
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental and theoretical studies of the [3,3]-sigmatropic rearrangement of prenyl azides†
Exequiel O. J. Portaa,
Margarita M. Vallejosb,
Andrea B. J. Bracca a and
Guillermo R. Labadie*ac
a and
Guillermo R. Labadie*ac
aInstituto de Química Rosario, UNR, CONICET, Suipacha 531, S2002LRK, Rosario, Argentina. E-mail: labadie@iquir-conicet.gov.ar; Fax: +54-341-4370477 ext. 112; Tel: +54-341-4370477 ext. 108
bLaboratorio de Química Orgánica, IQUIBA-NEA, Universidad Nacional del Nordeste, CONICET, FACENA, Av. Libertad 5460, Corrientes 3400, Argentina
cDepartamento de Química Orgánica, Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, S2002LRK, Rosario, Argentina
First published on 10th October 2017
Abstract
[3,3]-Sigmatropic rearrangement of isoprenyl azides has been extensively investigated in an experimental and theoretical level. Prenylazides with different chain lengths and phenyl prenylazide have been synthesized. NMR analysis of each azide has been made to determine the equilibrium composition, showing the predominance of primary allyl azides over the tertiary ones and E over Z isomer, regardless of the chain length, temperature, solvent or the regiochemistry of the precursor isoprenyl alcohol. It was determined that phenyl prenylazides do not experience [3,3]-sigmatropic rearrangement. In order to rationalize the experimental results and to gain insight in the mechanism of the [3,3]-sigmatropic rearrangement of prenylazides a theoretical study was performed using density functional theory and the QTAIM approach.
Introduction
In this century, an amazing increase in the number of applications of organic azide derivatives have been reported. Azides and alkynes chemical orthogonality has allowed unique applications in different fields including bioconjugation,1 material sciences,2 organic chemistry and medicinal chemistry.3 1,2,3-Triazoles synthesis became very valuable after the independent discoveries of Meldal's4 and Sharpless'5 groups that introduced the copper(I) accelerated Hüisgen 1,3-dipolar cycloaddition of alkynes and azides (CuAAC). That completely regioselective reaction has become one of the most commonly used organic synthesis.Allylic azides are versatile building blocks for the synthesis of natural products and nitrogen-containing heterocycles of pharmacological relevance. However, they undergo a spontaneous dynamic rearrangement at ambient temperature (Fig. 1). In 1960 Young and co-workers reported the study of [3,3]-sigmatropic rearrangement of allylic azides (known as Winstein's rearrangement).6 They observed that allylic azide exists as a regioisomeric mixture that interconverts rapidly at room temperature. Lately, VanderWerf and Heasley found that tertiary and secondary allylic azides rearrange faster than the primary ones7 with the regioisomer with the most substituted alkene being thermodynamically favoured. The effective application of this reaction in synthetic schemes has been difficult due to the spontaneous rearrangement. It is necessary to control the thermodynamic ratio of the regioisomeric azides in order to be synthetically useful and that generally depends on the substrate.8
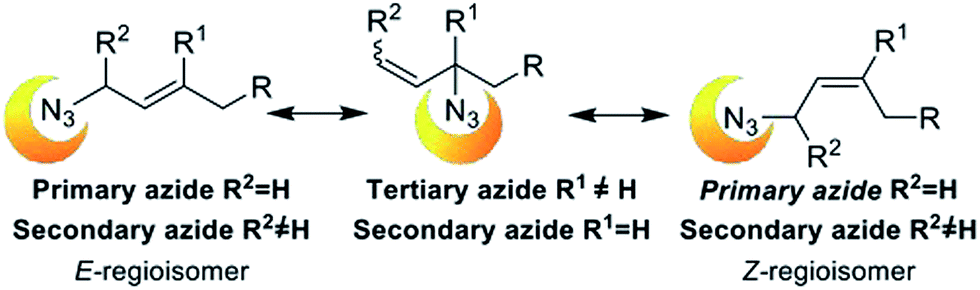 | ||
| Fig. 1 [3,3]-Sigmatropic rearrangement of allyl azides. | ||
Isoprenylazides have been used for protein labelling9 and to prepare 1,2,3-triazole libraries, generating phenyl triazoles 1 as antibiofilm,10 steryl derivatives 2 as antiparasites,11 bisphosphonates 3 as geranylgeranyl transferase II inhibitors12 and β-lactones 4 as fatty acid synthase inhibitors (Fig. 2).13 In those reports, prenyl azides in equilibrium reacted with alkynes to produce 1,2,3-triazoles as a mixture of regioisomers. Recently, our group has successfully prepared and chromatographically separated different regioisomers derived from prenyl-1,2,3-triazoles. The activity of those compounds is hardly dependent on the regiochemistry of the first isoprenic unit, playing an important role on the modulation of the activity between trypanosomatids.14
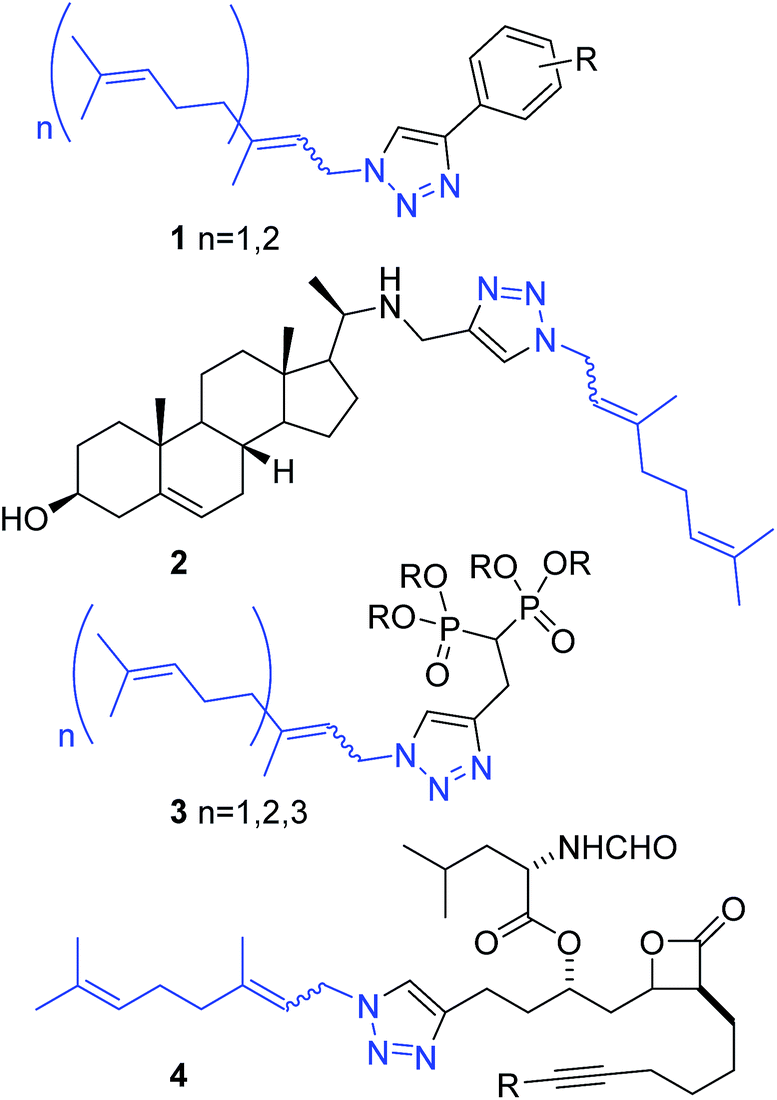 | ||
| Fig. 2 Bioactive prenyl 1,2,3-triazole derivatives. | ||
Besides the utility of this important scaffold, a complete study of the isoprenylazides equilibrium has not been conducted. That information will contribute to find factors that rule the equilibrium and the reactivity of this synthetically useful compounds. Therefore, in this paper we report a detailed experimental and theoretical study of isoprenylazides and their [3,3]-sigmatropic rearrangement.
