
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel strategy to immobilize enzymes on microporous membranes via dicarboxylic acid halides†
Cuijing Liu,
Daisuke Saeki and
Hideto Matsuyama*
and
Hideto Matsuyama*
Center for Membrane and Film Technology, Department of Chemical Science and Engineering, Kobe University, 1-1 Rokkodai, Nada, Kobe, Hyogo 657-8501, Japan. E-mail: matuyama@kobe-u.ac.jp
First published on 13th October 2017
Abstract
A major challenge in enhancing the acceptability of enzymes in enzymatic membrane bioreactors (EMBRs) for industrial processes lies in effectively immobilizing enzymes on supports while maintaining their conformation and activity. This work describes a novel methodology using dicarboxylic acid halides as a spacer for the surface-initiated, covalent immobilization of enzymes onto microporous membranes. One of the reactive carboxyl groups from the dicarboxylic acid halides was immobilized onto the membrane surface and the other was conjugated with an amino group of enzymes via an active ester method. Sebacoyl chloride (SC) and trypsin were used as models of the dicarboxylic acid halide and enzyme, respectively. The effects of the reaction conditions such as reaction temperature, trypsin concentration, reaction time, and SC concentration were investigated on the immobilization efficiency with respect to the surface density, specific activity, and activity retention of the immobilized trypsin. The optimum surface density was 36 μg cm−2, and the corresponding membrane showed an excellent specific activity of 22 U cm−2 with a high activity retention of 26% in a soaking mode. Furthermore, in a filtration mode, these values largely improved to 118 U cm−2 and 145% respectively due to the enhanced diffusion force, meaning that the immobilized trypsin on/in the membrane has higher activity than non-immobilized, native trypsin. In addition, the immobilized trypsin exhibited a remarkable improvement in thermal resistance, continuous operation capability, and reusability. Furthermore, the presented method was also applicable to lipase. This technique provides a practical and simple method to immobilize enzymes and offers a tool to design the membranes used in EMBRs.
Introduction
Enzymes are types of proteins that act as biological catalysts by recognizing target substrates. There are various different types of enzymes such as proteases, amylases, cellulases, and lipases that facilitate the decomposition or synthesis of organic matter in living organisms.1 Compared to ordinary catalysts, the reaction rates and selectivity of enzymes with respect to their substrates are exceptionally high because of their low activation energy and specific conformations for substrate recognition.2,3 Hence, enzymes have attracted constant attention in many fields including pharmaceuticals, food, fine chemicals, and energy.4–6However, enzymes are limited by several drawbacks such as their low thermal stability, complicated reusability, easy self-cleavage, and self-aggregation.1,7 The immobilization of enzymes onto solid supports is a promising method to overcome these disadvantages,1,8 vastly improving their performance—comparable to the free enzymes in solution.1 Therefore, the proper selection of supports is essential to ensure the complete use of these enzymes. Nanoparticles,9,10 microspheres,11,12 hydrogels,13–15 porous glass,16 and membranes17,18 are commonly used. Nanoparticles and microspheres have a high surface area effective for a large amount of immobilization; nevertheless, the reuse of them often relies on other separation processes like membrane filtration and centrifugation. Directly using microporous membranes as supports, often called as enzymatic membrane bioreactors (EMBRs), permits high surface area, easy reuse, low pressure drop, and relatively low diffusion resistance.
The important point on the enzyme immobilization is how to maintain their conformation and activity on the supports. Generally, enzyme immobilization onto membranes can be achieved by three methods: (a) physical adsorption,19–23 (b) entrapment,24–26 and (c) covalent attachment. Although physical adsorption (via electrostatic, hydrophobic, or hydrophilic interaction) is a simple process, immobilized enzymes are easily eluted when they are utilized due to the relatively weak interactions. The entrapment technique retains the activity of the enzyme as no chemical reaction takes place between the membrane and the enzyme. However, it suffers in performance due to the diffusion barrier and enzyme leakage.27 Covalent attachments simply form strong and stable linkages that prevent enzyme leakage, thereby giving rise to a robust biocatalyst. However, the formation of covalent bonds generally decreases enzyme activity and makes it difficult to efficiently immobilize a large amount of enzyme.28
The utilization of spacer-arms between the membrane surfaces and enzymes provides considerable mobility to the reactive sites and consequently the immobilized enzymes, resulting in the improvement of the immobilization efficiency and enzyme activity. The commonly reported spacers are hydrazine29 and diamine.30–33 Following the introduction of enzymes, one method primarily activates carboxyl groups on the enzymes and then the active esters are conjugated with amino groups on the spacers. However, once the enzymes are activated, the amino groups in the enzymes competitively react with themselves on the membrane surface. This self-crosslinking between enzymes negatively affects their activity, and especially in the case of membranes, causes membrane fouling due to the aggregate formation or multilayer accumulation on/in the membrane.34 Another commonly used method is inserting glutaraldehyde (GA) to conjugate amino groups on membrane surface and enzymes via the Schiff base reaction.20,34–38 However, GA also crosslinks enzymes each other and leads self-crosslinking. If the enzyme activity can be directly added to the end of the spacers on the membrane surfaces, enzyme immobilization efficiency can be improved by avoiding the above self-crosslinking. Although complicated, multi-step reactions were required, Sousa et al. immobilized pig liver esterase onto the surface of hollow fiber membranes using spacer molecules such as 1,4-butanediol diglycidyl ether and aminocaproic acid, which were activated to improve the enzyme activity.32 Since there are only a few reliable methods to immobilize enzymes on supports, only 20% of the industrial biocatalytic processes utilize immobilized enzymes.39
In this study, we report a novel method via dicarboxylic acid halides to immobilize enzymes on commercial microporous membranes (Fig. 1). Dicarboxylic acid halides with different molecular lengths are commercially available and exhibit a high reactivity with amino groups and hydroxyl groups, which are easily introduced onto membrane surfaces via plasma treatment, chemical treatment, polymer coating, and grafting. Trypsin and lipase were chosen as the enzyme model because they are commonly used for digestion of proteins and oils, respectively, as well as industrial production. Sebacoyl chloride (SC), a dicarboxylic acid halide, was used as a spacer and reacted with the surface of a commercial microfiltration membrane made from regenerated cellulose. The unreacted carboxylic end of SC was conjugated with trypsin via an active ester method. These surface-initiated, sequential reactions occur on the membrane surface without self-crosslinking. Therefore, the enzyme molecules are expected to be efficiently immobilized on the membrane surface, largely keeping their molecular conformation and activity. The immobilization parameters were investigated for performance optimization, and the potential application of the resulting membranes was also demonstrated.
