
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insight into the structure evolution and the associated catalytic behavior of highly dispersed Pt and PtSn catalysts supported on La2O2CO3 nanorods†
Fengjun Houab,
Huahua Zhao a,
Huanling Song*a,
Lingjun Chou*ac,
Jun Zhaoa,
Jian Yanga and
Liang Yana
a,
Huanling Song*a,
Lingjun Chou*ac,
Jun Zhaoa,
Jian Yanga and
Liang Yana
aState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, PR China. E-mail: ljchou@licp.cas.cn; songhl@licp.cas.cn; Fax: +86 931 4968 129; Tel: +86 931 4968 066
bUniversity of Chinese Academy of Sciences, Beijing 100049, PR China
cSuzhou Research Institute of LICP, Chinese Academy of Sciences, Suzhou 215123, PR China
First published on 16th October 2017
Abstract
The current work introduces highly dispersed Pt and PtSn catalysts supported on La2O2CO3 nanorods prepared via ultrasonic impregnation, which are used as probe catalysts for the liquid-phase crotonaldehyde hydrogenation. The physicochemical properties of the catalysts are assessed by means of various techniques, including XRD, TEM, XPS, H2-TPD, in situ CO-DRIFT and X-ray adsorption fine structure (XAS). A close combination of catalyst surface experiments and the reactive performances reveals that the distinct reactive performance of the Pt and PtSn catalysts is tentatively attributed to the composition-dependent architecture of Pt–lanthanum interfaces and bimetallic particles while excluding the particle size effect. Catalytic activity tests demonstrate that incorporation of Sn into Pt catalyst brings great significance to the selective hydrogenation of carbonyl groups as it results into the structure evolution of bimetallic particles. An optimization of Sn loading and reaction conditions achieves a 5-fold and 7-fold improvement in the selectivity and yield to crotyl alcohol over the parent Pt catalyst. Lastly, it is found from the catalyst reusability study that metal particles of PtSn catalysts suffers easily from particle migration and growth compared to the Pt catalyst, most likely resulting from a weaker metal–support interaction.
1. Introduction
Unsaturated alcohols are widely employed as important intermediates for the production of fragrances and pharmaceuticals, and can be attained by the chemoselective hydrogenation of unsaturated aldehydes.1 However, this selective hydrogenation process is challenging since the hydrogenation of the carbonyl group is thermodynamically unfavorable over the olefinic bond.2 To circumvent this issue, significant efforts have been devoted to the development of effective catalysts with superior carbonyl selectivity. For another, crotonaldehyde (CRAL) hydrogenation has been also studied as a model reaction to probe the catalyst surface and active sites of heterogeneous catalysts.3–8 Among them, platinum is one of the most exploited metals for its ability to easily capture and dissociate hydrogen. Regardless of the high activity, the limited selectivity to the desired product is one of the main problems for CRAL hydrogenation over the Pt catalysts.Heteroatoms of Pt and a transition metal (M = Fe, Co, Ge or Sn) are reported to perform high selectivity towards unsaturated alcohols and produce changes in the reactivity.9–14 The promotion effect of M on the catalytic behavior of Pt catalyst can be generally interpreted in terms of geometric (dilution of Pt sites by PtM alloys or MOx species) or electronic (the electron density of Pt) aspects. A plot of reaction selectivity to cinnamyl alcohol from cinnamaldehyde hydrogenation versus the first row of transition metals as dopers of Pt gave a typical volcano shape curve, which is proposed to be derived from the difference in d-band center position from the Fermi level when the Pt surface is electronically modified by the transition metal.15 In addition, the composition variation is reported to induce changes in the particle size and structure of bimetallic nanoparticles (NPs). Rong et al. reported the structure evolution of PtSn NPs from the SnO2−x-patched PtSn alloy to the SnO2−x-patched Pt cluster as the Sn/Pt ratio increases, accompanied by an increase in the particle size, improving the desired selectivity for CRAL hydrogenation.16 In this sense, various preparative methods, e.g. atomic layer deposition (ALD) and surface organometallic chemistry (SOMC), have been adopted to precisely control the architecture of bimetallic NPs.17,18 However, it is still difficult to obtain homogeneous composition and structure of bimetallic NPs in consideration of their sensitivity to the synthetic parameters.
Besides, support plays a crucial role in the chemical composition and structure of active metal, and therefore influences the catalytic behavior of its supported catalyst. In this case, developing of Pt or PtM crystals on an active support is one of the most common strategies adopted to improve the unsaturated alcohol yield. Novel carbon materials (CNTs, CNFs, OMCs, RGOs) and reducible oxides (TiO2, CeO2, FeOx), are usually employed as supports and demonstrated to enhance the selectivity towards the carbonyl hydrogenation.8,13,19–23 The new catalytic sites located at the metal–support interface, involving either reduced cations or oxygen vacancy sites, are reported to activate the carbonyl bond and increase the desired selectivity.22,23
Since studies on its coke inhibition and impressive activity for oxidative coupling of methane (OCM) are reported, lanthanum oxy-carbonate (La2O2CO3) receives considerable attention.24,25 As far as we know, La2O2CO3 materials with specific morphology are scarcely studied in the hydrogenation process, although they are widely employed as important ingredients of heterogeneous catalysts in oxygen reduction reaction, steam reforming of glycerol and biofuel production because of their unique hydrotalcite-like structure and abundant active oxygen species.26–29 In our previous work, preferential deposit of Pt on the {101} facets of La2O2CO3 nanorods (denoted as Pt/LOC) is accounted for its superior reactive performance for CRAL hydrogenation to the Pt randomly dispersed on the particle-shaped counterpart.30 Herein we carry out a systematic study on the physicochemical properties and the associated catalytic behavior of PtSn catalysts. An ultrasonic impregnation procedure is used to synthesize well-dispersed Pt and PtSn NPs on the support. The chemical composition and structure of the catalysts are elaborately analyzed by a series of techniques. Catalytic activity tests for CRAL hydrogenation are performed as a probe reaction to discern the structure-activity correlation of PtSn catalysts with varying Sn/Pt molar ratios.
2. Experimental
2.1 Catalysts preparation
La2O2CO3 powders and nanorods was obtained by annealing the lanthanum hydroxide nanostructures at 500 °C for 3 h.30 La2O2CO3 powders and nanorods has specific surface area of 103 m2 g−1 and 45 m2 g−1, which is calculated based on the Brunauer–Emmett–Teller (BET) method in the relative pressure range of 0.05–0.30.All catalysts were fabricated via an ultrasonic impregnation procedure. The Pt/LOC was obtained from the mixture of H2PtCl6·6H2O ethanol solution (1.13 × 10−3 gPt mL−1, 9 mL) and the support (1.0 g) with ultrasonic treatment for 30 min. This mixture was kept standing for 12 h, and then evaporated at 60 °C to remove the excessive solvent. Obtained powder was air-dried at 120 °C overnight, calcinated at 400 °C for 2 h, and thereafter reduced at 600 °C for 1 h under a flowing gas mixture of 20% H2/N2 (v/v). The nominal loading of Pt was 1.00 wt%.