Results and discussion
Chemistry
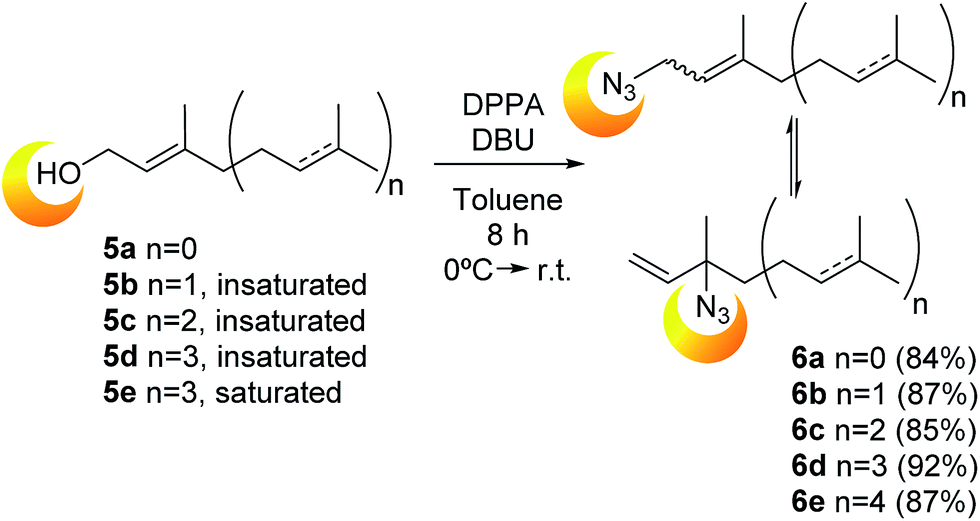 | ||
| Scheme 1 Synthesis of prenyl azides 6a–e from the corresponding alcohols (prenol, geraniol, farnesol, geranylgeraniol and phytol, respectively). | ||
All the attempts to obtain a single isomer from this mixture was doomed due to the dynamic interconversion. Presumably, the physicochemical properties are an additional factor that difficulted the separation based on their low polarity and similarity of the products. To rationalize these likeness, the physicochemical properties were calculated in silico using the Molinspiration software.26 As was expected, there are no substantial differences on the more relevant parameters of the isomers (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, volume and TPSA), explaining the impossibility of the experimental products separation. As an example, in the case of the mixture 6b: geranyl azide (6b-E), neryl azide (6b-Z) and linaloyl azide (6b–t) logP values, calculated as a lipophilicity parameter, were 4.54, 4.54 and 4.55, respectively. Those similarities, combined with the rapid interconversion at room temperature, made impossible to isolate any isomer.
P, volume and TPSA), explaining the impossibility of the experimental products separation. As an example, in the case of the mixture 6b: geranyl azide (6b-E), neryl azide (6b-Z) and linaloyl azide (6b–t) logP values, calculated as a lipophilicity parameter, were 4.54, 4.54 and 4.55, respectively. Those similarities, combined with the rapid interconversion at room temperature, made impossible to isolate any isomer.
Computational methods
The geometries of the reactants, the transition structures (TSs) and the products were optimized using MPWB1K27 global hybrid meta-GGA functional together with 6-311++G(d,p) basis set. The MPWB1K has shown to be appropriate to obtain reliable geometries and for describing kinetics of organic reactions. Solvent effects in toluene (solvent used experimentally) were taken into account through full optimizations using the SMD continuum solvation method.28 Frequency calculations were computed to verify the nature of the stationary points as true minima or as first order transition structure (TS) and to evaluate the thermal corrections. Free energies were calculated at 298.15 K and 1 atm in toluene. Intrinsic reaction coordinate (IRC) paths were traced using the Hessian-based predictor corrector (HPC)29,30 algorithm to verify the connectivity of the TSs with reactants and products. All calculations were performed with the Gaussian 09 package.31 The topological properties in the framework of the QTAIM theory were calculated using the AIMALL program.32Equilibrium composition studies
Given the limited precedents of the equilibrium for this system, a complete study was necessary. These analyzes included the nature of the substrates (number of isoprene units and unsaturations), regiochemistry of the starting alcohol, solvent and temperature. The composition of each mixture at room temperature was determined by 1H NMR spectra. First, a detailed analysis of the composition at the equilibrium of the mixture was carried out identifying the signals corresponding to each component of the mixture. In the case of 6b (6b-E, 6b-Z and 6b–t) the solution 1H NMR in CDCl3 (Fig. 3), shows a multiplet at 5.09 ppm assignable to the common olefinic proton H-6 (pink) shared by all the isomers, being taken as a reference. A double doublet at 5.78 ppm is observed for the olefinic proton H-2 (yellow) of linaloyl azide 6b–t. This azide is in a 13% in the mixture, as was expected, since it is the less substituted alkene. Finally, to calculate the E:Z ratio of the primary azides, nOe experiments selectively irradiating the triplet present at 5.33 ppm, which corresponds to the olefinic proton H-2 (blue) shown positive effect on the singlet present at 1.79 ppm. This signal is assigned to the methyl group of the Z isomer (6b-Z), providing a ratio E:Z of 5:3.
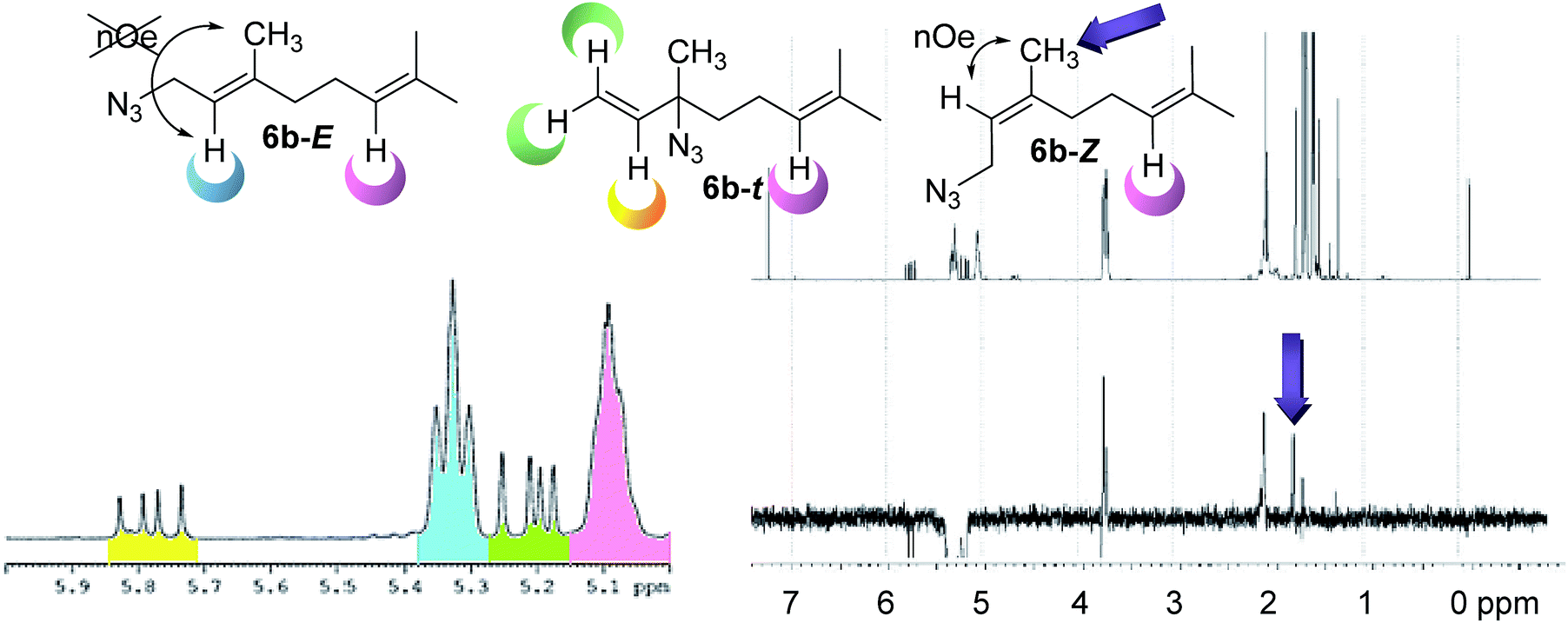 | ||
| Fig. 3 Left: 1H NMR spectrum of the double bond section of the mixture 6b. Right: nOe experiment. | ||
The NMR analysis of the other mixtures (6a, 6c, 6d and 6e) in CDCl3 have shown that the azide composition remains constant regardless the number of isoprenyl units or (un)saturation grade, suggesting that increasing the number of isoprene units does not influence the equilibrium. It was noticed that primary allyl-azides are the major compounds present in the mixture (87%), while tertiary allyl-azides represent only 13% at room temperature. Concerning the composition of primary allyl-azides, the E- and Z-isomers correspond to 54% and 33% of the mixture, respectively.
