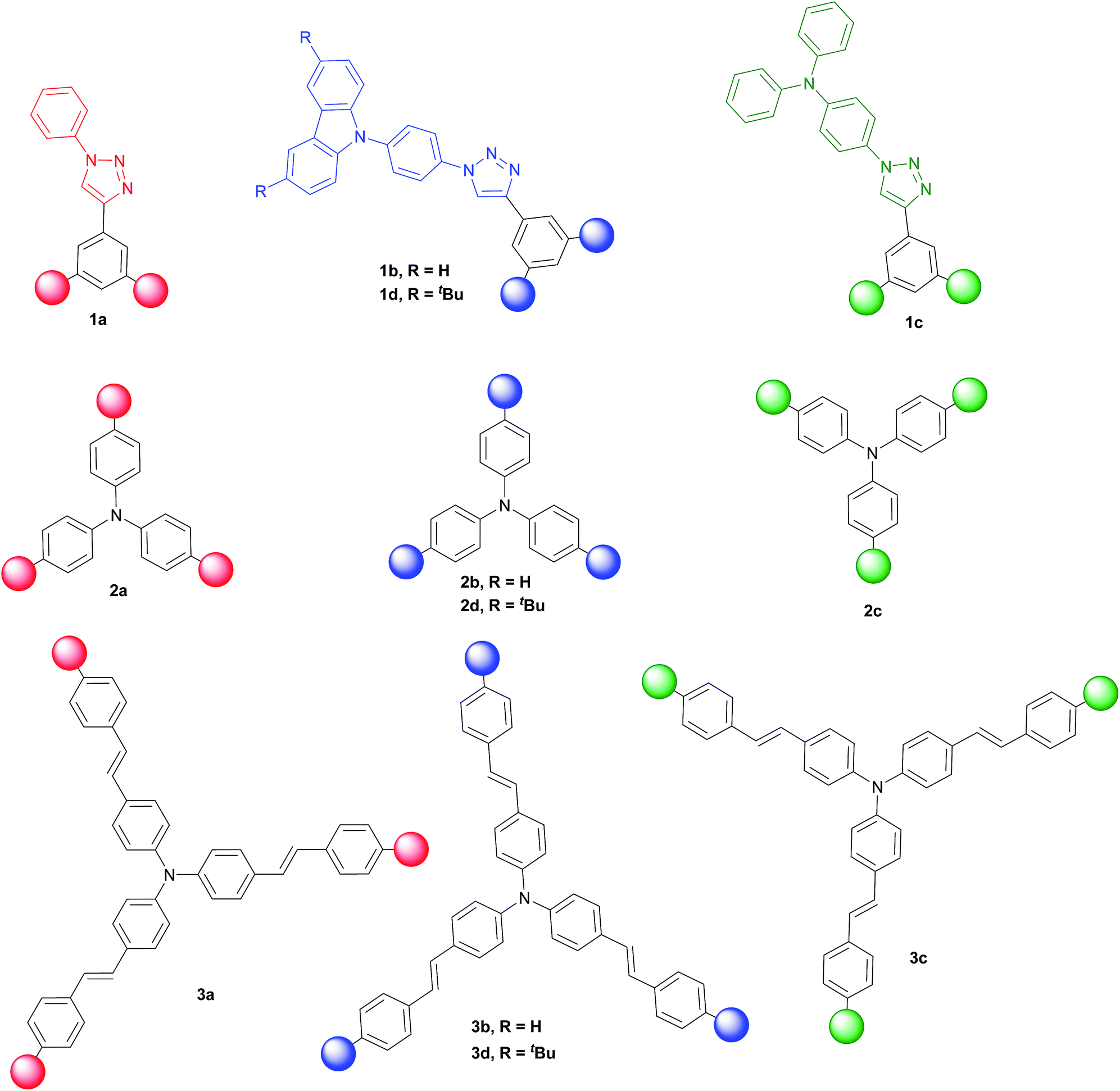
Three families of structurally related triazolic dendrimers were prepared by the combination of three different cores and four different aromatic azides, the structures of these compounds are depicted in Fig. 2. | ||
| Fig. 2 Chemical structures of cores and dendrons. | ||
With the exception of core 4 and dendron 7, the rest of the cores and dendrons contain electroactive moieties such as triphenylamine (cores 5 and 6, dendron 8) and carbazole (dendrons 9 and 10). The cores were synthesized from the corresponding aryl halide through a Sonogashira coupling reaction with trimethylsilylacetylene (TMSA) and further desilylation to get the free alkynes. In Scheme 1 are described the synthetic routes for every target core. The synthesis of core 4 and intermediate 15 has been previously reported by our laboratory.14 There are two common steps in each synthesis: Sonogashira coupling with TMSA and basic desilylation. Core 4 was obtained after these two steps with excellent yields. Synthesis of core 6 was accomplished from commercial triphenylamine 12 by formylation under Vilsmeier–Haack conditions15 in two steps furnishing tri-aldehyde derivative 13 in moderated yield. A Wadsworth–Horner–Emmons olefination reaction used to couple 13 with phosphonate 14 afforded tri-iodide 15 in good yield.16 Further Sonogashira