The PtSn catalysts with different Sn/Pt molar ratios were prepared by the impregnation of the air-dried Pt/LOC (1.0 g) with SnCl2·2H2O (0.0080 g, 0.0120 g or 0.0180 g) following an identical process. As determined by inductively coupled plasma optical emission spectrometry (ICP-OES) analyses (Table 1), Pt content of the prepared catalysts is in the range of 0.99–1.11 wt%; Sn content is 0.46 wt%, 0.57 wt%, and 0.87 wt%, corresponding to the Sn/Pt molar ratio of 0.69, 0.94, and 1.33; hence, the PtSn catalysts are denoted as PtSn0.69/LOC, PtSn0.94/LOC and PtSn1.33/LOC, respectively. In addition, the PtSn catalyst supported on the La2O2CO3 powders (PtSn0.94/LOC-NP) and the PtSn catalyst prepared by conventional impregnation method without ultrasonic treatment (PtSn0.94/LOC-CI) are fabricated for comparison.
| Catalyst | wt% Pta | wt% Sna | Sn/Pta | Amount of desorbed hydrogenb (μmol gPt−1) | |||
|---|---|---|---|---|---|---|---|
| Peak 1 | Peak 2 | Peak 3 | Total | ||||
| a From the ICP-OES analyses.b From the H2-TPD measurement. | |||||||
| Pt/LOC | 1.11 | — | — | 1.46(355) | 0.58(404) | 1.33(456) | 3.37 |
| PtSn0.69/LOC | 1.09 | 0.46 | 0.69 | 1.30(350) | 0.64(406) | 1.19(449) | 3.14 |
| PtSn0.94/LOC | 0.99 | 0.57 | 0.94 | 1.12(341) | 0.63(402) | 0.92(437) | 2.67 |
| PtSn1.33/LOC | 1.08 | 0.87 | 1.33 | 0.74(344) | 0.63(413) | 0.51(456) | 1.87 |
2.2 Catalyst characterization
The amount of Pt and Sn was determined by ICP-OES on an Agilent 725-ES.X-ray diffractograms (XRD) was performed on X'Pert Pro Multipurpose diffractometer (PANalytical, Inc.) with Cu Kα radiation (0.15406 nm) at room temperature from 10 to 80°. Measurements were conducted using a voltage of 40 kV, current setting of 20 mA, step size of 0.021, and count time of 4 s.
Sample morphology and dispersion was observed on the TECNAI G2 F20 high-resolution transmission electron microscopy under a working voltage of 200 kV. Sample preparation involved ultrasonic dispersion of the sample in ethanol and deposition of the mixture on a carbon-coated copper grid. The particle size distributions (PSD) were estimated by measuring at least 300 particles from several TEM micrographs for each sample.
Temperature programmed desorption of hydrogen (H2-TPD) was taken on a ChemBET Pulsar TPR/TPD analyzer (Quantachrome Instruments U.S.) equipped with a TCD detector. 50 mg of reduced catalyst was loaded in a U-tube quartz reactor and pretreated at 400 °C for 1 h with Ar flushing to remove the impurities. The TPD experiment was carried out by heating the furnace to 900 °C at a ramp of 20 °C min−1 in flowing Ar (40 mL min−1) after hydrogen-adsorption at 30 °C for 60 min.
X-ray photoelectron spectroscopy (XPS) was conducted on a Thermo Scientific ESCALAB250xi spectrometer to determine the surface atomic composition and chemical states of the catalysts. All binding energies were referenced to C 1s hydrocarbon peak at 284.8 eV. The spectra of each element were deconvoluted with a curve fitting routine in XPSPEAK41 software. Curve fitting of the Pt 4f, Sn 3d and O 1s spectra for the catalysts was performed after subtracting the background and fitting with the least squares best fitting routine, assuming an 80/20 Gaussian/Lorentzian product function.
The CO-DRIFT spectra of the samples were recorded on the BRUKER V70 Fourier transform infrared spectrometer equipped with an in situ infrared reaction device. Measurements were conducted using a spectral resolution of 4 cm−1, step size of 20 kHz, and accumulation of 64 scans. Briefly, self-supporting pellet was prepared from the reduced sample and placed directly in the IR quartz cell equipped with KBr windows. The reduced sample was pretreated at 300 °C for 90 min in flowing H2 followed by 30 min flushing in Ar. After pretreatment, the sample was cooling down to 40 °C. And then a gas mixture of 0.5% CO/Ar was admitted into the cell and left to equilibrate for 40 min. And then the CO-adsorption spectra of the sample were continuously recorded with 40 min flushing in Ar flow.
The Pt LIII-edge X-ray absorption spectroscopy (XAS) was measured at the 4W1B station of the Beijing Synchrotron Radiation Facility (BSRF). Experiments were performed at ambient temperature in transmission mode using a Si (111) solid state detector for selection of energy. The data reduction and process was performed using the ATHENA and ARTEMIS software.31 Each EXAFS function (χ) was attained by subtracting the post-edge background from the overall absorption and then normalized relating to the edge jump step. Then, k3-weighted EXAFS oscillation in the k-space for Pt LIII-edge, were Fourier transformed (FT) to the r-space to isolate the EXAFS contributions from different coordination shells. The Pt–Pt scattering path was used in the fitting of Pt foil; the Pt–O and Pt–Pt scattering paths were used in the fitting of the Pt/LOC; the Pt–O, Pt–Pt and Pt–Sn scattering paths were used in the fitting of the PtSnx/LOC samples. The structural parameters, such as the coordination number CN, the inter-atomic distance R, the Debye–Waller factor σ2, and the edge-energy shift ΔE0, were allowed to vary during the fitting process.
2.3 Catalytic test
The catalytic behavior for liquid-phase CRAL hydrogenation was operated in a 100 mL Parr reactor. 100 mg of catalyst, 1 mL of CRAL and 19 mL of ethanol was charged into the autoclave. The autoclave was purged three times and then pressurized with H2. The reaction was heated to the desired temperature for reaction with vigorous stirring. The compositions of reactants and products were identified and analyzed on Agilent 7890A-5975C equipped with an FID detector. The catalytic activity and selectivity of the products was determined by using the internal normalization method with n-hexanol as internal standard. CRAL conversion (Conv.CRAL) was defined as the adductive percentages of CRAL converted into different products. The selectivity to a given product (i) was calculated as the ratio between the moles of product i and the total moles of products.The catalyst reusability study was conducted under the optimization reaction conditions (T = 160 °C, p = 2.0 MPa, t = 60 min). Monometallic Pt/LOC and bimetallic PtSn0.94/LOC were selected as the catalysts for reutilization test. The spent catalysts were regenerated from the mixture of hydrogenation system by rinsing with absolute ethanol, and drying overnight at 120 °C before activation in air at 300 °C for 1 h.