To corroborate that the equilibrium composition is independent of the regiochemistry and the degree of substitution of the starting isoprenol, a set of reactions were conducted. Independently, nerol (5b-Z) and linalool (5b–t) were reacted under Thompson's conditions, providing the products with similar yield than that for geraniol (5b-E). In those cases, a similar isomeric ratio was obtained regardless the precursor alcohol. Geranyl azide (6b-E), neryl azide (6b-Z), linalooyl azide (6b–t) were obtained in a 54:33:13 ratio, respectively (Scheme 2). Also, in different reported procedures to generate 6b the same regioisomer proportions were obtained, regardless of reaction conditions used (temperature, solvents, reagents, catalysts).16,21,33,34 These results demonstrate that isoprenyl azides cannot be obtained as pure isomer and consequently not requiring regiopure isoprenols for their preparation, which results in a significant cost reduction. The relative tertiary azide proportion increases 15% (Fig. 4, from 11.2% to 13.2%).
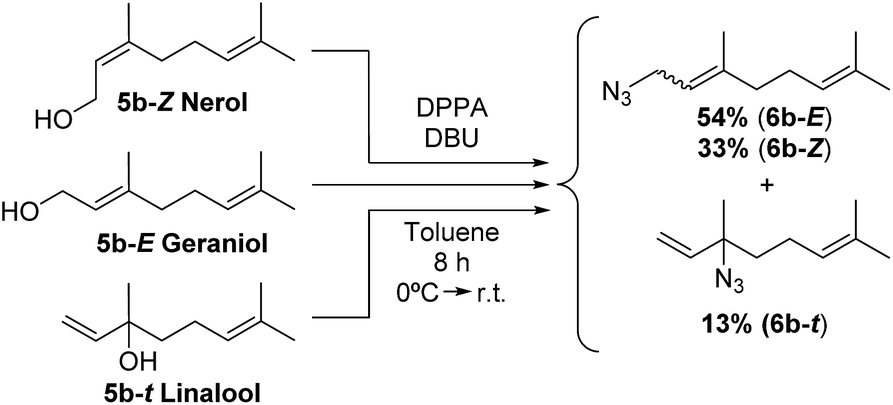 | ||
| Scheme 2 Synthesis of the azide mixture from nerol, geraniol and linalool. | ||
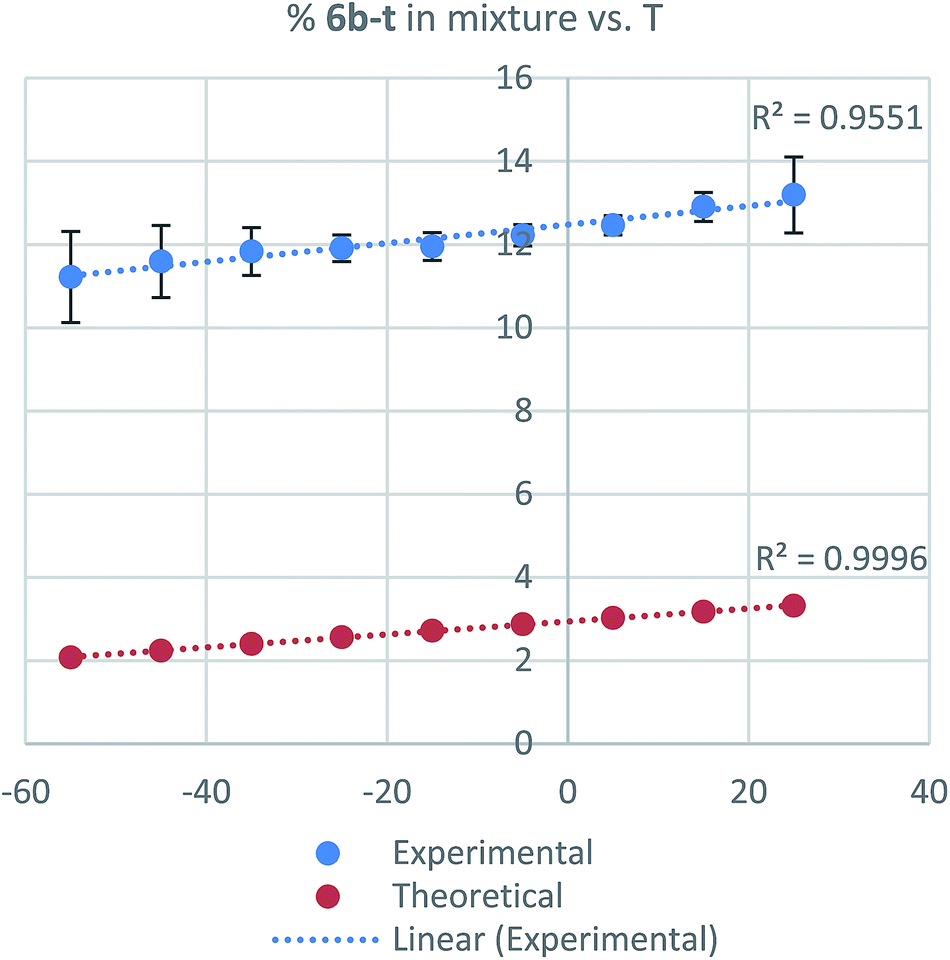 | ||
| Fig. 4 Dependence of tertiary azide population vs. temperature (T) (blue = experimental, red = calculated). | ||
Then, the temperature effect on the system was examined. NMR experiments were conducted in CD2Cl2 from −55 °C to 25 °C acquiring spectra every 10 °C. In that 80 degrees window, however, in the same experiment the primary regioisomer population composition change (6b-E and 6b-Z) is less appreciable (E-isomer increases only 7% at the same range of temperature, see ESI†). The same tendency was observed when the proportion of the tertiary azide on the equilibrium was analyzed theoretically (Fig. 4).
Strikingly, when the same experiment was conducted on DMSO-d6 to increase the temperature beyond 80 °C, the integrity of the compound was affected based on the wide change on the 1H NMR spectra. The decomposition of the azide was confirmed by the disappearance of the characteristic azide stretching band at 2200 to 2100 cm−1 in the IR spectrum. That unexpected reaction avoids expanding the equilibrium shift beyond that temperature.
The next step was to determine how the solvent polarity affects the equilibrium. 1% v/v solution of the 6b in different deuterated solvents, including non-polar (benzene, toluene, chloroform), polar aprotic (THF, acetone, DCM, DMSO) and a protic solvent (MeOH). Those solvents cover a wide spectrum of dielectric constant (from 2.27 for benzene to 46.83 for DMSO). The analysis of the spectra revealed that the nature of solvents does not appreciably affect the azide population. The proportion of tertiary azide goes from 12.5% in chloroform to 13.7% in methanol, having a distribution between those values for the rest of the solvents. The theoretical calculations agree with the experimental results showing a small population difference among the solvents, demonstrating the low effect of the type of solvent on the population distribution.
It has previously been reported for other allyl azides systems that solvent exchange and temperature can be used to control the equilibrium composition. Thevenet et al. efficiently modulated the reactivity of allylazides controlling the [3,3]-sigmatropic rearrangement changing the solvent and the temperature of the reaction.35 Our results showed that prenyl azides are insensitive to those changes, leaving equilibrium distribution solely dependent on the substrate structure.
Finally, to study how the structure affects the equilibrium, a set of new analogues were proposed. The methyl group should be exchange by a phenyl group (changing electronic and steric factors). Aromatic compounds containing isoprenyl substituents have interesting pharmacological profiles including antimicrobial, antifungal, anti-carcinogenic and anti-HIV activities.36–38 The proposed phenyl derivatives 8 and 15, inspired by prenyl azide mixtures 6a were prepared. In both compounds one methyl group was replaced by an aromatic ring, keeping the other methyl group in one analog or being replace by a hydrogen (Fig. 5).
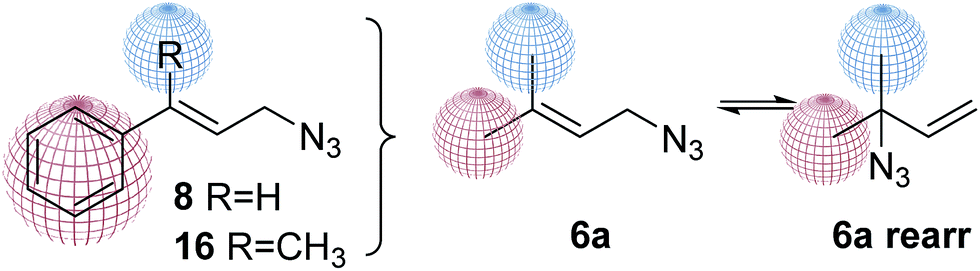 | ||
| Fig. 5 Phenyl prenylazides proposed. | ||
To prepare the phenyl substituted allylic azide 8, cinnamyl alcohol 7 was transformed into the corresponding azide by Thompson's reaction with DPPA and DBU in toluene providing the expected product with 78% yield after purification (Scheme 3). Cinnamylazide 8 was obtained as the only product, maintaining the same regiochemistry of the starting material. This was confirmed by 1H NMR visualization of the signal corresponding to the olefinic proton adjacent to the aromatic ring at 6.66 ppm with a J = 15.8 Hz, distinctive for the E-isomer. The other olefinic signal appears at 6.25 ppm as a double triplet (J = 15.8 and 6.6 Hz).