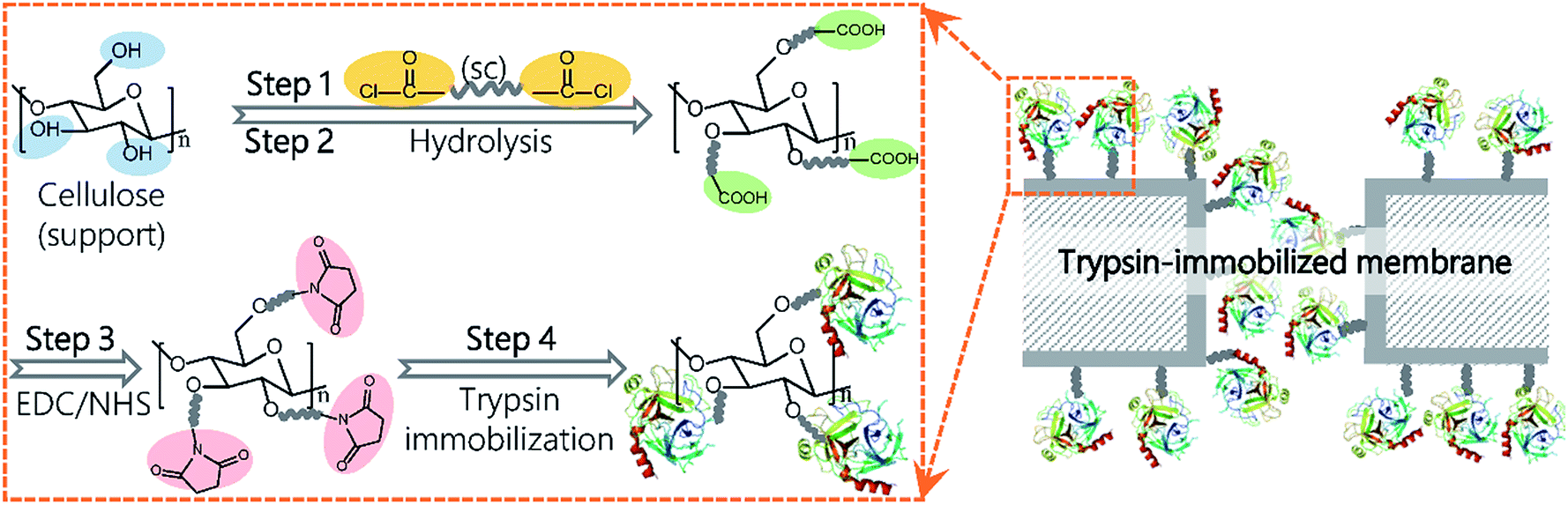 | ||
| Fig. 1 The schematic process for trypsin immobilization on regenerated cellulose membranes. | ||
Experimental
Materials
Regenerated cellulose membranes with 0.2 μm pores were obtained from Merck Millipore (Darmstadt, Germany). Sebacoyl chloride (SC) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) were supplied by Tokyo Chemical Industry (Tokyo, Japan). N-Hydroxysuccinimide (NHS), p-nitrophenyl palmitate (p-NPP) and trichloroacetic acid were provided by Wako Pure Chemical Industries (Osaka, Japan). Benzoyl-L-arginine ethyl ester (BAEE) was obtained from Peptide Institute (Osaka, Japan). Fluorescein isothiocyanate-labeled albumin from bovine serum (FITC-BSA, 67 kDa), trypsin from bovine pancreas (24 kDa), and lipase from Candida rugosa were purchased from Sigma-Aldrich (St. Louis, MO, USA). Bicinchoninic acid (BCA) protein assay kit was purchased from Nacalai Tesque (Kyoto, Japan). Deionized water was produced in a Millipore Milli-Q water purification system. Other chemicals were purchased from Wako Pure Chemical Industries. All reagents were used as received without further purification.Trypsin immobilization
A schematic representation of the enzyme immobilization process is shown in Fig. 1. A regenerated cellulose membrane was immersed in n-hexane containing SC for 10 min and then washed in n-hexane to remove unreacted SC molecules (step 1). Following the evaporation of n-hexane, the membrane was submerged in deionized water for 30 min at 37 °C to completely hydrolyze the acyl chloride groups on the membrane (step 2). The carboxylic groups on the SC-reacted membrane were conjugated with enzymes via an active ester method using EDC and NHS. To activate the carboxylic groups, the membrane was immersed into a 50 mM solution of phosphate buffered saline (PBS, pH 6.0) containing EDC (0.1 M) and NHS (0.1 M) with gentle shaking for 60 min at 25 °C and further washed with PBS (pH 7.6) three times (step 3). Next, the activated membrane was submerged in PBS (pH 7.6) containing trypsin or lipase and incubated in a shaker at 60 rpm for a given amount of time (step 4). Finally, the membrane was washed with PBS (pH 7.6) containing 3 M NaCl for 60 min followed by PBS (pH 7.6) for another 60 min to completely remove the physically and loosely adsorbed enzyme molecules. The membrane was stored in PBS (pH 7.6) at 4 °C for further use.Membrane characterization
To analyze the chemical property, attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy (Nicolet iS5, Thermo Fisher Scientific, Waltham, MA, USA) and X-ray photoelectron spectroscopy (XPS; JPS-9010 MC, JEOL, Tokyo, Japan) were performed. FTIR spectra recorded wavenumbers ranging between 1300–3500 cm−1. Thirty-two scans were taken for each spectrum at a resolution of 2 cm−1. The XPS spectra were recorded with the Al Kα radiation source at a take-off angle of 45°. The whole (0–1000 eV) and narrow spectra of all elements containing high resolutions were recorded. The hydrophilicity of the membrane surface was characterized based on the static contact angle measurement using a contact angle goniometer (Drop Master 300, Kyowa Interface Science, Saitama, Japan). A drop of Milli-Q water (4 μL) was released onto the membrane surface and the contact angle was measured using the images of the water drop on the membrane. At least six measurements were taken from different positions. The surface morphology of the membranes was observed by a field emission scanning electron microscope (FE-SEM; JSF-7500F, JEOL). The samples were freeze-dried (FD-1000, Tokyo Rikakikai, Tokyo, Japan) and then coated with a 5–10 nm osmium layer (Neco-STB, Meiwafosis, Tokyo, Japan). To verify the protein immobilization ability, FITC-BSA was immobilized onto the EDC/NHS activated membrane and the obtained membrane was observed by a confocal laser scanning microscope (CLSM; FV1000, Olympus, Tokyo, Japan). Pure water permeability was measured with a lab-made filtration cell (see Fig. S1†) in a gravity drive mode. The effective area of the used membrane was 3.14 cm2.Trypsin amount quantification
The membrane was analyzed regarding the amount of the immobilized trypsin via BCA assay.40,41 The membrane sample was immersed into BCA working solution, incubated for 30 min at 37 °C, and then agitated for 5 min at 25 °C. The absorbance of this solution at 562 nm was measured using a UV-vis spectrophotometer (V-630, Jasco, Tokyo, Japan). For calibration, free trypsin concentrations of 100, 200, 400, 600, 800, 1000, and 1500 ppm were used. The surface density (D) of the trypsin immobilized on the membrane was calculated from the equation below:| D (μg cm−2) = A/S | (1) |
Activity determination
The trypsin activity was determined by measuring the kinetic hydrolysis of BAEE at 37 °C in a stirred cuvette reactor. Briefly, 75 μL of free trypsin or a trypsin-immobilized membrane (9 mm diameter) was added into a cuvette containing 3 mL of 0.086 mg mL−1 BAEE in 67 mM PBS (pH 7.6) and 125 μL of 1 mM HCl solution. The increase in absorbance at 253 nm (ΔAbs253 nm) was recorded for approximately 20 min by the UV-vis spectrometer. As an index of enzymatic activity, a BAEE unit (U) was used, which is defined as the amount of trypsin required to produce a ΔAbs253 nm of 0.001 per minute with a pH 7.6 BAEE substrate at 25 °C in a reaction volume of 3.2 mL.42,43 We calculated the specific activity (SA) of the membrane at 37 °C as follows:| SA (U cm−2) = 1000[ΔAbs253 nm (sample) − ΔAbs253 nm (blank)]/(tS) | (2) |
For the specific activity of free trypsin at 37 °C:
| SAfree (U μg−1) = 1000[ΔAbs253 nm (sample) − ΔAbs253 nm (blank)]/(tAfree) | (3) |
Meanwhile, the activity retention (R) was calculated as follows:
| R (%) = 100(SA/D)/SAfree | (4) |
Each result was obtained from the average of five parallel tests.