3. Results
3.1 Catalysts characterization
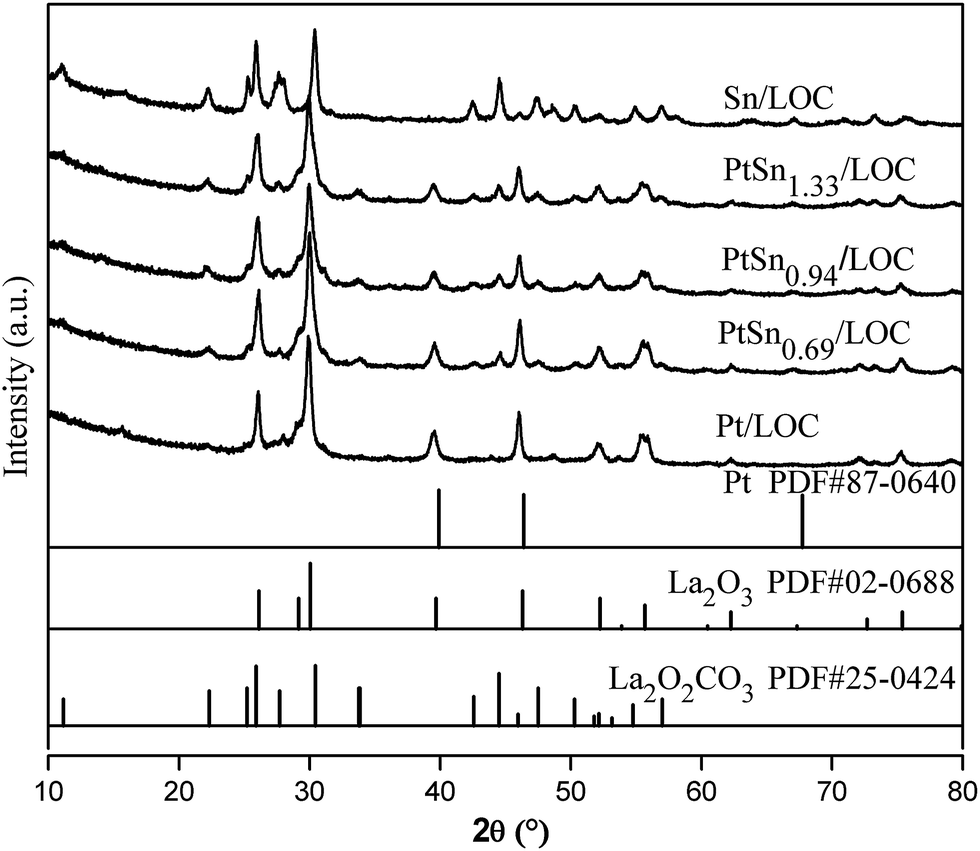 | ||
| Fig. 1 Standard patterns of hexagonal La2O2CO3, hexagonal La2O3 and cubic Pt crystalline. XRD patterns of the Pt/LOC, Sn/LOC and PtSnx/LOC series catalysts. | ||
 | ||
| Fig. 2 Typical TEM images of (a) LOC, (b) Pt/LOC, (c) PtSn0.69/LOC, (d) PtSn0.94/LOC and (e) PtSn1.33/LOC, (f) histograms showing the size distributions of metal particles over the catalysts. | ||
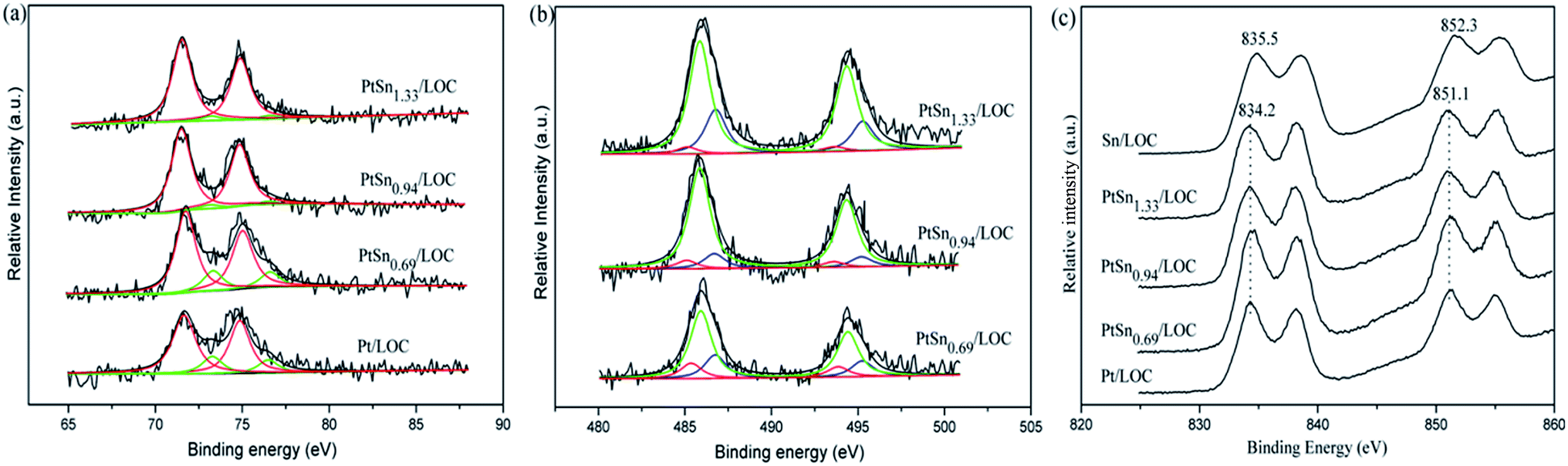 | ||
| Fig. 3 XPS spectra of (a) Pt 4f, (b) Sn 3d, (c) La 3d for the samples. The curve fitting of the Pt 4f and Sn 3d spectra for the samples are performed using the XPSPEAK41 software according to the uniform constraints on the fitting parameters after subtracting the background. | ||
| Sample | Species | Binding energy (eV) | Pt2+/Pt0 or Sn0/(Sn2+ + Sn4+)a | Pt/Lab | Sn/Ptc | O/Lad | |
|---|---|---|---|---|---|---|---|
| a Pt2+/Pt0: the atomic ratio of oxidized platinum (Pt2+) to metallic platinum (Pt0); Sn0/(Sn2+ + Sn4+): the atomic ratio of metallic tin (Sn0) to oxidized tin (Sn2+ + Sn4+).b The molar ratio of Pt to La.c The molar ratio of Sn to Pt.d The molar ratio of O to La. | |||||||
| Pt/LOC | Pt 4f7/2 | Pt0 | 71.7 | 0.26 | 0.020 | 0.00 | 3.19 |
| Pt2+ | 73.3 | ||||||
| PtSn0.69/LOC | Pt 4f7/2 | Pt0 | 71.6 | 0.25 | 0.014 | 1.60 | 2.60 |
| Pt2+ | 73.3 | ||||||
| Sn 3d5/2 | Sn0 | 485.4 | 0.17 | ||||
| Sn2+ | 485.9 | ||||||
| Sn4+ | 486.8 | ||||||
| PtSn0.94/LOC | Pt 4f7/2 | Pt0 | 71.5 | 0.05 | 0.016 | 1.95 | 2.59 |
| Pt2+ | 73.2 | ||||||
| Sn 3d5/2 | Sn0 | 485.1 | 0.07 | ||||
| Sn2+ | 485.9 | ||||||
| Sn4+ | 486.7 | ||||||
| PtSn1.33/LOC | Pt 4f7/2 | Pt0 | 71.5 | 0.06 | 0.015 | 2.67 | 2.50 |
| Pt2+ | 73.2 | ||||||
| Sn 3d5/2 | Sn0 | 485.2 | 0.04 | ||||
| Sn2+ | 485.9 | ||||||
| Sn4+ | 486.8 | ||||||
As shown in Fig. 3b, the Sn 3d5/2 peaks can be deconvoluted into three features assigned to Sn0, Sn2+ and Sn4+ species. It is found that large quantities of tin exist as oxidative status according to the curve-fitting results of the Sn 3d5/2 spectra. As expected, with higher Sn/Pt molar ratio, the Sn 3d5/2 signals are evidently intensified. The Sn/Pt atomic ratio for the PtSn catalysts assessed by XPS analyses is 1.60, 1.95 and 2.67, much higher than that from ICP results. This demonstrates a significant surface enrichment of tin presumably because the impregnation order leads to the partially covered Pt NPs by tin. Besides, the atomic ratio of Sn0/(Sn2+ + Sn4+) is higher for PtSn0.69/LOC (0.17) than that for PtSn0.94/LOC (0.07) and PtSn1.33/LOC (0.04), suggesting that most of tin is hardly reduced to metallic Sn0 and exists as SnOx entities at high Sn loading.