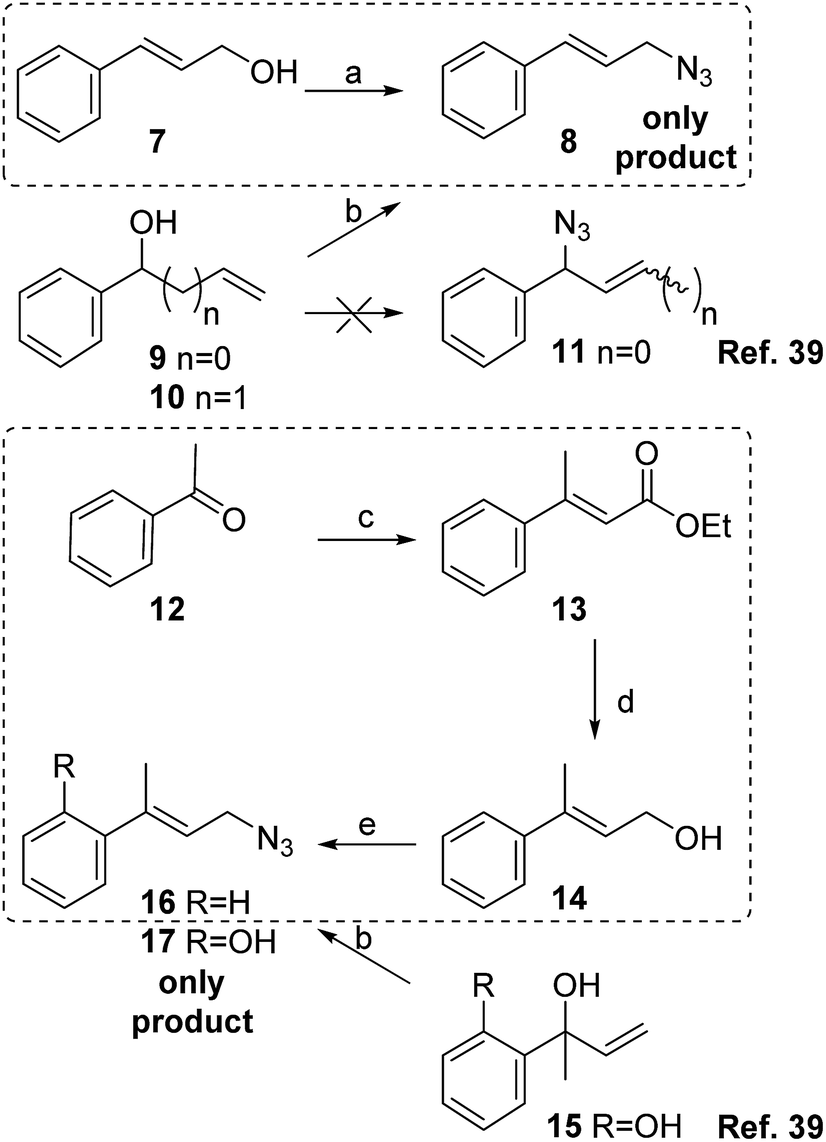 | ||
| Scheme 3 Reagents and conditions: (a) DPPA, DBU, toluene, 12 h, 0 °C → rt, 78%; (b) BF3·Et2O, TMSN3, DCM (ref. 39); (c) triethylphosphonoacetate, NaH, dimethoxyethane, 3 h, r.t., 60%; (d) LiAlH4, THF, 1 h, rt, 80%; (d) DPPA, DBU, toluene, 12 h, 0 °C → r.t., 91%. In dotted lines, the synthetic routes developed in the present work. | ||
Srinu et al. have obtained cinnamyl azide 8 from vinylbenzyl alcohol 9 using BF3·Et2O and TMSN3 without isolating the expected product 11.39 These authors proposed a SN2′ with the azide attacking the terminal olefinic carbon and releasing a TMSO as possible mechanism for that reaction. Different authors have also reported the same results using substituted phenyl propenols,16,21,23,40 and different reagents and reaction conditions. The reaction of homoallylic alcohols using TMSN3 under PdCl2 catalysis also produced the conjugated products.41
In order to synthesize the isomeric mixture 16, acetophenone 12 was submitted to a Horner–Wadsworth–Emmons reaction with triethylphosphonoacetate providing the α,β-unsaturated ester 13 in 3 hours with 60% yield (E:Z, 92:8, Scheme 3).
The ester was reduced with LiAlH4 in THF in 1 hour, to get the alcohol 14 (80% yield). In this step, the E-isomer was separated by chromatography. Lastly, the alcohol was transformed into azide 16 by reaction with DPPA and DBU in toluene with 91% yield.
The 1H NMR spectrum of product 16 shows a triplet at 5.88 ppm with a J = 6.7 Hz for the olefinic proton coupled with the protons of the allylic methylene of 3.99 ppm (J = 6.7 Hz). These signals are a clear indication that only one isomer has been formed without experiencing the [3,3]-sigmatropic rearrangement. Those results are also in agreement with the work reported by Srinu et al. where only the primary azide 16 was obtained starting from the alcohol 14 or an isomeric alcohol 1539 (Scheme 3).
In summary, our results and the literature precedents clearly showed that, in any case starting from conjugated allylic alcohols or their isomers phenethyl vinyl alcohols, only one conjugated product is obtained.
Products from the reactions with cinnamyl alcohol 7 and its methyl substituted derivative 14 retain their regiochemistry demonstrating that the presence of the aromatic ring disfavours the [3,3]-sigmatropic rearrangement due to stereoelectronic and inductive effects.
To rationalize the experimental evidences that showed a high dependency of the double bond substitution on the equilibrium of allylic alcohols, a theoretical study was performed. In contrast to the large number of experimental and theoretical investigations aimed to understand sigmatropic rearrangements of different systems,35,42,43 azides has received scarce attention, especially from the theoretical point of view. In addition, an analysis based on the quantum theory of atoms in molecules (QTAIM)44,45 was performed. Such studies provide information of the electron charge distribution involved in the [3,3]-sigmatropic rearrangements of 6a, 8 and 12. That approach is based on the topological analysis of the electron charge density allowing a clear insight into structure and the stability of a chemical species.
Theoretical studies of 6a, 8 and 16
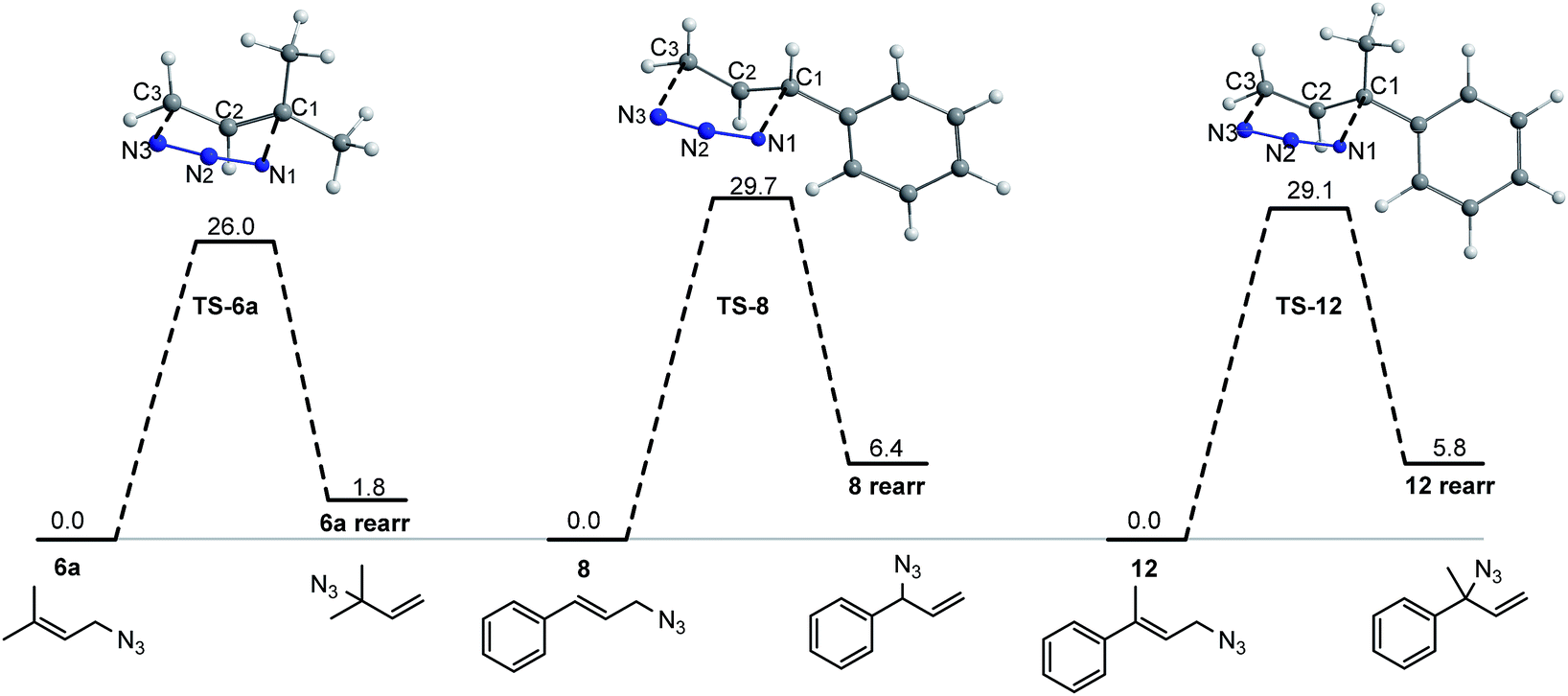 | ||
| Fig. 6 MPWB1K/6-311++G(d,p) free energy profiles for the [3,3]-sigmatropic rearrangement of allylic azides 6a, 8 and 16. Relative free energies in toluene are in kcal mol−1. The optimized geometries (in 3D) for the transition structures are showed. | ||
Several topological properties evaluated at a bond critical point (bcp) are used to characterize the nature of a bonding interaction (calculated properties at the bcp are labelled with the subscript “b”).