The kinetic parameters were determined according to the Michaelis–Menten equation:
| V = Vmax[S]/(Km + [S]) | (6) |
The turnover number (Kcat) was calculated via the equation:
| Kcat = Vmax/[E] | (7) |
The method for activity determination of lipase was described in the ESI.†
Stability of the immobilized trypsin
The thermal resistance of immobilized trypsin and free trypsin was evaluated by measuring the relative activity at different catalytic temperatures. The specific activity at 37 °C was selected as the benchmark and the corresponding value was 100%. The relative activity was defined as:| Relative activity (%) = 100SAT/SA37 °C | (5) |
The continuous operation capability was studied by measuring the relative activity after incubation in a water bath at 37 °C for various periods of time. The initial specific activity at 37 °C was selected as the benchmark.
The durability of the immobilized trypsin after repeated use was determined as follows: after one operation cycle for 20 min, the membrane was washed twice in 50 mM PBS (pH 7.6) and then soaked in a fresh reaction mixture to assay the enzymatic activity for ten cycles. The specific activity in cycle 1 was selected as the benchmark.
Potential application in enzymatic membrane bioreactors (EMBRs)
To use membranes in EMBRs, the effect of BAEE concentration in the soaking mode and the flow rate in the filtration mode on the activity of the trypsin-immobilized membrane were investigated to obtain a better performance in bioreactors. The apparatus and process for the filtration mode are shown in Fig. S1.†In order to demonstrate the potential application of the trypsin-immobilized membrane in a more practical situation, the catalytic performance against protein was investigated. BSA was chosen as a substrate model. Briefly, a 25 mm diameter membrane was immersed in 6 mL of BSA solution (5000 ppm) in 50 mM PBS (pH 7.6). After incubation at 37 °C for different time periods with stirring, the reaction was terminated by the addition of 1 mL trichloroacetic acid (50 wt%). The trichloroacetic acid denatured and precipitated BSA, leaving the peptide products in the solution. The solution was subsequently centrifuged at 8000 rpm for 3 min and the concentration of the peptides produced in the supernatant by the enzymatic reaction was quantified by measuring the absorbance at 280 nm.
Results & discussion
Membrane characterization
Firstly, the chemical properties of the modified membranes were analyzed by a combination of ATR-FTIR and XPS. Fig. 2(A) shows the FTIR spectra of the membrane surfaces. A new peak appeared at 1740 cm−1 for the SC-immobilized membrane, which verified the presence of carboxyl groups. The peak at 1539 cm−1 existing in the spectrum of the trypsin-immobilized membrane was the characteristic peak of amino groups from trypsin. Table 1 shows the chemical composition of the membrane surface measured by XPS. The C/O ratio increased along with the reaction process (pristine membrane: 1.32; SC-reacted membrane: 1.67; trypsin-immobilized membrane: 2.25).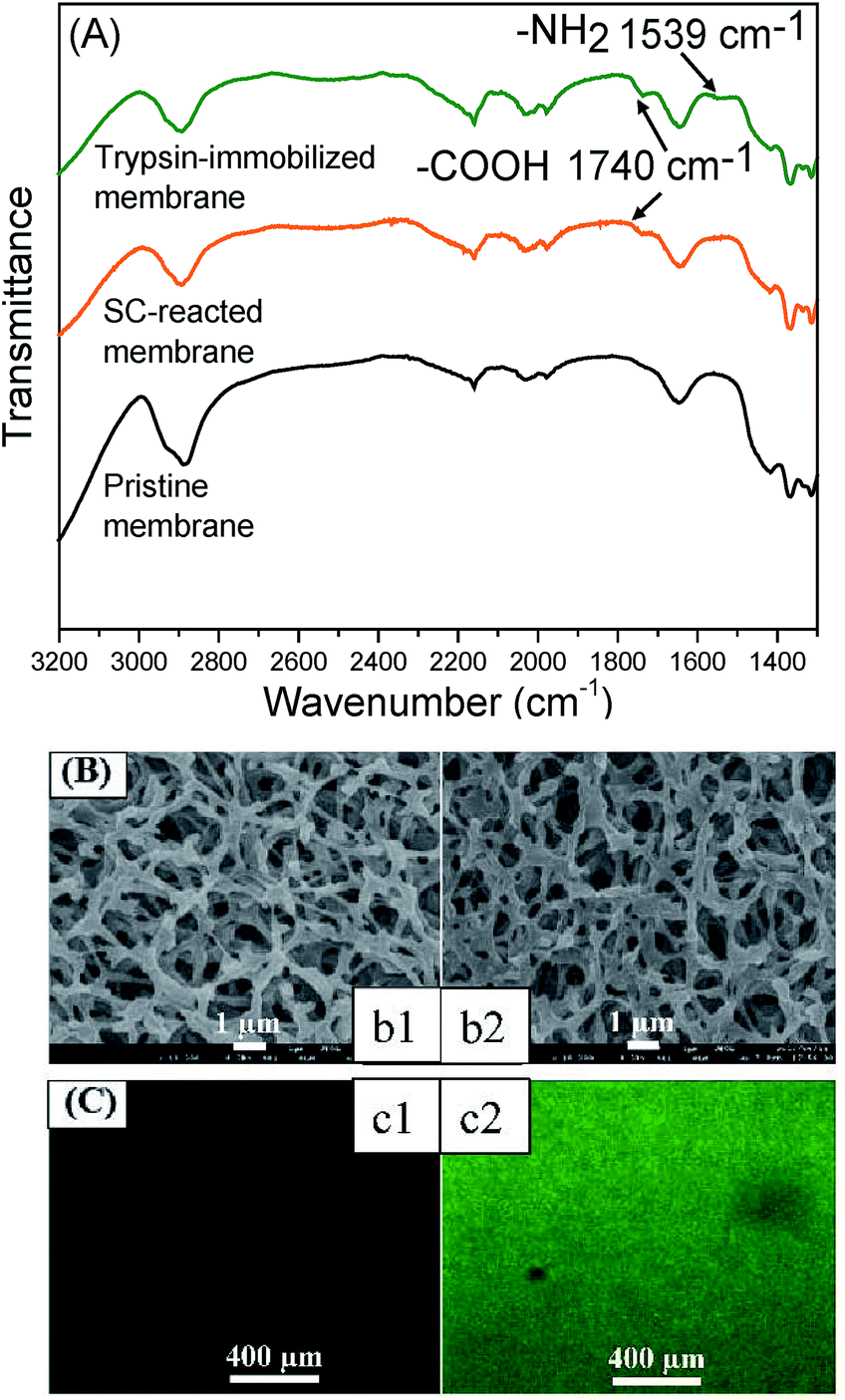 | ||
| Fig. 2 Characterization of the pristine and modified membrane. (A) The FTIR spectra of pristine, SC-reacted, trypsin-immobilized membranes. (B) Surfaces of the (b1) pristine membrane and (b2) trypsin-immobilized membrane. (C) CLSM images of the (c1) pristine membrane and (c2) FITC-BSA-immobilized membranes. | ||
| Sample | Elemental ratio (rel. atom%) | C/O ratio | Contact angle (o) | ||
|---|---|---|---|---|---|
| C 1s | O 1s | N 1s | |||
| Pristine membrane | 56.86 | 43.14 | 0 | 1.32 | 20.4 ± 2 |
| SC-reacted membrane | 62.51 | 37.49 | 0 | 1.67 | 30.7 ± 3 |
| Trypsin-immobilized membrane | 64.50 | 28.60 | 6.89 | 2.25 | 75.2 ± 3 |
Theoretically, molecules of hydrolyzed SC and trypsin have higher C/O ratios than pristine cellulose membrane and thus, the increase in C/O ratio can be explained by the introduction of carboxyl acid groups on the SC-reacted membrane surface and trypsin on the trypsin-immobilized membrane surface. Additionally, the appearance of nitrogen on the trypsin-immobilized membranes also demonstrated the presence of trypsin. The contact angle of the membrane surface, shown in Table 1, was also changed by each modification process. After SC introduction, the contact angle value (30.7 ± 3°) became higher than that of the pristine membrane (20.4 ± 2°), which can be explained by the relative hydrophobicity of the long carbon chain of SC molecules. The trypsin immobilization further changed the contact angle to a more hydrophobic orientation (75.2 ± 3°), which corresponded to some hydrophobic chains in trypsin.