coupling reaction with TMSA followed by desilylation procedure furnished core 6 in very good yield. For the synthesis of 5, triphenylamine 12 was treated with a mixture of KI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :KIO3 in glacial acetic acid to give tri-iodide derivative 16 in moderated yield.17 After Sonogashira and desilylation reactions sequence, core 5 was obtained in good yield.18 Cores 4, 5 and 6 present three terminal alkyne functionalities arranged in a “star-shape” architecture which it will ultimately define the shaped of the resulting dendrimers.
:KIO3 in glacial acetic acid to give tri-iodide derivative 16 in moderated yield.17 After Sonogashira and desilylation reactions sequence, core 5 was obtained in good yield.18 Cores 4, 5 and 6 present three terminal alkyne functionalities arranged in a “star-shape” architecture which it will ultimately define the shaped of the resulting dendrimers.
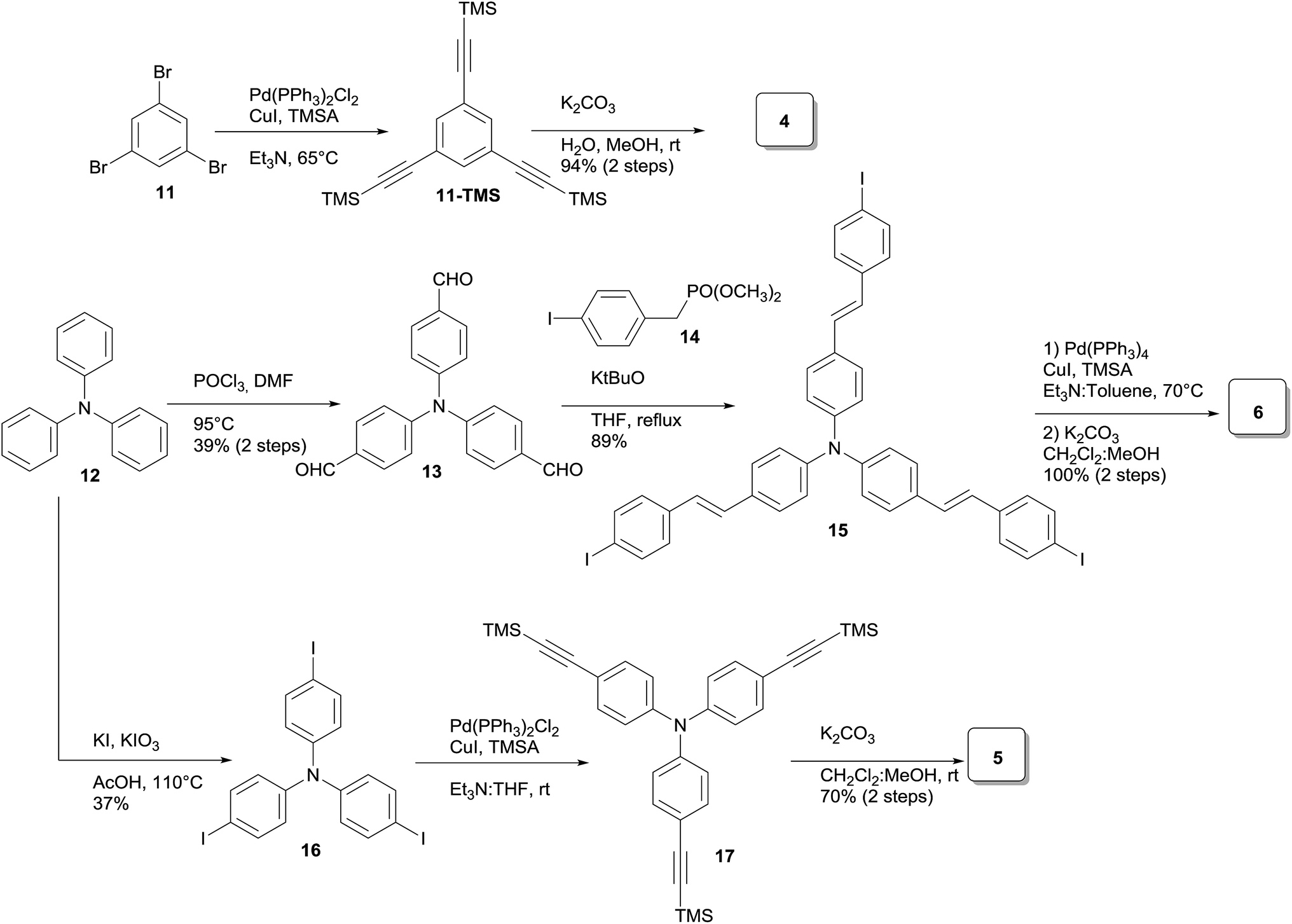 | ||
| Scheme 1 Synthesis of cores. | ||
The synthesis of azides 7–10 are described in Scheme 2. All azides derivatives were obtained in very good yields through a diazotization reaction to transform the amino group into the azide one. It is worth to mention that azides showed to be highly photosensitive, therefore all the azides were used as crude products without purification and handled protected from light. Products 7, 8, 9 and 10 were characterized by 1H and 13C NMR and FT-IR spectroscopy. The last one was very useful to show the distinctive signal of azide group between 2160–2120 cm−1.19 Phenyl azide 7 was prepared according to literature protocol20 via diazotization of freshly distilled aniline 18 and further treatment with sodium azide affording 7 in excellent yield. Triphenyl azide 8 was prepared by nitration of triphenylamine 12,21 the mono nitrate derivative was reduced using hydrazine monohydrate and Pd/C to furnish amine 20,22 subsequently diazotization and treatment with sodium azide afforded the expected product 8.21 The preparation of azides 9 and 10 were achieved from commercial carbazole 21a and 3,6-di-tert-butyl carbazole 21b which was previously synthesized in our laboratory.23 Ullmann coupling of 21a and 21b24 with 4-nitroiodobenzene afforded 22a and 22b. The nitro derivatives were reduced with hydrazine22 yielding amines 23a and 23b, which were submitted to diazotization and further treatment with sodium azide to give the corresponding azides 9 and 10.25 It is noteworthy to mention that all the reaction steps were accomplished with excellent yields and only one column chromatography purification was needed for the nitro derivatives 22a and 22b, all the others compounds were solids and purified by crystallization.