The La 3d spectra for Pt-involved catalysts, as shown in Fig. 3c, contain peak at 834.2 eV and its accompanying satellite peaks that can be attributed to the electrons of La 3d5/2 for La2O3. The peak at higher value (835.5 eV) for Sn/LOC corresponds to the electrons of La 3d5/2 for La2O2CO3. These results verify the phase evolution of lanthanum catalyzed by Pt atoms at the Pt–lanthanum interfaces, which is consistent with the previously discussed XRD patterns. It is noteworthy that the O/La ratio decreases from 3.19 to 2.50 as Sn/Pt value increases. This suggests that a proportion of oxygen species on the catalyst surface or subsurface is consumed, producing more oxygen vacancies by the promotion effect of tin. According to above results, it can be inferred that the tin addition induces an interaction between both metals, which affects the redox property of Pt and Sn, generating the Pt–SnOx entities; at the same time, more surface oxygen vacancies are produced due to the formation of Pt–SnOx species.
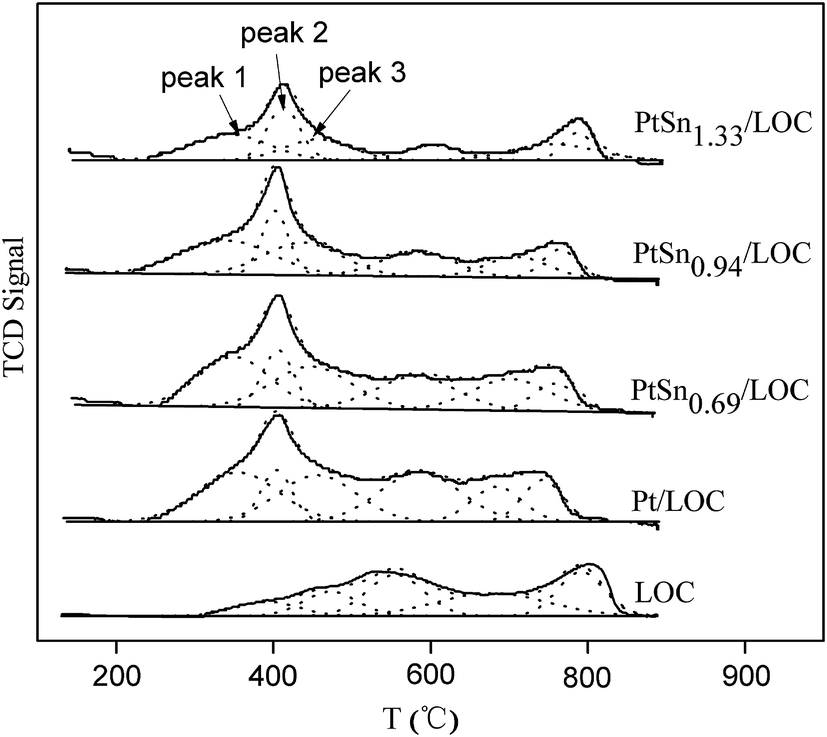 | ||
| Fig. 4 H2-TPD profiles of the samples. | ||
In the case of the Pt/LOC, three overlapped desorption peaks centered at 355, 404 and 455 °C, are assigned to weak, medium and strong bonding sites with hydrogen, respectively. Table 1 summarizes the H2 desorption capacity (in μmol gPt−1) of the prepared catalysts calculated on the categorized temperature below 500 °C. The hydrogen desorption capacity for the samples is followed in a decreasing order of PtSn0.69/LOC (3.14 μmol gPt−1) > PtSn0.94/LOC (2.67 μmol gPt−1) > Pt/LOC (2.30 μmol gPt−1) > PtSn1.33/LOC (1.87 μmol gPt−1). It is known that Pt is the effective sites for hydrogen adsorption and dissociation, whereas tin oxides has a relatively low capacity for hydrogen activation, and therefore the hydrogen desorption behaviors can mostly be ascribed to the noble metal Pt. The calculated hydrogen desorption capacity reveals that tin deposit changes the exposure degree of surface Pt sites available for hydrogen chemisorption. At low Sn content, the dilution effect of Pt sites by tin prevails, favoring the exposure of Pt sites; while at high Sn content, the blocking effect of tin predominates and decreases the number of exposed Pt sites. Besides, incorporation of tin lowers the overall peak intensity, in particular for the mid-temperature peak, confirming the strong passivation of catalyst surface by tin.