Fig. 2(B) and (C) show the membrane surface morphologies observed by FE-SEM and CLSM, respectively. The SEM images in Fig. 2(B) showed no obvious morphological change or pore blocking after the trypsin immobilization. Furthermore, the water permeability of the trypsin-immobilized membrane was measured to be around 85% of that of the pristine membrane, which indicated that the layer of immobilized trypsin on the membrane surfaces and inner pores was thin enough to not largely affect the permeability. It was consistent with previous studies in which very few enzyme molecules were immobilized.28,41 The CLSM images in Fig. 2(C) revealed that the fluorescence from the immobilized FITC-BSA was uniformly distributed on the membrane surface, indicating that the presented method could be applied to uniformly immobilize various biomolecules.
Trypsin immobilization
The pivotal parameters include reaction temperature, concentrations of SC and trypsin, and reaction time. In this section, the effects of these four factors on membrane performance were investigated. In order to calculate the activity retention (R) of immobilized trypsin, the specific activity of free trypsin (SAfree) was measured and the value was determined to be 2.256 U μg−1. Table 2 shows the effect of reaction temperature on trypsin immobilization. The surface density (D) and specific activity (SA) of the immobilized trypsin at 25 °C were six and two times higher than those at 4 °C respectively, due to the high reactivity between active ester and trypsin at high temperature. The value of D at 25 °C in this study was estimated to be 60 μg cm−2, which was in good agreement with those reported values, ranging from several μg cm−2 to several tens μg cm−2.41,44–47 Most of the previously reported values for SA were low such as 0.1 (ref. 43) or 1 U cm−2.44 However, in this study, SA reached to around 27 U cm−2 due to the high immobilization efficiency and uniform distribution of immobilized trypsin on the membrane surface. The R value of membranes modified at 4 °C was higher than that at 25 °C due to the better mobility of trypsin on the membranes owing to its low density. This corresponded to the rule reported by Koki et al.48 The R value modified at 25 °C was around 20%, which was comparable to the previous reports.49,50 From the comprehensive analysis of D, SA and R at 25 °C supplemented with the nearly unchanged water permeability result, we can conclude that the immobilization efficiency in this study was higher than other previously reported studies. The enzyme immobilization was usually performed at 4 °C to maintain the enzyme activity during the immobilization process.46,48 However, it needs a long reaction time such as 24 h.45 Actually, the trypsin activity did not undergo a major change at 25 °C for several hours, which was verified in the following section. Although more high temperature may improve the immobilization efficiency, it affects the catalytic activity, as described the “Stability of the immobilized trypsin” section. Therefore, 25 °C was chosen for all further experiments.| Reaction temperature (°C) | D (μg cm−2) | SA (U cm−2) | R (%) |
|---|---|---|---|
| 4 | 8 ± 2 | 9 ± 3 | 45 ± 2 |
| 25 | 60 ± 6 | 27 ± 2 | 20 ± 4 |
Fig. 3(A) shows the effects of trypsin concentration on its immobilization. Increasing the trypsin concentration highly facilitated the reaction between trypsin and the membrane surface, thereby increasing D, which reached a maximum threshold of 75 μg cm−2 at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm of the trypsin concentration. However, SA almost achieved its maximum value at 7000 ppm and then nearly approached a plateau. Meanwhile, R decreased continuously from 30% at 100 ppm to 16% at 10000 ppm. This proved that “saturation region” existed for catalysis, implying that extremely high amounts of trypsin did not correlate to higher trypsin activity on the whole membrane. While the number of trypsin active sites on the membrane surface increased with its immobilized amount, the increasing trypsin density contrastingly hindered its activity. The balance of these two opposite phenomena resulted in a steady value for SA. The membrane with a trypsin concentration of 1000 ppm showed the best integrated performance and thus, this concentration was chosen for all following experiments.
000 ppm of the trypsin concentration. However, SA almost achieved its maximum value at 7000 ppm and then nearly approached a plateau. Meanwhile, R decreased continuously from 30% at 100 ppm to 16% at 10000 ppm. This proved that “saturation region” existed for catalysis, implying that extremely high amounts of trypsin did not correlate to higher trypsin activity on the whole membrane. While the number of trypsin active sites on the membrane surface increased with its immobilized amount, the increasing trypsin density contrastingly hindered its activity. The balance of these two opposite phenomena resulted in a steady value for SA. The membrane with a trypsin concentration of 1000 ppm showed the best integrated performance and thus, this concentration was chosen for all following experiments.