The charge density, ρb, reflects the bond strength and its Laplacian, ∇2ρb gives information about the local charge concentration (∇2ρb < 0) or depletion (∇2ρb > 0).44,45 The ellipticity, ε, provides information about the charge distribution around the bond path and can be used to determine the bond stability and its π character.46 The total energy density, Hb, (Hb = Gb + Vb, being Gb and Vb the potential and the kinetic energy densities at the bcp, respectively), which designs the density of the total electronic energy at a bcp, is used to analyse the covalent character of an interaction.47 Also, the delocalization index, DI, is an important parameter that indicates the amount of the electron pairs number shared by two atoms.48,49
The molecular graphs of the TSs under study are shown in Fig. 7 and the topological properties evaluated at the selected bcps are listed in Table 1 along with the bond distances (such properties for another bcps are given in the ESI†).
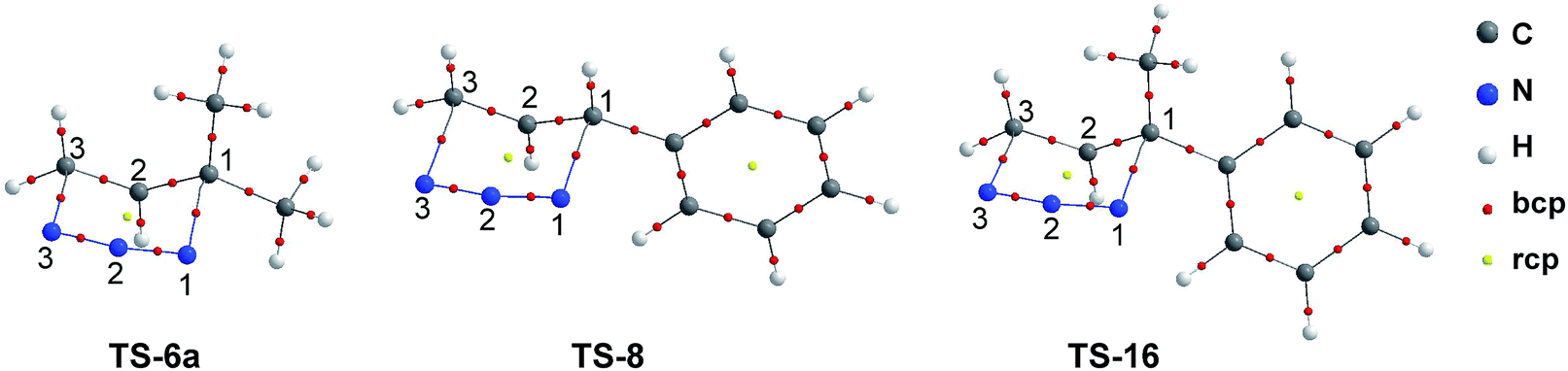 | ||
| Fig. 7 Molecular graphs of the TSs corresponding to [3,3]-sigmatropic rearrangement of allylic azides 6a, 8 and 12. | ||
| Species | Bonds | R | ρb | ∇2ρb | ε | Hb | DI |
|---|---|---|---|---|---|---|---|
| a Bond distances (R) in Å; ρb, ∇2ρb, Hb in au. ε and DI are dimensionless.b To identify the atoms, see Fig. 7. | |||||||
| TS-6a | C1–N1 | 2.14 | 0.058 | 0.091 | 0.142 | −0.072 | 0.32 |
| C3–N3 | 2.11 | 0.059 | 0.095 | 0.104 | −0.009 | 0.37 | |
| TS-8 | C1–N1 | 2.08 | 0.063 | 0.092 | 0.133 | −0.011 | 0.37 |
| C3–N3 | 2.15 | 0.054 | 0.096 | 0.120 | −0.007 | 0.34 | |
| TS-16 | C1–N1 | 2.13 | 0.059 | 0.089 | 0.138 | −0.009 | 0.33 |
| C3–N3 | 2.17 | 0.052 | 0.095 | 0.123 | −0.006 | 0.33 | |
The molecular graphs of TSs show the bcps associated with forming C1–N1 and breaking C3–N3 bonds and a ring critical point (rcp) related to a half-chair cyclic structure.
In TS-6a, the values at the C1–N1 bcp of ρb (0.058 au.), ∇2ρb (0.091 au.), and DI (0.32) are slightly lower than these calculated at the C3–N3 bcp (ρb = 0.059 au., ∇2ρb = 0.095 au., and DI = 0.37). In contrast, in TS-8 and TS-16, the values of ρb and DI at C1–N1 bcp are slightly higher than these calculated at the C3–N3 bcp (except DI in TS-16, that in both bcps take the same value). Therefore, in TSs with aromatic ring substituent, the formation/rupture of C1–N1/C3–N3 bonds are more advanced than in TS with methyl substituents. These facts are also reflected toward the bond distances.
The topological properties evaluated at C1–C2 and C2–C3 bcps (see the ESI†) indicate that these bonds have an intermediate character between single and double covalent bond, due to the delocalization of the “π” electrons upon the C1–C2–C3 fragment. The double bond character is higher for the C2–C3 bond, being more notable in TS-8 and TS-16.
The distances and topological properties of N1–N2 and N2–N3 bonds are quite similar, however, these show that the “π” electron density is slightly more localized on the N2–N3 bond particularly in TS-8 and TS-16. The geometric and topological results support the existence of π-electron delocalization in the TSs and strongly suggest that the effect of the aromatic substituent is strictly electronic, thus the contribution of steric factor to the stability of the systems must be scarce or null.
In addition, an analysis of the variation of the topological properties at the selected bcps (C1–N1, C3–N3, C1–C2, C2–C3, N1–N2, N2–N3) along the reaction courses was carried out (see the ESI†). Such analysis has been successfully applied to rationalize the mechanism of a wide variety of chemical transformations.46,50–58 The profiles of topological properties evaluated at the selected bcps showed similar patterns for all reaction pathways. The evolution of the topological properties of the pairs of bcps C1–N1 and C3–N3, C1–C2 and C2–C3, and N1–N2 and N2–N3, are complementary demonstrating that a continuous electron transfer occurs. These results reinforce the fact that the effect of the aromatic substituent is strictly electronic.
Fig. 8 displays the changes in atomic population ΔN(Ω) and atomic energy ΔE(Ω) in the TSs respect to the corresponding reactants (6a, 8 and 16). The changes in such properties in the group of atoms, R1 and R2 that form the substituent fragments are also included. The net charges calculated for selected atoms in the TSs and azides 6a, 8 and 16 are showed in Table 2.