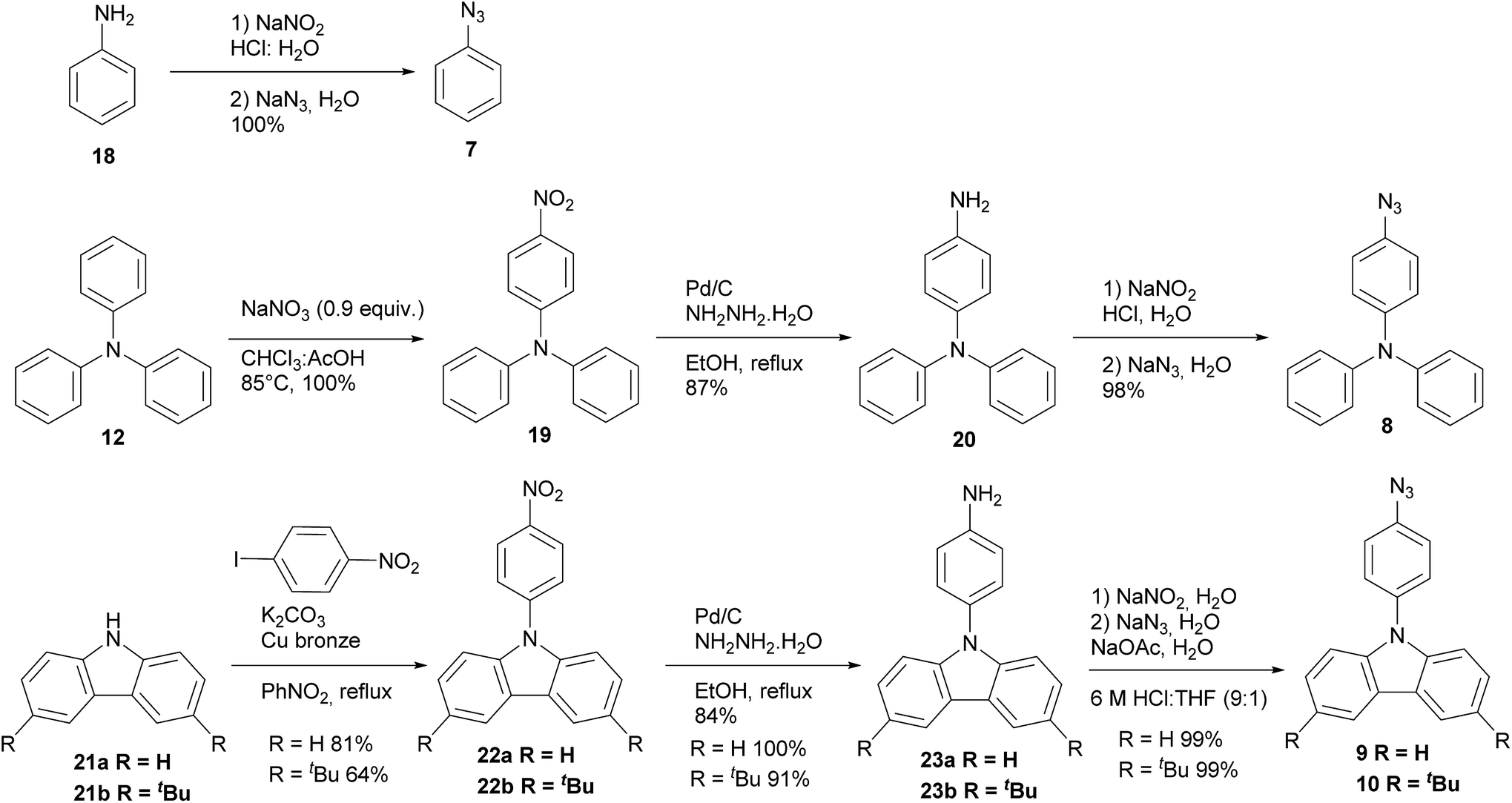 | ||
| Scheme 2 Synthesis of dendrons. | ||
Once all the components needed for the click reaction were synthesized, we looked for a general coupling condition that could allow us to attach all the cores with the corresponding azides in a convergent manner, regardless their chemical structures, to obtain triazolic dendrimers with star-shaped architecture. In a typical procedure for a CuAAC reaction, the Cu(I) cation used as catalyst was generated in situ from a copper(II) salt and a reducing agent (most often sodium ascorbate). These systems are efficient but limited to substrates with high water tolerance.26 Sometimes, in order to apply milder reaction conditions or increase the reaction applicability, addition of ligands to protect Cu(I) centers is a common strategy. In our case, the use of sodium ascorbate and CuSO4 as source of Cu(I) in different proportions, afforded mixtures of products and partial recovery of starting materials. We assumed that incomplete reactions were a consequence of only partial solubility of starting material in the aqueous solvents mixture. Hence, we looked for homogeneous conditions for the dipolar cycloadditions and we found that the used of Cu(PPh3)3Br and DiPEA as organic base in tetrahydrofuran allowed us to obtain the expected products in very good yields. Cu(PPh3)3Br is a crystalline solid, stable to air and ambient moisture and it is prepared by the addition of triphenylphosphine to a hot methanolic solution of CuBr2.27 This procedure avoids the need to reflux moisture-sensitive CuBr with triphenylphosphine. Furthermore, this complex is soluble in several organic solvents and can be stored under air for prolonged times, without any visible decomposition.28 In a general procedure, 0.10–0.12 mmol of core were dissolved in THF (3 to 4 mM), azide (3.6 equivalents), 0.06 equivalents of Cu(I) catalyst and 3 equivalents of DiPEA were used to afforded the expected products showed in Fig. 3.