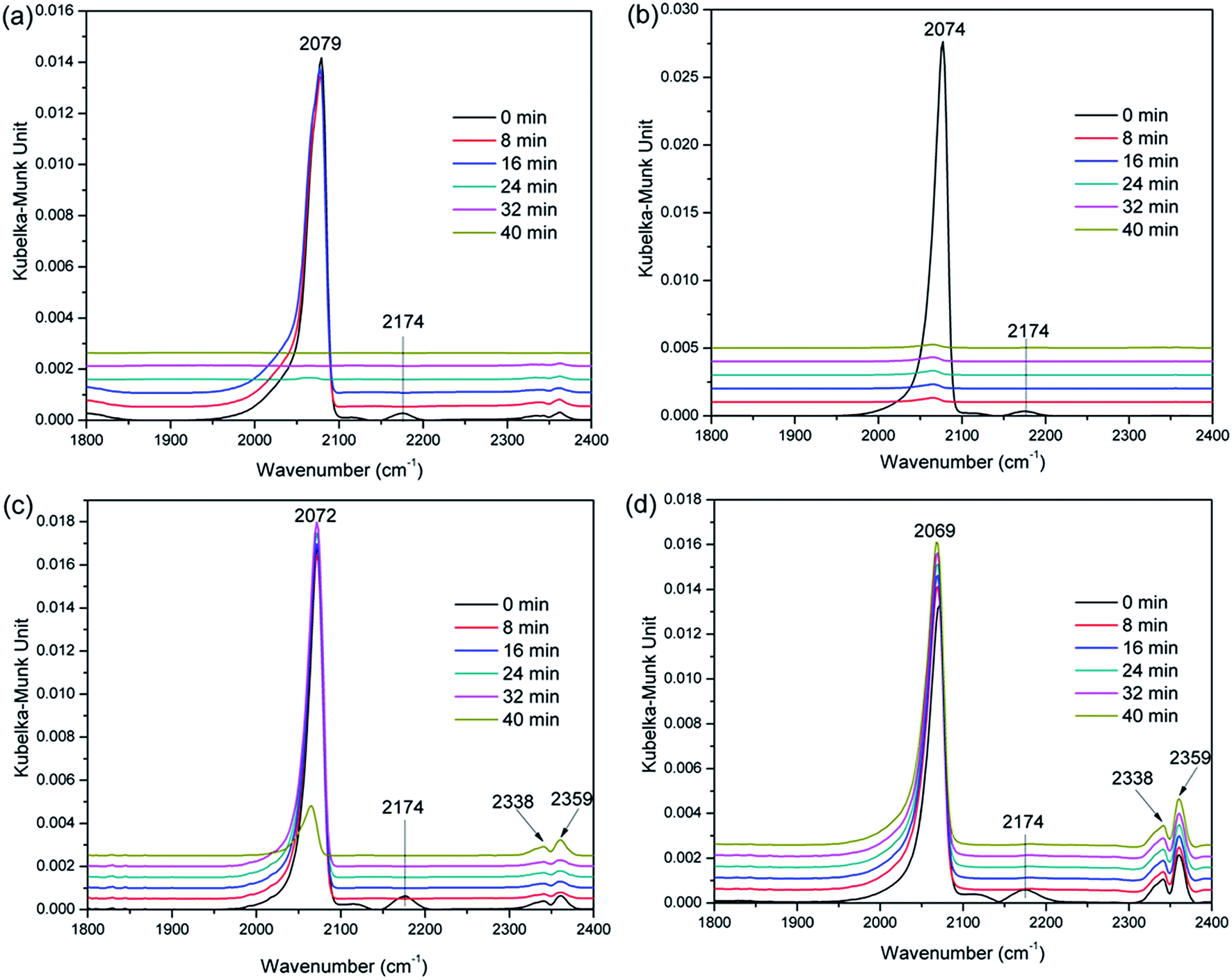 | ||
| Fig. 5 DRIFT spectra of CO adsorbed on the prepared (a) Pt/LOC, (b) PtSn0.69/LOC, (c) PtSn0.94/LOC and (d) PtSn1.33/LOC samples. The spectra were continuously recorded on the samples (50 mg) at 40 °C during Ar flushing for 40 min after introducing 0.5 Ar/CO for 40 min. | ||
It is proposed that the binding strength of CO on the Pt sites can somewhat reflect the adsorption of carbonyl group and desorption of the product.37,38 The linear-CO adsorption on fresh Pt/LOC seems to be completely reversible as this band disappears when flushing with Ar for 24 min. By contrast, the linear-CO adsorption on the PtSn0.69/LOC can be easily removed when flushing with Ar for 8 min; the linear-CO adsorption on the PtSn0.94/LOC seems to be mostly desorbed on Ar flushing for 40 min; however, high tin concentration in PtSn1.33/LOC does not result into noticeable changes of the linear adsorbed CO signals even after Ar flushing for 40 min. These results further manifest that the interaction between CO and Pt is greatly enhanced at high tin loadings. This enhanced binding force, on one hand, results into a chemisorption mode favoring the activation and hydrogenation of carbonyl groups on the metal Pt. On the other hand, it inhibits the product desorption and the recovery of active sites. In summary, the CO-DRIFT studies reveal that the coordination environment and exposure degree of Pt atoms in the catalysts is greatly changed upon tin addition; further, metal particles consist of Pt and Sn atoms leads to the composition-dependent variations in the interaction between CO and Pt. These two factors work together to determine the CO chemisorption behavior on Pt centers.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 570.3 eV ascribes to the 2p3/2 to 5d transition in Pt foil and is very sensitive to the average oxidation state of Pt. The white line intensities of the Pt-involved samples are higher than that of Pt foil, verifying the existence of positively charged Pt atoms that consists with the XPS results.39,40 In the case of Pt-involved samples (the inset of Fig. 6a), XANES shifts to higher energy by 0.5 eV, and the region between 11578 eV and 11600 eV shows a different fine structure, which is indicative of a different geometry around the Pt atoms depending on the Sn/Pt ratios.
570.3 eV ascribes to the 2p3/2 to 5d transition in Pt foil and is very sensitive to the average oxidation state of Pt. The white line intensities of the Pt-involved samples are higher than that of Pt foil, verifying the existence of positively charged Pt atoms that consists with the XPS results.39,40 In the case of Pt-involved samples (the inset of Fig. 6a), XANES shifts to higher energy by 0.5 eV, and the region between 11578 eV and 11600 eV shows a different fine structure, which is indicative of a different geometry around the Pt atoms depending on the Sn/Pt ratios.