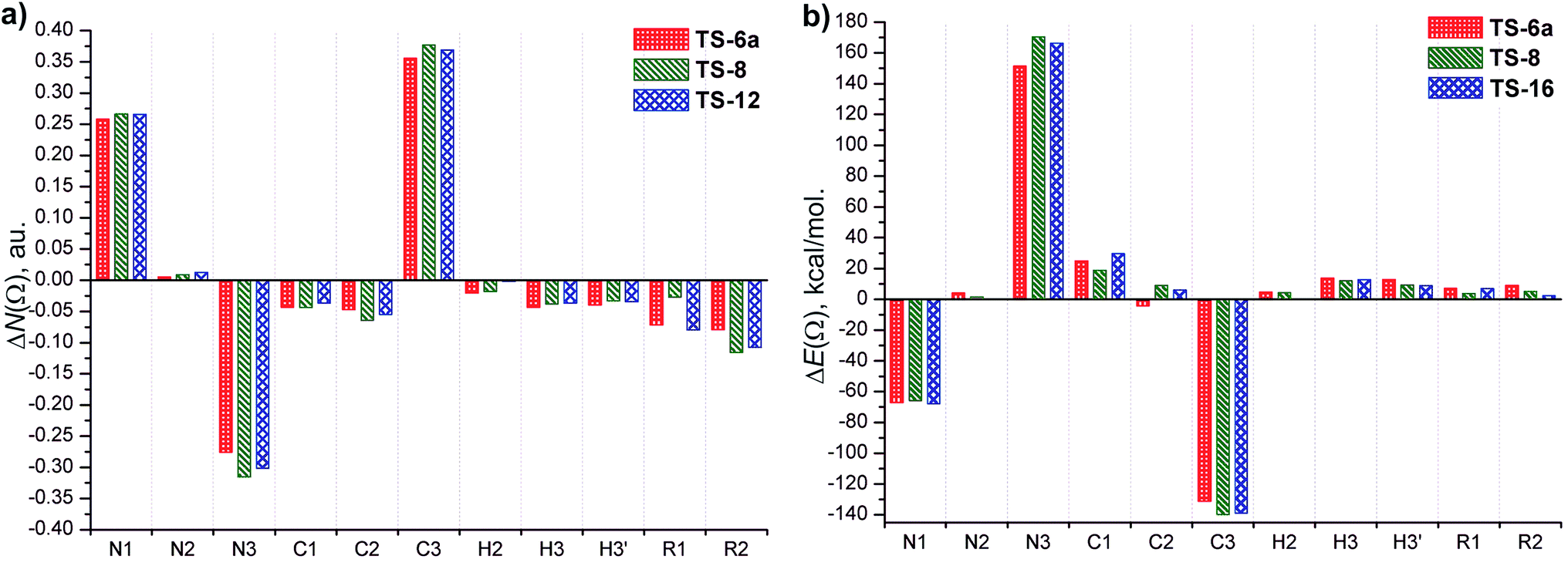 | ||
| Fig. 8 Variation of (a) electron population, ΔN(Ω), and (b) energy, ΔE(Ω), for atoms and atom groups at the TSs with respect to the corresponding reactants. ΔN(Ω) < 0 and ΔN(Ω) > 0 indicate that the atom loses and gains electron population. ΔE(Ω) < 0 and ΔE(Ω) > 0 indicate that the atom is stabilized and destabilized at TS regarding to the corresponding reactants, respectively. R1 = Me, H, Me and R2 = Me, Ph, Ph in TS-6a, TS-8 and TS-16, respectively. See Fig. 7 to identify atoms label. | ||
| Species | q(C1) | q(C2) | q(C3) | q(N1) | q(N2) | q(N3) |
|---|---|---|---|---|---|---|
| 6a | −0.012 | −0.035 | +0.352 | +0.124 | −0.213 | −0.402 |
| TS-6a | +0.031 | +0.012 | −0.004 | −0.134 | −0.219 | −0.127 |
| 8 | −0.022 | −0.045 | +0.374 | 0.138 | −0.211 | −0.420 |
| TS-8 | +0.022 | +0.019 | −0.003 | −0.129 | −0.220 | −0.105 |
| 16 | −0.010 | −0.038 | +0.359 | +0.136 | −0.213 | −0.414 |
| TS-16 | +0.027 | +0.017 | −0.010 | −0.131 | −0.226 | −0.112 |
The atoms in TSs whose atomic population and atomic energy were significantly affected respect to the reactant 6a, 8 and 16 (i.e., |ΔN(Ω)| > 0.02 e, and |ΔE(Ω)| > 20 kcal mol−1) are N1, N3 and C3. The C1 atom shows a considerable variation in its atomic energy, however, the change in its atomic population is less important. Thus, the largest changes of atomic properties to reach the TSs occur in the atoms involved in the forming and breaking bonds, particularly in the latter one.
The atomic properties of the remaining atoms and the group of atoms, R1 and R2, show relative lower changes in achieving the TSs. Overall, these atoms lose electron population and are destabilized (except N2, which increases its electron population and is stabilized, and C2 in TS-6a, which decreases its atomic energy).
N(Ω) of C1 atom decrease taking its net charge values positive, +0.031, +0.022 and +0.027 e for TS-6a, TS-8 and TS-16, respectively (Table 2). The destabilization of C1 increases in the order TS-8 (19 kcal mol−1) < TS-6a (25 kcal mol−1) < TS-16 (30 kcal mol−1). The decrease of N(Ω) and the destabilization of C1 are related with the loss of “s” character.
Because of the C3–N3 bond is breaking, an increase of “s” over “p” character in C3 occurs and in consequence, it gains electron population. The largest amount of electron population gained by C3 comes mainly from all atoms of allylic fragment and R1 and R2 groups, which lose electron population. The π-electrons of aromatic substituent in TS-8 and TS-16 may be better delocalized than the electrons donated by the methyl group in TS-6a, and the electron transfer toward C3 may occur due to the delocalization of the charge density of the double bond C1–C2. Consequently, the C3 atom in TS-8 (0.37 e) and TS-16 (0.36 e) gains a slightly greater electron population than in TS-6a (0.35 e), and it is more stabilized in the former TSs (ΔE(C3) = −140 and −138 kcal mol−1 for TS-8 and TS-16, and −131 kcal mol−1 for TS-6a). In addition, a good linear correlation between ΔN(C3), ΔE(C3) and ΔG#(R2 = 0.891 and 0.995, respectively) is obtained (see the ESI†). Thus, ΔN(C3) reflects the effect of the different substituents upon the electron delocalization but the stabilization of C3 is inverse to the values of ΔG#, and therefore does not explain the high energy barriers.
The loss of electron population of N3 increases in the order TS-6a (−0.27 e) > TS-16 (−0.30 e) > TS-8 (−0.31 e), and its net charge becomes less negative (Table 2) due to the partial breaking of C3–N3 bond. This trend is contrary to the increase of electron population of C3, and reflects the effect of the substituents. In addition, a good linear correlation between ΔN(N3) and ΔG# (R2 = 0.921), is obtained (see the ESI†). N3 is destabilized due to the loss of a great amount of electron population in achieving the TSs. The increase of ΔE(N3) follows the order TS-16 (170 kcal mol−1) > TS-8 (166 kcal mol−1) > TS-6a (151 kcal mol−1), which is in line with the increase of free activation energy calculated. Accordingly, a strong linear dependence of ΔE(N3) on ΔG# is established (R2 = 0.994, see the ESI†). These results clearly demonstrate that the relatively high free activation energy for the [3,3]-sigmatropic rearrangement under study come from the great destabilization of N3.
In a related reaction, the Cope rearrangement of 1,5-hexadiene, the great destabilization of two ethylenic terminal carbons (C1 and C6) was considered as the “source” of the high barrier for this reaction. In the case of the sigmatropic rearrangement of isoprenylazides, the particular redistribution of the electron charge density which makes the destabilization of N3, is attributed to the singular electronic characteristic of the azide group.
The charge loss by N3 achieving the TSs is mainly transferred to N1, which increased its electron and is stabilized, but less than C3. The stabilization of the N1 and C3 atoms in the TSs contributes to lowering the energy barrier although the destabilization of N3 is more relevant.
Additionally, an analysis of the evolution of ΔN(Ω) and ΔE(Ω) along the course of reaction was performed (see the ESI†). Such analysis demonstrates that the atomic properties of C1, C3, N1 and N3 are significantly affected along the reaction pathways. Interestingly, the profiles of ΔE(N3) have a similar pattern but show larger differences in quantitative sense which is in agreement with the energy barriers of the reaction under study.