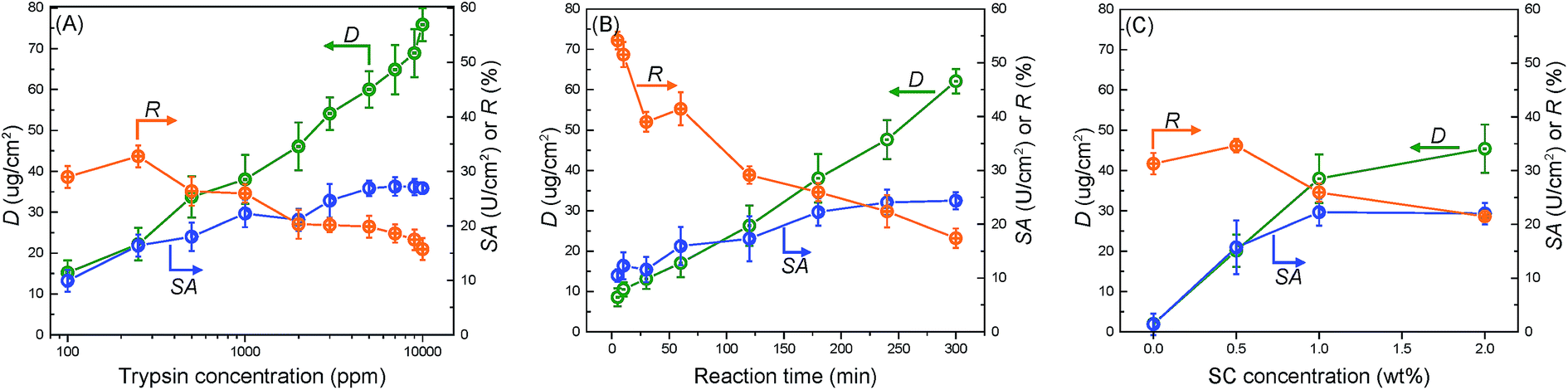 | ||
| Fig. 3 The effect of (A) trypsin concentration, (B) reaction time, and (C) SC concentration on membrane performance factors such as surface density of immobilized trypsin (D), specific activity (SA), and activity retention (R). | ||
Fig. 3(B) and (C) show the effects of the reaction time and SC concentration on trypsin immobilization, respectively. Increasing the reaction time increased D and SA while decreasing R. The SA value reached its stable value at 180 min. Furthermore, increasing the SC concentration improved D (Fig. 3(C)). The high SC concentration probably facilitated the introduction of carboxyl acid groups on the membrane surfaces. An extremely high acyl chloride concentration (2.0 wt%) was not so effective in improving trypsin immobilization because the carboxyl acid groups on the membrane reached saturation.
Fig. 4 summarizes the relationship between the surface density (D) and specific activity (SA). The “critical point” represents the optimum performance values for membranes. The corresponding values for D, SA, and R were around 36 μg cm−2, 22 U cm−2, and 26% respectively. From the results in Fig. 3, the “critical point” was obtained under optimum immobilization condition, which was decided as follows: 25 °C reaction temperature, 1000 ppm trypsin concentration, 180 min reaction time, and 1.0 wt% SC concentration. “Saturation region” represented membranes possessing the immobilized enzyme with the largest surface density and highest specific activity.
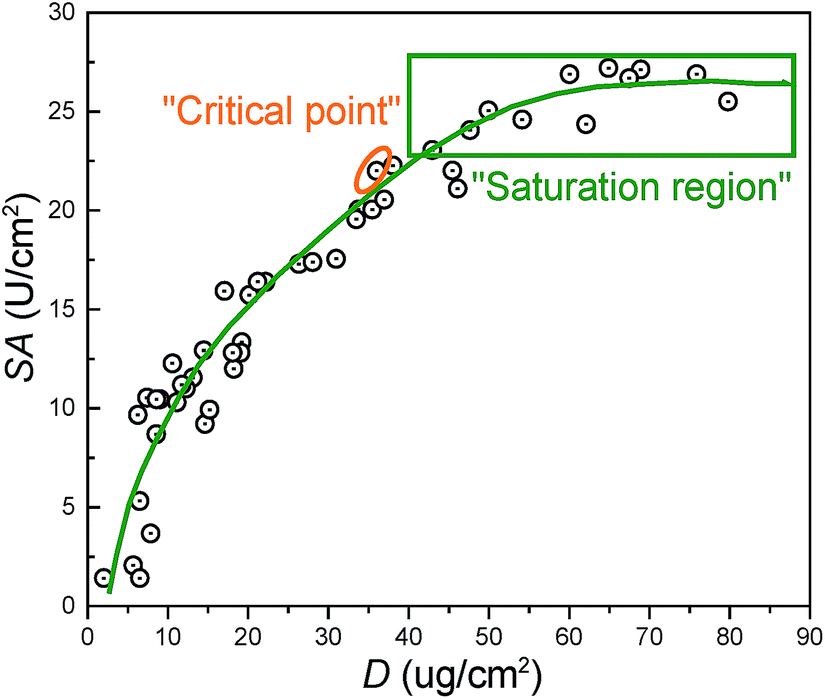 | ||
| Fig. 4 The relationship between the surface density (D) and specific activity (SA) of the immobilized trypsin on the membranes. | ||
Table 3 lists various immobilized enzymes, particularly trypsin, that used polymeric membranes as supports and compared the related performance. The values of SA and R for immobilized trypsin in this work were higher than the trypsin in the other reports, while D was comparable. The immobilization method in this work could efficiently retain the enzymatic activity due to the prevention of self-crosslinking of enzymes, insertion of flexible spacers between enzymes and membrane surfaces, and uniform distribution of enzymes on membranes.
| Enzyme/membraneb | D (μg cm−2) | SAa (U cm−2) | R (%) | Ref. |
|---|---|---|---|---|
| a All the values of SA here were measured at 25 °C.b The “n.d.” in the table represents no reported data. m-PE: modified polyethylene, PE: polyethylene, PVDF: polyvinylidene fluoride, p(HEMA–GMA): poly((2-hydroxyethyl methacrylate)-co-(glycidyl methacrylate)). | ||||
| Trypsin/cellulose | 36 | 13.2 | 26 | This study |
| Trypsin/cellulose | 81 | 0.2 | 11 | 49 |
| Trypsin/m-PE | n.d. | 0.1 | n.d. | 43 |
| Trypsin/PE | 14 | n.d. | 25 | 50 |
| Trypsin/PVDF | 3 | n.d. | n.d. | 28 |
| Lipase/cellulose | 18 | 25 | 119 | This study |
| Gox/cellulose | 40 | n.d. | 19 | 51 |
| Pig liver esterase/nylon | 2 | n.d. | 22 | 32 |
| Urease/p(HEMA–GMA) | 16 | n.d. | 27 | 52 |
The kinetic parameters of trypsin were determined using Lineweaver–Burk plots (Fig. S2†) to evaluate the catalytic performance. The maximum reaction rate (Vmax) and the turn over number (Kcat) of the immobilized trypsin were lower than that of the free trypsin because of the lower diffusion rate in the membrane pores, resulting in the decrease of the reaction rate of the immobilized trypsin in the pores. The Michaelis constant (Km) were higher for the immobilized trypsin due to the introduction of stable covalent bonding and restriction of conformation change.
To demonstrate the applicability for various types of enzymes, lipase was also immobilized via the same method. The performance of free lipase and immobilized lipase was shown in Table 3 and Fig. S3.† The relative activity of immobilized lipase to free lipase was 119%, and the thermal stability of lipase was also improved after immobilization.