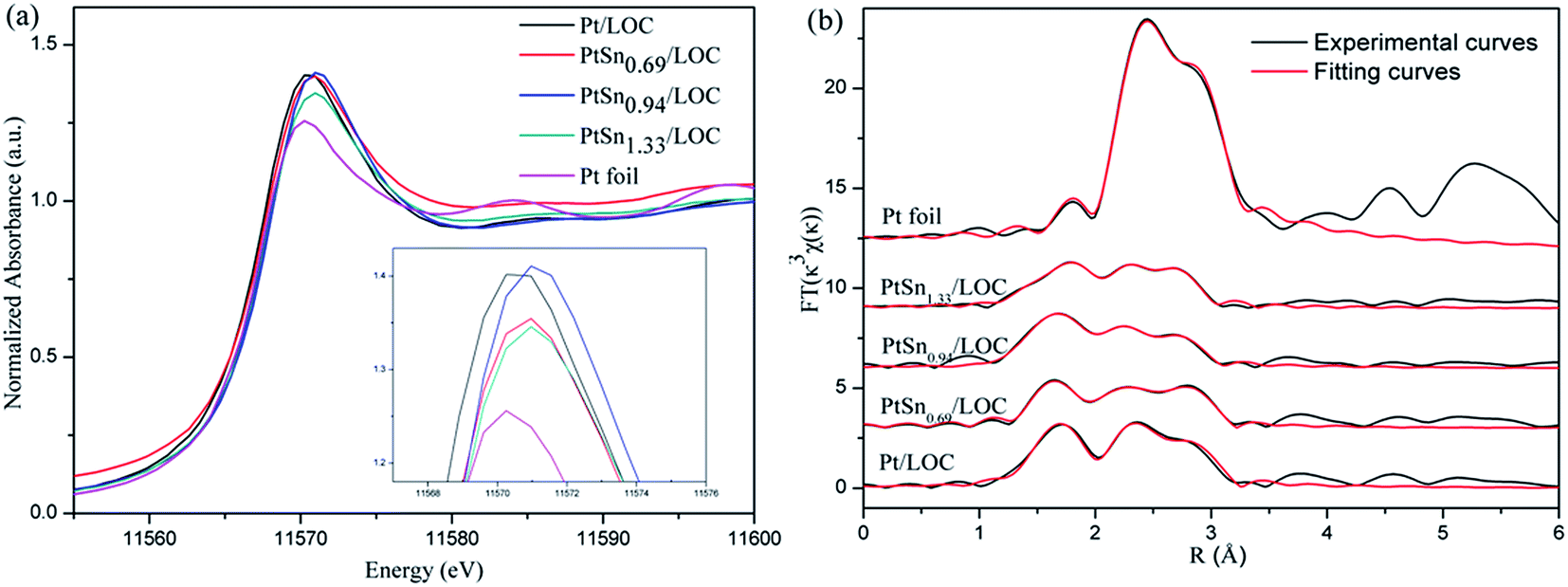 | ||
| Fig. 6 (a) Normalized XANES spectra and (b) Fourier transforms (FTs) of Pt LIII-edge k3-weighted EXAFS oscillations for Pt foil, Pt/LOC and PtSnx/LOC catalysts. | ||
Fig. 6b shows the Fourier transforms (FTs) of k3-weighted extended X-ray absorption fine structure (EXAFS) oscillations at the Pt LIII-edge. The main peaks of FTs for the catalysts located in the range of 2–3 Å can be assigned to the Pt–O, Pt–Pt or Pt–Sn contributions.41 The structural parameters can be attained by curve-fitting the EXAFS data and summarized in Table 3. There is Pt–O bond at a distance of 2.02 Å for the Pt/LOC; its coordination number (CN) is 1.5. The contribution of Pt–O shell gives further evidence on the presence of oxidized Pt coordinated by oxygen atoms of lanthanum. In the case of bimetallic PtSn catalysts, the CN of Pt–O bond decreases to around 1.0, suggesting that some Pt atoms segregated from lanthanum surface are greatly reduced after the decoration of tin, in consistence with higher Pt0/Pt2+ ratios for tin-promoted samples from XPS results.
| Sample | Shell | CNa | R (Å)b | ΔE0 (eV)c |
σ2 × 10−3 (Å2)d |
R factor | ΣNPt-ie |
|---|---|---|---|---|---|---|---|
| a N: the average coordination number.b R: the interatomic distance.c ΔE0: the inner potential shift.d σ2: the Debye–Waller factor.e ΣNPt-i: the overall coordination number. The data range used for data fitting in k-space (Δk) and R-space (ΔR) are 2.0–10.7 Å−1 and 1.2–3.5 Å, respectively. | |||||||
| Pt foil | Pt–Pt | 12.0 | 2.76 | 8.0 | 4.57 | 0.001 | 12.0 |
| Pt/LOC | Pt–O | 1.5 | 2.02 | 10.9 | 3.47 | 0.008 | 7.4 |
| Pt–Pt | 5.9 | 2.72 | 8.6 | 9.36 | |||
| PtSn0.69/LOC | Pt–O | 1.1 | 2.04 | 15.4 | 2.80 | 0.004 | 7.2 |
| Pt–Sn | 1.1 | 2.66 | −5.2 | 11.46 | |||
| Pt–Pt | 5.0 | 2.75 | 7.1 | 10.0 | |||
| PtSn0.94/LOC | Pt–O | 1.0 | 1.96 | 2.6 | 2.00 | 0.008 | 6.5 |
| Pt–Sn | 0.3 | 2.57 | 14.3 | 2.32 | |||
| Pt–Pt | 5.3 | 2.62 | 2.2 | 9.00 | |||
| PtSn1.33/LOC | Pt–O | 1.0 | 2.03 | 12.0 | 5.00 | 0.006 | 5.4 |
| Pt–Sn | 0.2 | 2.62 | 10.1 | 5.07 | |||
| Pt–Pt | 4.2 | 2.66 | 1.8 | 9.00 | |||
For Pt/LOC, there is a Pt–Pt contribution at a distance of 2.72 Å, with an average CN of 5.9. By contrast, the CN of Pt–Pt bond for tin-promoted samples is continuously decreased to 4.2. The Pt–Pt bond of PtSn0.94/LOC (2.62 Å) and PtSn1.33/LOC (2.65 Å) shows shorter distance than that of Pt/LOC (2.72 Å) and PtSn0.69/LOC (2.75 Å), suggesting the contraction of the bond distances owing to the formation of small Pt clusters at high Sn loading. In addition, the best fit for the third shell of PtSn0.69/LOC is a Pt–Sn contribution at a distance of 2.66 Å, with an average CN of 1.1. The Pt–Sn contribution is greatly decreased to 0.3 for PtSn0.94/LOC and 0.2 for PtSn1.33/LOC, implying that a phase segregation of PtSn alloys occurs at high Sn loading. These changes give important clues on the structure evolution of bimetallic NPs from PtSn alloys to small Pt clusters with increasing Sn/Pt ratios.