Conclusions
Since the advent of click chemistry, azides play a key role in the synthesis of 1,2,3-triazoles. Isoprenyl azides can be considered an important synthetic intermediate to generate new chemotherapeutic entities. Their use has been disfavoured since they tend to produce Winstein's rearrangements, producing a mixture of chromatographically inseparable regioisomers.In this work, isoprenylic azides were synthesized, using NMR spectroscopy to identify the regioisomers' ratio. It was observed that the ratio remains unchanged at room temperature regardless the number of isoprene units, regiochemistry and the substitution of the starting isoprenol. Additionally, when isoprene double bond was decorated with aromatic rings, the azide do not experiment the [3,3]-sigmatropic rearrangement. The equilibrium composition dependence with the temperature and the solvent were also analysed. From −55 to 25 °C, the tertiary azide proportion increases only 15% (11.2% to 13.2%). The study curry out on eight different solvents showed that the equilibrium is not appreciably affected. Those results were confirmed by theoretical calculation where the energy difference for the isomers on different solvents has low impact on the equilibrium population distribution. Together, those results proved that isoprenyl azides cannot be experimentally obtained as pure isomer.
To rationalize the experimental results, theoretical studies were performed using density functional theory and the QTAIM approach. The free energy barrier and the endothermicity of the reaction involving aromatic azides were higher than that of methyl azides. Thus, Winstein' rearrangement of aromatic systems is kinetically and thermodynamically disfavoured which is in agreement with the experimental results. The topological analysis of the electron charge density revealed that the rearrangement takes place via a concerted mechanism involving a half-chair TS in which a significant redistribution of the charge density occurs. Additionally, it was demonstrated that the effect of the aromatic substituent is purely electronic.
The analysis of atomic properties allowed pinpoint the atoms or region that experience the greatest changes in electron population and energy in achieving the TSs. Although the effect of the substituent affects the properties of the whole systems, it was found that the destabilization of N3 plays a key role in the larger energy barrier, and clearly reflects the degree of electron delocalization caused by the substituent effect.
Therefore, the formation of the tertiary regioisomers through [3,3]-sigmatropic rearrangement may be controlled by modifying the electronic structure of isoprenylazides.
Experimental
General information
1H and 13C NMR spectra were acquired on a BrukerAvance II 300 MHz (75.13 MHz) using CDCl3 as solvent. Chemical shifts (δ) were reported in ppm downfield from tetramethylsilane (TMS) at 0 ppm as internal standard and coupling constants (J) are in hertz (Hz). Chemical shifts for carbon nuclear magnetic resonance (13C NMR) spectra are reported in parts per million relative to the center line of the CDCl3 triplet at 76.9 ppm. The following abbreviations are used to indicate NMR signal multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, p = pentuplet, m = multiplet, br = broad signal. Electrospray and Atmospheric Pressure Chemical Ionization High-resolution mass spectra (ESI-HRMS, APCI-HRMS) were recorded on a BrukerMicroTOF II with lock spray source. IR spectra were obtained using an FTIR Shimadzu spectrometer and only partial spectral data are listed. Chemical reagents were purchased from commercial suppliers and used without further purification, unless otherwise noted. Solvents were analytical grade or were purified by standard procedures prior to use. Yields were calculated for material judged homogeneous by thin layer chromatography (TLC) and nuclear magnetic resonance (1H-NMR). All reactions were monitored by thin layer chromatography performed on silica gel 60 F254 pre-coated aluminum sheets, visualized by a 254 nm UV lamp, and stained with an ethanolic solution of 4-anisaldehyde. Column flash chromatography was performed using silica gel 60 (230–400 mesh).General procedure for prenyl azides synthesis
One equivalent of the prenyl alcohol substrate was dissolved in anhydrous toluene at 0 °C. Then, 1.3 equivalents of diphenylphosphorylazide (DPPA) were added followed by the addition of 1.3 equivalents of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). After 2 h of continuous stirring at 0 °C, the reaction was warmed to room temperature. Finally, the mixture of reaction was quenched with water. The aqueous layer was extracted 3 times with ethyl acetate and the combined organic layers concentrated under vacuum. The residue was purified on a silica gel column (hexane).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 2928 (–CH2–), 2884 (–CH2–C–N–), 2098 (–N3), 1664 (–CC–).
CH–), 2928 (–CH2–), 2884 (–CH2–C–N–), 2098 (–N3), 1664 (–CC–).The spectra of 1H and 13C coincide with spectra obtained of mixture of allylic azides from the geraniol.
CH–), 2926 (–CH2–), 2856 (–CH2–C–N–), 2097 (–N3), 1665 (–CC–).CH–), 2928 (–CH2–), 2882 (–CH2–C–N–), 2096 (–N3), 1660 (–CC–).:ethyl acetate) to afford 205 mg of colourless oil (isolated yield: 60%, ratio E/Z 92:8). 1H NMR (CDCl3): δ 7.47 ppm (m, 2H, meta aromatics, C3–H and C5–H); 7.37 (m, 3H, ortho and para aromatics, C2–H, C4–H and C6–H); 6.14 (dd, 1H, olefinic, C8–H, 4JH8–H10 = 1.3 Hz); 4.22 (q, 2H, 3JH11–H12 = 7.1 Hz, C11–H); 2.58 (d, 3H, 4JH10–H8 = 1.2 Hz, C10–H) and 1.32 (t, 3H, 3JH12–H11 = 7.1 Hz, C12–H). 13C NMR (CDCl3): δ 166.9 (C9, carbonyl); 155.5 (C7, quaternary olefinic); 142.3 (C1, ipso aromatic); 129.0 (C4, para aromatic); 128.5 (C3 and C5, meta aromatics); 126.3 (C2 and C6, ortho aromatics); 117.2 (C8, CH, olefinic); 59.8 (C11, O–CH2–); 17.9 (C10, CH3) and 14.3 (C12, CH3). HRMS: mass calculated for C12H15O2+ (M + H+), 191.10666; found, 191.10670. IR (film): υmax (cm−1) 2978 (CH–), 2926 (–CH2–), 1713 (CO), 1628 (CC).CH–), 2926 (–CH2–), 1713 (CO), 1628 (CC).:water (90:10), followed by the addition of 5 mL of a NH4Cl solution (10% w/w). A partial evaporation of the mixture is performed to remove interfering solvent. The inorganic phase was extracted 3 times (3 × 15 mL) with ethyl ether and the combined organic layers concentrated under vacuum. The residue was purified on a silica gel column (hexane:ethyl acetate) to afford 34 mg of colourless oil (isolated yield: 80%). 1H NMR (CDCl3): δ 7.42 ppm (m, 2H, meta aromatic, C3–H and C5–H); 7.37 (t, 2H, ortho aromatics, C2–H and C6–H); 7.21 (d, 1H, para aromatic, C4–H); 5.98 (dt, 1H, 3JH8–H9 = 6.7 Hz, 4JH8–H10 trans = 1.3 Hz, C8–H); 4.37 (q, 2H, 3JH9–H8 = 6.7 Hz, C9–H) and 2.09 (d, 3H, 4JH10–H8 = 1.3 Hz, C10–H). 13C NMR (CDCl3): δ 142.9 ppm (C1, ipso aromatic); 128.5 (C7, quaternary olefinic); 128.3 (C3 and C5, meta aromatics); 127.0 (C4, para aromatic); 125.8 (C2 and C6, ortho aromatics); 60.0 (C9, O–CH2–) and 16.0 (C10, CH3). HRMS (ESI): mass calculated for C10H13O+ (M + H+), 149.09609; found, 149.09610. IR (film): υmax (cm−1) = 3402 (O–H), 2969 (CH–), 2921 (–CH2–).C).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to express their gratitude to UNR (Universidad Nacional de Rosario), Fundación Josefina Prats, CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas, PIP 2009-11/0796 and PIP 2012-14/0448), Agencia Nacional de Promoción Científica y Tecnológica (PICT-2011-0589). MMV thanks UNNE, SECYT-UNNE (PI F005-2013), and ANPCyT (PICT-2015-2635). This investigation also received financial support (through GRL) from the UNICEF/UNDP/WORLD BANK/WHO Special Programme for Research and Training in Tropical Diseases (TDR). GRL, MMV and ABJB are members of the Research Career of CONICET. EOJP thanks CONICET for the award of a Fellowship.Notes and references
- T. Zheng, S. H. Rouhanifard, A. S. Jalloh and P. Wu, Top. Heterocycl. Chem., 2012, 28, 163–183 CAS.
- S. Schricker, M. Palacio, B. T. Thirumamagal and B. Bhushan, Ultramicroscopy, 2010, 110, 639–649 CrossRef CAS PubMed.
- D. Dheer, V. Singh and R. Shankar, Bioorg. Chem., 2017, 71, 30–54 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- A. Gagneuz, S. Winstein and W. G. Young, J. Org. Chem., 1969, 1, 5956–5957 Search PubMed.
- C. A. VanderWerf and V. L. Heasley, J. Org. Chem., 1966, 31, 3534 CrossRef CAS.