Stability of the immobilized trypsin
The thermal resistance of the immobilized enzymes is one of the most important criteria in actual application.53 Fig. 5(A) shows the effect of the catalytic temperature on the relative activity. Generally, the effect of the thermal resistance of immobilized enzymes, especially for the covalently bound system, is higher than that of free enzymes.2 The optimum temperature of free trypsin was observed to be about 37 °C, whereas the immobilized trypsin was observed to be approximately 45 °C. The increase in the optimum temperature indicates that the thermal resistance of trypsin was improved by the immobilization onto the membrane surfaces. The covalent bond formation between trypsin and the membrane surfaces suppressed the conformational change of trypsin, which resulted in a higher activation energy needed for the enzyme to reorganize itself into the proper conformation in order to bind to the substrate molecules.12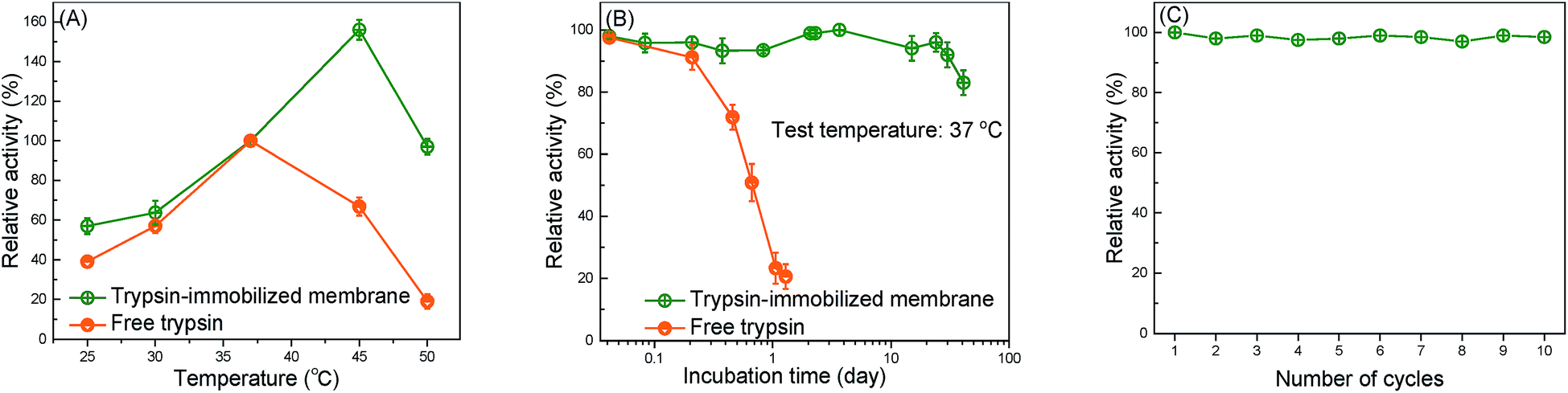 | ||
| Fig. 5 Evaluation of the stability of the immobilized trypsin (A) the effect of the catalytic temperature on the relative activity of the trypsin-immobilized membrane and free trypsin. (B) Time courses of the relative activity of the trypsin-immobilized membrane and free trypsin at 37 °C. (C) The relative activity of trypsin-immobilized membrane during ten operation cycles. | ||
The capability of the trypsin-immobilized membrane to continuously operate at 37 °C was investigated and the result is shown in Fig. 5(B). At 37 °C, the activity of free trypsin reduced to 20% of the initial value only during the 20 h incubation. However, the activity of trypsin-immobilized membrane kept the initial value even after the 24 day incubation, suggesting excellent durability over a long-term operation. The durability of the enzyme-immobilized membranes was reported to be not so effective in many previous studies. For example, a lipase immobilized membrane lost 20% of its original activity after storing at 4 °C for 15 days.36 However, our immobilized enzyme showed better durability. Fig. 5(C) shows the reusability. Its activity hardly decreased even after ten cycles. This result indicated no leakage and denaturation of the immobilized trypsin during repeated catalysis and washing.
Potential application in enzymatic membrane bioreactors (EMBRs)
The application of the trypsin-immobilized membrane in EMBRs was briefly investigated to demonstrate the potential for its practical application. We performed a continuous catalytic reaction under the filtration mode. Fig. 6(A) shows the influence of the flow rate on the specific activity. The value of SA and R were surprisingly improved from 22 to 118 U cm−2 and from 26% to 145%, when the flow rate was increased from 0 to 1.52 mL min−1. In the soaking mode (see Fig. S4†), SA increased with increasing the substrate concentration for the trypsin-immobilized membrane, suggesting that the catalytic reaction at low substrate concentration was restricted by the diffusion of substrates,54 and especially the trypsin in the pores didn't react effectively. In the filtration mode, the convective flow in the membrane pores offered an enhanced mass transfer of substrates, which led to higher accessibility between substrates and immobilized trypsin on/in the membrane, and thus brought about much higher SA and R than the reported values. This high R value, 145%, means that immobilized trypsin has higher catalytic activity than the non-immobilized, native trypsin due to probably the stabilization of the molecular conformation by the immobilization onto the surface. The flow mode operation of the modified membranes was effective to enhance the catalytic activity as EMBRs.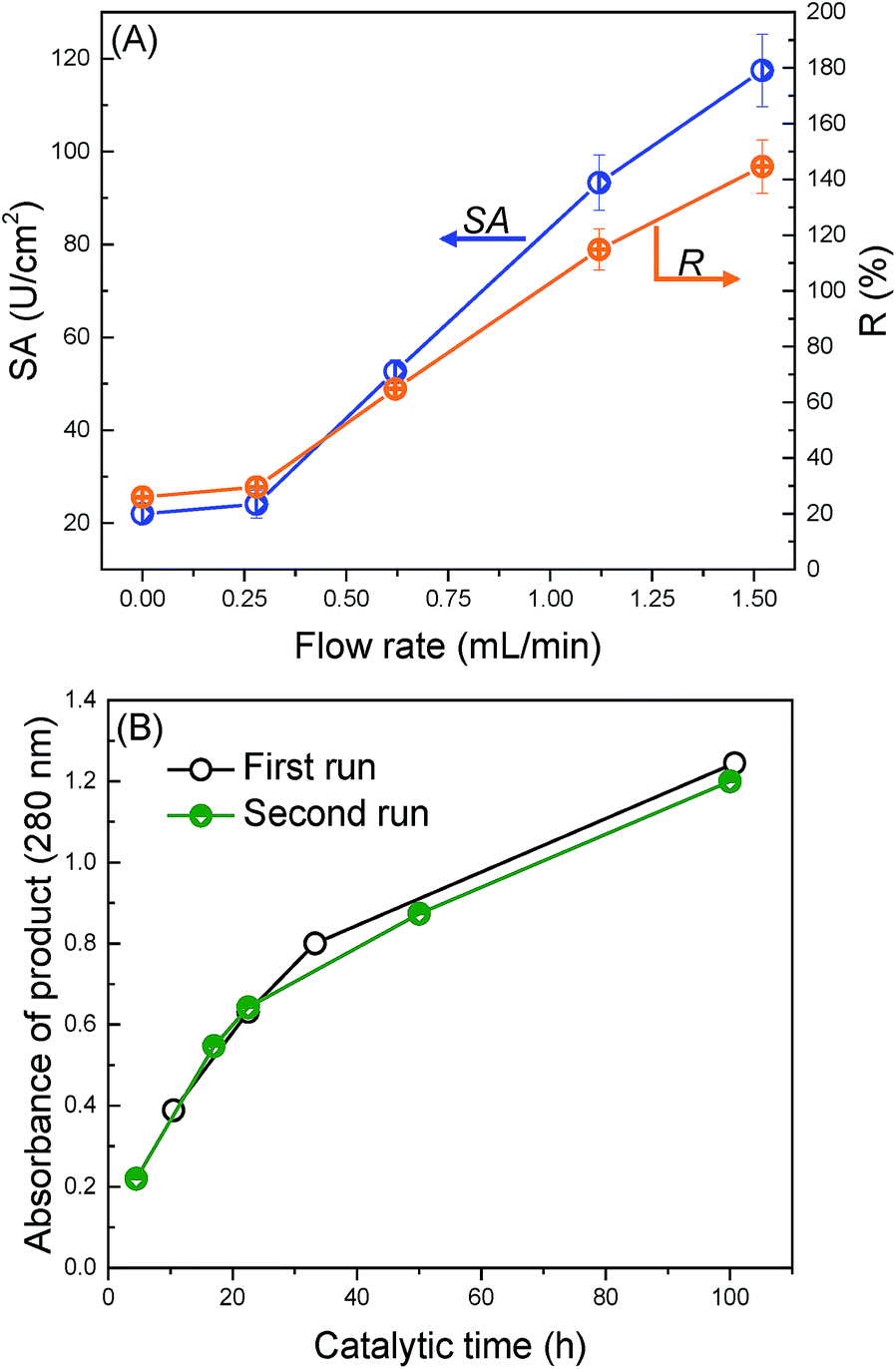 | ||
| Fig. 6 Demonstration of the trypsin-immobilized membrane as EMBRs. (A) Effect of the flow rate on the SA and R of the trypsin-immobilized membrane in the filtration mode. The plot at 0 mL min−1 were estimated from the result of the soaking mode. (B) Digestion kinetics of BSA in the soaking mode at 37 °C. | ||
Finally, we applied the modified membranes to catalyze BSA as a protein substrate model (Fig. 6(B)). The amount of the peptide products was observed to continuously increase with the catalytic time. Furthermore, no obvious decrease in the reaction rate was observed during the second digestion run, suggesting good reusability of the modified membranes. This digestion ability was comparable to the pepsin membranes reported by Michiel, which required 30 h to obtain large amounts of products when BSA was used as a substrate.55 In conclusion, the trypsin-immobilized membrane exhibits a great potential application in EMBRs.