It is found that the total CN of absorbing platinum atoms (ΣNPt-i) for Pt/LOC is 7.4, much higher than that for PtSn0.69/LOC (7.2), PtSn0.94/LOC (6.5), and PtSn1.33/LOC (5.4). The decreased ΣNPt-i values for tin-dopped samples lead to a hypothesis that a number of Pt atoms are neither directly interacted with the Sn species nor with the support, presumably instead by forming the Pt–SnOx entities through Pt–O–Sn bond. As revealed by the XPS results, more SnOx entities with oxygen vacancy sites are produced at high tin loadings, which could be the probable reason for lower ΣNPt-i values for the PtSn catalysts. Analogous results are previously discussed by Taniya on the tin-modified SiO2-coated Pt catalyst.38 In summary, the EXAFS analyses throw light on the structure evolution of bimetallic PtSn NPs with increasing Sn content. At low Sn concentration, the structure of PtSn0.69/LOC is PtSn alloys patched by SnOx entities; whereas at high Sn concentration, the structure of PtSn0.94/LOC and PtSn1.33/LOC is Pt clusters patched by SnOx entities. The composition-dependent structure evolution of bimetallic NPs has also been observed on unsupported PtSn NPs and PtNi NPs.41,42 As shown below, these Pt atoms diluted by PtSn alloy or SnOx species exhibit distinct reactive behaviors for CRAL hydrogenation.
3.2 Reactive performance
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydrogenation for bimetallic systems. When the Sn/Pt ratio increases to 0.94, it reaches a maximum value of CROL selectivity (70.7%) at the expense of a slight decrease in the reactivity (0.51 mmol gPt−1 s−1) and CRAL conversion (91.7%). Further increasing Sn content leads to a sharp drop in reactivity and CROL selectivity for PtSn1.33/LOC. The CRAL conversions over the catalysts follows a decreasing order of PtSn0.69/LOC > PtSn0.94/LOC > Pt/LOC > PtSn1.33/LOC. These results coincide with the desorbed hydrogen/CO capacity determined by H2-TPD and CO-DRIFT analyses, which most likely result from the composition-dependent variations in the exposure degree of active Pt sites and the interaction between carbonyl groups and Pt centers. By contrast, as shown in Table S1,† the selectivity to crotyl alcohol (62.0%) at a conversion of 81.2% is obtained on PtSn0.94/LOC-CI, and the value is decreased to 54.3% at a conversion of 79.6% for PtSn0.94/LOC-NP, much lower than that obtained on the PtSn0.94/LOC, highlighting the superiority of La2O2CO3 nanorods and sonochemical approach in this work.
O hydrogenation for bimetallic systems. When the Sn/Pt ratio increases to 0.94, it reaches a maximum value of CROL selectivity (70.7%) at the expense of a slight decrease in the reactivity (0.51 mmol gPt−1 s−1) and CRAL conversion (91.7%). Further increasing Sn content leads to a sharp drop in reactivity and CROL selectivity for PtSn1.33/LOC. The CRAL conversions over the catalysts follows a decreasing order of PtSn0.69/LOC > PtSn0.94/LOC > Pt/LOC > PtSn1.33/LOC. These results coincide with the desorbed hydrogen/CO capacity determined by H2-TPD and CO-DRIFT analyses, which most likely result from the composition-dependent variations in the exposure degree of active Pt sites and the interaction between carbonyl groups and Pt centers. By contrast, as shown in Table S1,† the selectivity to crotyl alcohol (62.0%) at a conversion of 81.2% is obtained on PtSn0.94/LOC-CI, and the value is decreased to 54.3% at a conversion of 79.6% for PtSn0.94/LOC-NP, much lower than that obtained on the PtSn0.94/LOC, highlighting the superiority of La2O2CO3 nanorods and sonochemical approach in this work.
 | ||
| Fig. 7 The reaction diagram of CRAL hydrogenation. | ||
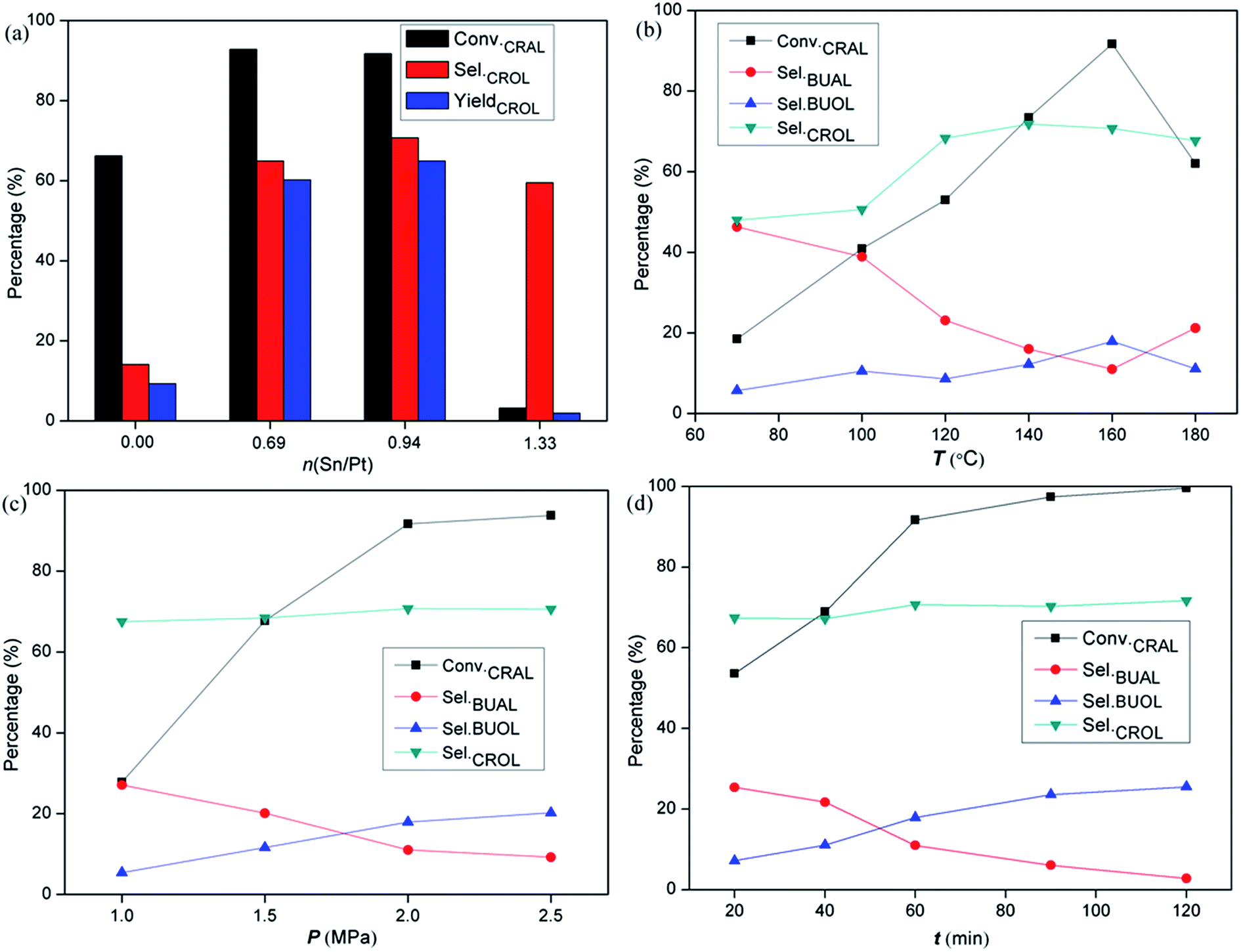 | ||
| Fig. 8 (a) Liquid phase hydrogenation results over the PtSnx/LOC catalysts with different Sn/Pt molar ratios. Reaction conditions: 1 mL CRAL catalyzed by 100 mg of the catalyst in 19 mL ethanol, T = 160 °C, p = 2.0 MPa, t = 60 min. Liquid phase hydrogenation results of CRAL over the PtSn0.94/LOC as a function of: (b) temperature, (c) H2 pressure and (d) time. | ||
Reaction parameters, e.g. hydrogen pressure, temperature and time, can affect the kinetics of competitive adsorption and hydrogenation of the reactant. Reaction parameters are tested in order to evaluate their influence on the activity and selectivity for liquid-phase CRAL hydrogenation. Reaction parameters have been optimized by correlating reaction rates with product distributions over the PtSn0.94/LOC. As can be seen from Fig. 8b, high hydrogenation rate is attainable with increasing reaction temperature, mainly from the formation of alcohol products coupled with a decreased yield of saturated aldehyde. In a period reaction of 60 min, the CROL selectivity presents a “parabola” curve: it ascends to 71.5% as the temperature rises to 160 °C, and then descends slightly at higher temperature possibly due to the distortion of the catalyst structure. When reaction temperature is 160 °C, increasing hydrogen pressure and reaction time displays an analogous promotion effect on the reactivity, while the CROL selectivity retains at the initial level (around 70%) with additional formation of BUOL, resulting from the deep hydrogenation of BUAL. Noticeably, an optimization of Sn content and reaction conditions gives rise to a 1.5-fold and 5-fold improvement in reactivity and CROL selectivity over the Pt/LOC. The extremely high activity and CROL selectivity over the PtSn0.94/LOC clearly demonstrate that the coexistence of Pt and the partially reduced SnOx species is essential to achieve high efficiency in carbonyl hydrogenation.