 | ||
| Fig. 3 Triazolic star-shaped dendrimers structures. | ||
The twelve triazolic dendrimers (Fig. 3) can be classified in three families according to the identity of the core. For the dendrimers 1a–d the common core was benzene, 2a–d triphenylamine and 3a–d conjugated triphenylamine. All the products were obtained applying the same reaction conditions with high yields. The coloured solid dendrimers were purified by suspension of the solids in two different solvents or a mixture of both: ethanol and methyl-tert-butyl ether. This treatment allowed removing excess of azides, copper salts and any other by-product formed in the reaction and avoids the use of chromatography column step to isolate pure triazoles. All these compounds are very polar and therefore soluble in polar aprotic solvents such as dimethylsulfoxide, tetrahydrofuran and slightly soluble in dichloromethane. The last one is better to dissolve tert-butylated derivatives 1d, 2d and 3d. As far as we know, there are not literature references related to the preparation of structurally related families of aromatic triazoles. A few of triazolic dendritic structures has been published and they showed that this type of compounds have a high potential on new material design.29,30 Additionally, the purification procedure was simple and similar in each case with no need for column chromatography and afforded pure samples.
Conclusions
In this work we were able to synthesize twelve dendritic aromatic triazoles structurally related using four different aromatic azides as dendrons and varying the chemical identity of the core. A standard reaction procedure was developed and applied in all cases with very good results. The homogeneous CuAAC reaction afforded solid compounds and their purification only included a suspension in different washing solvents avoiding any chromatography column technique. All the dendrimers presented a starburst molecular shape and includes triarylamines as electroactive moiety which turns them into excellent candidates for the development of novel organic materials. The structural changes introduced in every family of compounds were selected in order to establish the relationship between chemical structure–optoelectronic properties. The optoelectronic studies are being carried out and will be published in due course.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET – Argentina), Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT – Argentina), Fundación Nuevo Banco de Santa Fe (Santa Fe – Argentina) and Universidad Nacional de Rosario. M. I. M. and R. A. S are scientific members of CONICET.Notes and references
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., 2001, 113, 2056 (Angew. Chem., Int. Ed., 2001, 40, 2004) CrossRef.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1025 CrossRef PubMed.
- (a) C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987 CrossRef CAS; (b) S. Mongkhontreerat, M. V. Walter, O. C. J. Andren, Y. Cai and M. Malkoch, Adv. Funct. Mater., 2015, 25, 4837 CrossRef CAS; (c) X. Wang, B. Huang, X. Liu and P. Zhan, Drug Discovery Today, 2016, 21, 118 CrossRef CAS PubMed; (d) E. Haldon, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528 RSC.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Folkin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tornøe, M. Meldal and C. Christensen, J. Org. Chem., 2002, 67, 3057 CrossRef.
- R. Huisgen, R. Knorr, L. Moebius and G. Szeimies, Chem. Ber., 1965, 98, 4014 CrossRef CAS.
- (a) P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Fréchet, K. B. Sharpless and V. V. Folkin, Angew. Chem., 2004, 116, 4018 CrossRef; (b) P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Fréchet, K. B. Sharpless and V. V. Folkin, Angew. Chem., Int. Ed., 2004, 43, 3928 Search PubMed.
- (a) W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572 CrossRef CAS; (b) M. Arseneault, C. Wafer and J.-F. Morin, Molecules, 2015, 20, 9263 CrossRef CAS PubMed; (c) A. Marrocchi, A. Facchetti, D. Lanari, S. Santoro and L. Vaccaro, Chem. Sci., 2016, 7, 6298 RSC; (d) M. Sowinska and Z. Urbanczyk-Lipkowska, New J. Chem., 2014, 38, 2168 RSC.
- C.-H. Wong and S. C. Zimmerman, Chem. Commun., 2013, 49, 1679 RSC.
- K. C. Majumdar and K. Ray, Synthesis, 2011, 3767 CrossRef CAS.