- B. M. Trost and S. R. Pulley, Tetrahedron Lett., 1995, 36, 8737–8740 CrossRef CAS.
- A. J. DeGraw, C. Palsuledesai, J. D. Ochocki, J. K. Dozier, S. Lenevich, M. Rashidian and M. D. Distefano, Chem. Biol. Drug Des., 2010, 76, 460–471 CAS.
- Y. Blache, O. Bottzek, J.-F. Briand, L. Dombrowsky and A. Praud-Tabaries, Tetrahedron Lett., 2009, 50, 1645–1648 CrossRef.
- E. O. Porta, P. B. Carvalho, M. A. Avery, B. L. Tekwani and G. R. Labadie, Steroids, 2014, 79, 28–36 CrossRef CAS PubMed.
- J. P. Smits, D. F. Wiemer, X. Zhou, E. J. Born, S. V. Hartman, S. A. Holstein and D. F. Wiemer, Bioorg. Med. Chem. Lett., 2013, 23, 764–766 CrossRef PubMed.
- M. H. Ngai, P. Y. Yang, K. Liu, Y. Shen, M. R. Wenk, S. Q. Yao and M. J. Lear, Chem. Commun., 2010, 46, 8335–8337 RSC.
- E. O. J. Porta, S. N. Jager, I. Nocito, G. I. Lepesheva, E. C. Serra, B. L. Tekwani and G. R. Labadie, MedChemComm, 2017, 8, 1015–1021 RSC.
- G. Papeo, H. Posteri, P. Vianello and M. Varasi, Synthesis, 2004, 2004, 2886–2892 CrossRef.
- A. Jayanthi, V. K. Gumaste and A. R. A. S. Deshmukh, Synlett, 2004, 979–982 CAS.
- T. Kanai, Y. Kanagawa and Y. Ishii, J. Org. Chem., 1990, 55, 3274–3277 CrossRef CAS.
- M. Arimoto, H. Yamaguchi, E. Fujita, M. Ochiai and Y. Nagao, Tetrahedron Lett., 1987, 28, 6289–6292 CrossRef CAS.
- S. Murahashi, Y. Taniguchi, Y. Imada and Y. Tanigawa, J. Org. Chem., 1989, 54, 3292–3303 CrossRef CAS.
- E. J. Alvarez-Manzaneda, R. Chahboun, E. Cabrera Torres, E. Alvarez, R. Alvarez-Manzaneda, A. Haidour and J. M. Ramos Lopez, Tetrahedron Lett., 2005, 46, 3755–3579 CrossRef CAS.
- Y. Uozumi, T. Suzuka, R. Kawade and H. Takenaka, Synlett, 2006, 2006, 2109–2113 CrossRef.
- M. Mizuno, T. Shioiri and M. Mizuno, Chem. Commun., 1997, 2165–2166 RSC.
- M. Rueping, C. Vila and U. Uria, Org. Lett., 2012, 14, 768–771 CrossRef CAS PubMed.
- A. S. Thompson, G. R. Humphrey, A. M. DeMarco, D. J. Mathre and E. J. J. Grabowski, J. Org. Chem., 1993, 58, 5886–5888 CrossRef CAS.
- G. R. Labadie, R. Viswanathan and C. D. Poulter, J. Org. Chem., 2007, 72, 9291–9297 CrossRef CAS PubMed.
- Molinspiration chemoinformatics.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908–6918 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Phys., 2004, 120, 9918–9924 CrossRef CAS PubMed.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61–69 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. A. Keith, AIMAll, Version 11.12.19 edn, TK Gristmill Software, Overland Park KS, USA, 2011 Search PubMed.
- A. Padwa and M. M. Sá, Tetrahedron Lett., 1997, 38, 5087–5090 CrossRef CAS.
- S. Murahashi, Y. Taniguchi, Y. Imada and Y. Tanigawa, J. Org. Chem., 1989, 54, 3292–3303 CrossRef CAS.
- N. Thevenet, V. de la Sovera, M. A. Vila, N. Veiga, D. Gonzalez, G. Seoane and I. Carrera, Org. Lett., 2015, 17, 684–687 CrossRef CAS PubMed.
- K.-i. Hayashi, F.-R. Chang, Y. Nakanishi, K. F. Bastow, G. Cragg, A. T. McPhail, H. Nozaki and K.-H. Lee, J. Nat. Prod., 2004, 67, 990–993 CrossRef CAS PubMed.
- T. D. Stark, M. Salger, O. Frank, O. B. Balemba, J. Wakamatsu and T. Hofmann, J. Nat. Prod., 2015, 78, 234–240 CrossRef CAS PubMed.
- M. Iinuma, H. Tosa, T. Ito, T. Tanaka and M. Aqil, Phytochemistry, 1995, 40, 267–270 CrossRef CAS.
- G. Srinu and P. Srihari, Tetrahedron Lett., 2013, 54, 2382–2385 CrossRef CAS.
- J. Le Bras and J. Muzart, Tetrahedron Lett., 2011, 52, 5217–5219 CrossRef CAS.
- P. Surendra Reddy, V. Ravi and B. Sreedhar, Tetrahedron Lett., 2010, 51, 4037–4041 CrossRef CAS.
- S. M. So, L. Mui, H. Kim and J. Chin, Acc. Chem. Res., 2012, 45, 1345–1355 CrossRef CAS PubMed.
- R. Koch, J. J. Finnerty, S. Murali and C. Wentrup, J. Org. Chem., 2012, 77, 1749–1759 CrossRef CAS PubMed.
- C. F. Matta and R. J. Boyd, The Quantum Theory of Atoms in Molecules: from solid state to DNA and drug design, Wiley-VCH, Weinheim, 2007 Search PubMed.
- R. F. W. Bader, Atoms in Molecules. A Quantum Theory, Oxford Science Publications, Clarendon Press, London, 1990 Search PubMed.
- C. S. López, O. N. Faza, F. P. Cossío, D. M. York and A. R. de Lera, Chem.–Eur. J., 2005, 11, 1734–1738 CrossRef PubMed.
- D. Cremer and E. Kraka, Angew. Chem., Int. Ed. Engl., 1984, 23, 627–628 CrossRef.
- X. Fradera, M. A. Austen and R. F. W. Bader, J. Phys. Chem. A, 1999, 103, 304–314 CrossRef CAS.
- G. Merino, A. Vela and T. Heine, Chem. Rev., 2005, 105, 3812–3841 CrossRef CAS PubMed.
- N. H. Werstiuk and W. Sokol, Can. J. Chem., 2008, 86, 737–744 CrossRef CAS.
- E. C. Brown, R. F. W. Bader and N. H. Werstiuk, J. Phys. Chem. A, 2009, 113, 3254–3265 CrossRef CAS PubMed.
- G. Wagner, T. N. Danks and V. Vullo, Tetrahedron, 2007, 63, 5251–5260 CrossRef CAS.
- M. M. Vallejos, N. M. Peruchena and S. C. Pellegrinet, Org. Biomol. Chem., 2013, 11, 7953–7965 CAS.
- M. M. Vallejos and S. C. Pellegrinet, RSC Adv., 2015, 5, 70147–70155 RSC.
- J. E. Rode and J. C. Dobrowolski, J. Phys. Chem. A, 2006, 110, 3723–3737 CrossRef CAS PubMed.
- J. E. Rode and J. C. Dobrowolski, J. Phys. Chem. A, 2006, 110, 207–218 CrossRef CAS PubMed.
- J. E. Rode and J. C. Dobrowolski, Chem. Phys. Lett., 2007, 449, 240–245 CrossRef CAS.
- C. S. López and Á. R. d. Lera, Curr. Org. Chem., 2011, 15, 3576–3593 CrossRef.
- C. F. Matta, A. A. Arabi and T. A. Keith, J. Phys. Chem. A, 2007, 111, 8864–8872 CrossRef CAS PubMed.
- A. A. Arabi and C. F. Matta, J. Phys. Chem. A, 2009, 113, 3360–3368 CrossRef CAS PubMed.
- J. L. López, A. M. Graña and R. A. Mosquera, J. Phys. Chem. A, 2009, 113, 2652–2657 CrossRef PubMed.
- M. M. Vallejos, E. L. Angelina and N. M. Peruchena, J. Phys. Chem. A, 2010, 114, 2855–2863 CrossRef CAS PubMed.
- M. M. Vallejos and N. M. Peruchena, J. Phys. Chem. A, 2012, 116, 4199–4210 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of all synthesized compounds. Distances, topological properties and electron population along the Cartesian coordinates of the stationary points under study. See DOI: 10.1039/c7ra09759j |
| This journal is © The Royal Society of Chemistry 2017 |