The method presented in this study successfully immobilized a large amount of enzyme while effectively maintaining their catalytic activity by preserving their three-dimensional conformation. Besides enzyme immobilization, this method could also be applied to immobilize various biomolecules such as peptides, proteins, antibodies, and DNA onto various supports containing hydroxyl or amino groups. These composite materials have a wide range of applications in separation, adsorption, catalysis, and sensors using their molecular recognition function.
Conclusions
Here, we presented a novel and facile strategy that used dicarboxylic acid halides as a functional spacer to immobilize enzymes onto porous membranes. The high reactive affinity between the acyl chlorides of the dicarboxylic acid halides and the hydroxyl groups on the membrane surface rapidly produced efficient introduction spacers on the membrane surfaces. During enzyme immobilization, the carboxyl groups on the ends of the spacers avoided the undesirable self-crosslinking of enzymes to ensure good catalytic activity and permeability. The optimum surface density of trypsin was reported to be 36 μg cm−2, which showed a high specific activity of 22 U cm−2 and a high activity retention of 26%. In addition, the thermal stability, continuous operation capability, and reusability of the modified membranes were considered to be favorable for practical application. To demonstrate as EMBRs, the catalytic reaction in a filtration mode was attempted, and the resulted value of the activity retention was greatly improved to 145%, showing the catalytic activity of the immobilized trypsin was higher than that of non-immobilized, native trypsin. Furthermore, the efficient decomposition ability against BSA verified its excellent application potential in EMBRs. In addition, this method was effective for not only trypsin but also lipase, thus, it would be applicable to the immobilization of various kinds of enzymes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by a Special Coordination Fund for Promoting Science and Technology, Creation of Innovation Centers for Advanced Interdisciplinary Research Areas (Innovative Bioproduction, Kobe) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.Notes and references
- R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernandez-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- P. Jochems, Y. Satyawali, L. Diels and W. Dejonghe, Green Chem., 2011, 13, 1609–1623 RSC.
- M. B. Rao, A. M. Tanksale, M. S. Ghatge and V. V. Deshpande, Microbiol. Mol. Biol. Rev., 1998, 62, 597–635 CAS.
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- J. P. Rasor and E. Voss, Appl. Catal., A, 2001, 221, 145–158 CrossRef CAS.
- S. M. Thomas, R. DiCosimo and A. Nagarajan, Trends Biotechnol., 2002, 20, 238–242 CrossRef CAS PubMed.
- Y. R. Qiao, M. Gumpertz and T. Van Kempen, J. Food Biochem., 2002, 26, 355–375 CrossRef CAS.
- P. Torres-Salas, A. del Monte-Martinez, B. Cutino-Avila, B. Rodriguez-Colinas, M. Alcalde, A. O. Ballesteros and F. J. Plou, Adv. Mater., 2011, 23, 5275–5282 CrossRef CAS PubMed.
- D. F. M. Neri, V. M. Balcao, F. O. Q. Dourado, J. M. B. Oliveira, L. B. Carvalho and J. A. Teixeira, J. Mol. Catal. B: Enzym., 2011, 70, 74–80 CrossRef CAS.
- Y. H. Ren, J. G. Rivera, L. H. He, H. Kulkarni, D. K. Lee and P. B. Messersmith, BMC Biotechnol., 2011, 11, 63–70 CrossRef CAS PubMed.
- C. Chen, X. Y. Zhu, Q. L. Gao, F. Fang, L. W. Wang and X. J. Huang, J. Mol. Catal. B: Enzym., 2016, 132, 67–74 CrossRef CAS.
- M. Y. Arica, G. Bayramoglu and N. Bicak, Process Biochem., 2004, 39, 2007–2017 CrossRef CAS.
- Y. C. Qian, P. C. Chen, G. J. He, X. J. Huang and Z. K. Xu, Molecules, 2014, 19, 9850–9863 CrossRef PubMed.
- D. Kubac, A. Cejkova, J. Masak, V. Jirku, M. Lemaire, E. Gallienne, J. Bolte, R. Stloukal and L. Martinkova, J. Mol. Catal. B: Enzym., 2006, 39, 59–61 CrossRef CAS.
- A. C. Pierre, Biocatal. Biotransform., 2004, 22, 145–170 CrossRef CAS.
- W. Limbut, P. Thavarungkul, P. Kanatharana, P. Asewatreratanakul, C. Limsakul and B. Wongkittisuksa, Biosens. Bioelectron., 2004, 19, 813–821 CrossRef CAS PubMed.
- H. Kawakita, K. Sugita, K. Saito, M. Tamada, T. Sugo and H. Kawamoto, J. Membr. Sci., 2002, 205, 175–182 CrossRef CAS.