- Y. Fu, Y. Liu, X. Fu, L. Zou, H. Li, M. Li, X. Chen and J. Qin, Chin. J. Chem., 2010, 28, 2226 CrossRef CAS.
- S. S. Bag and R. Kundu, J. Org. Chem., 2011, 76, 3348 CrossRef CAS PubMed.
- Z. Ning and H. Tian, Chem. Commun., 2009, 5483 RSC.
- H. Nonami, S. Fukui and R. Erra-Balsells, J. Mass Spectrom., 1997, 32, 287 CrossRef CAS.
- M. I. Mangione, R. A. Spanevello, A. Rumbero, D. Heredia, G. Marzari, L. Fernandez, L. Otero and F. Fungo, Macromolecules, 2013, 46, 475 CrossRef.
- T. Malegol, S. Gmouh, M. A. A. Meziane, M. Blanchard-Desce and O. Mongin, Synthesis, 2005, 11, 1771 Search PubMed.
- (a) H. Shao, X. Chen, Z. Wang and P. Lu, J. Lumin., 2007, 127, 349 CrossRef CAS; (b) H. F. Kung, P. M. Kung and Z. Zhuang, U.S. Pat., 7,297,820, 2007.
- T. Khanasa, N. Jantasing, S. Morada, N. Leesakul, R. Tarsang, S. Namuangruk, T. Kaewin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, Eur. J. Org. Chem., 2013, 2608 CrossRef CAS.
- (a) S. P. McIlroy, E. Cló, L. Nikolajsen, P. K. Frederiksen, C. B. Nielsen, K. V. Mikkelsen, K. V. Gothelf and P. R. Ogilby, J. Org. Chem., 2005, 70, 1134 CrossRef CAS PubMed; (b) P. Hrobárik, V. Hrobáriková, I. Sigmundová, P. Zahradník, M. Fakis, I. Polyzos and P. Persephonis, J. Org. Chem., 2011, 76, 8726 CrossRef PubMed; (c) K. Vongnam, T. Vilaivan and M. Sukwattanasinitt, J. Fluoresc., 2014, 24, 197 CrossRef CAS PubMed.
- E. Lieber, C. M. Ramachandra Rao, T. S. Chao and C. W. W. Hoffman, Anal. Chem., 1939, 19, 55 Search PubMed.
- H. J. Lucas and E. R. Kennedy, Org. Synth., 1939, 19, 55 CrossRef CAS.
- L. C. Raiford and A. L. LeRosen, J. Am. Chem. Soc., 1944, 66, 1872 CrossRef CAS.
- (a) W. Y. Lee, T. Kurosawa, S. T. Lin, T. Higashihara, M. Ueda and W. C. Chen, Chem. Mater., 2011, 23, 4487 CrossRef CAS; (b) Y. Liu, D. Chao and H. Yao, Org. Electron., 2014, 15, 1422 CrossRef CAS.
- M. I. Mangione and R. A. Spanevello, Tetrahedron Lett., 2015, 56, 465 CrossRef CAS.
- S. Tripathi, M. Mohan, R. K. Pandey and K. Misra, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1999, 38, 317 Search PubMed.
- B. Schulze, D. Escudero, C. Friebe, R. Siebert, H. Görls, S. Sinn, M. Thomas, S. Mai, J. Popp, B. Dietzek, L. González and U. S. Schubert, Chem.–Eur. J., 2012, 18, 4010 CrossRef CAS PubMed.
- S. Lal and S. Diez-González, J. Org. Chem., 2011, 76, 2367 CrossRef CAS PubMed.
- J. Germain, F. Svec and J. M. J. Fréchet, Chem. Mater., 2008, 20, 7069 CrossRef CAS.
- R. Gujadhur and D. Venkataraman, Synth. Commun., 2001, 31, 2865 CrossRef CAS.
- Q. Zhang, Z. Ning and H. Tian, Dyes Pigm., 2009, 81, 80 CrossRef CAS.
- Q. Zhang, Z. Ning, Y. Yan, S. Qian and H. Tian, Macromol. Rapid Commun., 2008, 29, 193 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and complete characterization data are depicted in ESI for each compound described in Schemes 1 and 2. See DOI: 10.1039/c7ra09558a |
| This journal is © The Royal Society of Chemistry 2017 |