- S. V. Ebadi, A. Fakhrali, S. O. Ranaei-Siadat, A. A. Gharehaghaji, S. Mazinani, M. Dinari and J. Harati, RSC Adv., 2015, 5, 42572–42579 RSC.
- S. J. Bian, K. Gao, H. J. Shen, X. H. Jiang, Y. F. Long and Y. Chen, J Mater Chem B, 2013, 1, 3267–3276 RSC.
- Z. G. Wang, J. Q. Wang and Z. K. Xu, J. Mol. Catal. B: Enzym., 2006, 42, 45–51 CrossRef CAS.
- C. Mateo, O. Abian, R. Fernandez-Lafuente and J. M. Guisan, Biotechnol. Bioeng., 2000, 68, 98–105 CrossRef CAS PubMed.
- S. Sakai, Y. P. Liu, T. Yamaguchi, R. Watanabe, M. Kawabe and K. Kawakami, Biotechnol. Lett., 2010, 32, 1059–1062 CrossRef CAS PubMed.
- X. Y. Ji, P. Wang, Z. G. Su, G. H. Ma and S. P. Zhang, J. Mater. Chem. B, 2014, 2, 181–190 RSC.
- X.-Y. Zhu, C. Chen, P.-C. Chen, Q.-L. Gao, F. Fang, J. Li and X.-J. Huang, RSC Adv., 2016, 6, 30804–30812 RSC.
- D. H. Chen, J. C. Leu and T. C. Huang, J. Chem. Technol. Biotechnol., 1994, 61, 351–357 CrossRef CAS PubMed.
- S. Gupta, J. Environ. Chem. Eng., 2016, 4, 1797–1809 CrossRef CAS.
- W. Zhang, A. Abbaspourrad, D. Chen, E. Campbell, H. Zhao, Y. Li, Q. Li and D. A. Weitz, Adv. Funct. Mater., 2017, 27, 1700975–1700981 CrossRef.
- S. Starke, M. Went, A. Prager and A. Schulze, React. Funct. Polym., 2013, 73, 698–702 CrossRef CAS.
- C. Jolivalt, S. Brenon, E. Caminade, C. Mougin and M. Pontie, J. Membr. Sci., 2000, 180, 103–113 CrossRef CAS.
- S. Georgieva, T. Godjevargova, D. G. Mita, N. Diano, C. Menale, C. Nicolucci, C. R. Carratelli, L. Mita and E. Golovinsky, J. Mol. Catal. B: Enzym., 2010, 66, 210–218 CrossRef CAS.
- S. Georgieva, T. Godjevargova, M. Portaccio, M. Lepore and D. G. Mita, J. Mol. Catal. B: Enzym., 2008, 55, 177–184 CrossRef CAS.
- H. A. Sousa, C. Rodrigues, E. Klein, C. A. M. Afonso and J. G. Crespo, Enzyme Microb. Technol., 2001, 29, 625–634 CrossRef CAS.
- A. De Maio, M. M. El-Masry, M. Portaccio, N. Diano, S. Di Martino, A. Mattei, U. Bencivenga and D. G. Mita, J. Mol. Catal. B: Enzym., 2003, 21, 239–252 CrossRef CAS.
- D. Y. Koseoglu-Imer, N. Dizge and I. Koyuncu, Colloids Surf., B, 2012, 92, 334–339 CrossRef CAS PubMed.
- O. Barbosa, C. Ortiz, A. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, RSC Adv., 2014, 4, 1583–1600 RSC.
- J. Zhu and G. Sun, React. Funct. Polym., 2012, 72, 839–845 CrossRef CAS.
- S. Tembe, B. S. Kubal, M. Karve and S. F. D'Souza, Anal. Chim. Acta, 2008, 612, 212–217 CrossRef CAS PubMed.
- S. L. Chen, X. J. Huang and Z. K. Xu, Cellulose, 2012, 19, 1351–1359 CrossRef CAS.
- A. J. J. Straathof, S. Panke and A. Schmid, Curr. Opin. Biotechnol., 2002, 13, 548–556 CrossRef CAS PubMed.
- P. K. Smith, R. I. Krohn, G. Hermanson, A. Mallia, F. Gartner, M. Provenzano, E. Fujimoto, N. Goeke, B. Olson and D. Klenk, Anal. Biochem., 1985, 150, 76–85 CrossRef CAS PubMed.
- A. Schulze, D. Breite, Y. Kim, M. Schmidt, I. Thomas, M. Went, K. Fischer and A. Prager, Polymers, 2017, 9, 97–106 CrossRef.
- G. W. Schwert and Y. Takenaka, Biochim. Biophys. Acta, 1955, 16, 570–575 CrossRef CAS.
- M. Ghasemi, M. J. G. Minier, M. Tatoulian, M. M. Chehimi and F. Arefi-Khonsari, J. Phys. Chem. B, 2011, 115, 10228–10238 CrossRef CAS PubMed.
- R. Sternberg, D. S. Bindra, G. S. Wilson and D. R. Thevenot, Anal. Chem., 1988, 60, 2781–2786 CrossRef CAS PubMed.
- M. Y. Arica, S. Senel, N. G. Alaeddinoglu, S. Patir and A. Denizli, J. Appl. Polym. Sci., 2000, 75, 1685–1692 CrossRef CAS.
- D. Saeki, S. Nagao, I. Sawada, Y. Ohmukai, T. Maruyama and H. Matsuyama, J. Membr. Sci., 2013, 428, 403–409 CrossRef CAS.
- A. Kumari and S. Datta, J. Membr. Sci., 2017, 539, 43–51 CrossRef CAS.
- K. Itoyama, H. Tanibe, T. Hayashi and Y. Ikada, Biomaterials, 1994, 15, 107–112 CrossRef CAS PubMed.
- A. H. M. Cavalcante, L. B. Carvalho and M. G. Carneiro-da-Cunha, Biochem. Eng. J., 2006, 29, 258–261 CrossRef CAS.
- K. Yamada, T. Nakasone, R. Nagano and M. Hirata, J. Appl. Polym. Sci., 2003, 89, 3574–3581 CrossRef CAS.
- U. Bora, P. Sharma, K. Kannan and P. Nahar, J. Biotechnol., 2006, 126, 220–229 CrossRef CAS PubMed.
- M. Y. Arica, J. Appl. Polym. Sci., 2000, 77, 2000–2008 CrossRef CAS.
- T. C. Cheng, K. J. Duan and D. C. Sheu, J. Chem. Technol. Biotechnol., 2006, 81, 233–236 CrossRef CAS.
- C. Garcia-Galan, A. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- M. J. T. Raaijmakers, T. Schmidt, M. Barth, M. Tutus, N. E. Benes and M. Wessling, Angew. Chem., Int. Ed., 2015, 54, 5910–5914 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Activity determination method for lipase. Experimental setup for the permeability measurement and activity determination in filtration mode for EMBR (Fig. S1). Lineweaver–Burk plots to determine kinetic parameter of trypsin (Fig. S2). Evaluation of the stability of the lipase-immobilized membrane and free lipase (Fig. S3). Effect of the substrate (BAEE) concentration on the enzymatic activity of the trypsin-immobilized membrane (Fig. S4). See DOI: 10.1039/c7ra10012d |
| This journal is © The Royal Society of Chemistry 2017 |