To explore the reason for catalyst deactivation, characterizations including XRD patterns, HAADF-STEM images and XPS spectra on the cycled catalysts (denoted as Pt/LOC-C and PtSn0.94/LOC-C) are elaborately performed. As shown in Fig. S3,† the diffraction patterns of the cycled catalysts can be indexed to hexagonal La2O2CO3 (JCPDS No. 25-0424). HAADF-STEM images in Fig. S4† reveal that the cycled catalysts consist of highly dispersed metal NPs with narrow size distributions, which can explain why no diffraction signals related to metals are detected. The almost unchanged Pt particle size of Pt/LOC-C relative to the Pt/LOC, provide direct evidence that the working catalyst is rather stable probably due to the strong anchoring sites on the lanthanum surfaces, which inhibits the migration and growth of Pt atoms even after five reduction–oxidation cycles. While for PtSn0.94/LOC-C, an obvious increase in metal particle size is observed, showing an average size of 2.2 nm, higher than that for PtSn0.94/LOC (1.3 nm).
As is clear from XPS analyses in Fig. S5 and Table S2,† Pt in the cycled catalysts still exists as a mixture of Pt0 and Pt2+, showing a higher Pt2+/Pt0 value than the fresh ones presumably resulting from calcinations in air to regenerate the catalysts. No metallic tin is detected for PtSn0.94/LOC-C and tin exists as ionic states (Sn2+ or Sn4+), suggesting a complete phase segregation of PtSn alloys. In addition, the Sn/Pt value of PtSn0.94/LOC-C is much lower than the fresh one, implying that a small amount of tin species leaches out during the reaction process. According to the previously discussed XRD and XPS results, the decreased phase composition of La2O3 for fresh PtSn0.94/LOC compared to the Pt/LOC clearly illustrates that the tin addition much weakens the Pt–lanthanum interfaces, resulting into more Pt atoms segregated from lanthanum surfaces to interact with the tin species. Therefore, it is plausible that the heat-treatment in air can easily induce the phase segregation of PtSn alloys and further result into the loss of SnOx layers, assuming that a weak interaction between lanthanum and tin oxides.43 Moreover, ICP-OES results in Table S2† clearly indicate a decrease of Sn content (0.46 wt%) and Sn/Pt molar ratio (0.73) in PtSn0.94/LOC-C compared to the fresh PtSn0.94/LOC, which provides further evidence on the leaching of Sn during the cycle experiment. Above results lead us to a conclusion that the phase segregation of PtSn alloys and the loss of SnOx patches in the vicinity of Pt centers during the cycle experiments, may be the primary reason responsible for the catalyst deactivation of the PtSn0.94/LOC.
4. Discussion
According to the reactive performance over the resultant catalysts, bimetallic PtSn systems are highly active and selective for CRAL hydrogenation to the desired CROL compared to the parent Pt/LOC. It seems that the enhanced catalytic behaviors of tin-modified catalysts most likely result from the interaction between Pt and the tin species, which affects the Pt–support interfaces and the bimetallic structure. Therefore, it is worthwhile to study the interaction between both metals and lanthanum surfaces on the basis of detailed characterization data.4.1 Pt–lanthanum interfaces
The electronic and structural properties of the Pt/LOC are estimated to probe the metal–support interaction. XRD patterns in Fig. 1 suggest that Pt atoms onto the lanthanum surfaces catalyze the phase transformation of La2O2CO3 to La2O3, which is supported by the variations of La 3d spectra in Fig. 3d. As shown in Fig. 3a, the split of Pt 4f spectra into metallic Pt0 and oxidized Pt2+ clearly demonstrates that two kinds of platinum species coexist in the catalyst. On account of the high-temperature reduction, Pt2+ species can be ascribed to Pt atoms that are strongly interacted with lanthanum through the Pt–O–La bond. The Pt–O contribution to the Pt LIII-edge EXAFS oscillation provide further evidence on the existence of O-coordinated Pt atoms at the Pt–lanthanum interfaces. On one hand, the strong metal–support interaction (SMSI) plays a decisive role in retaining the high platinum dispersion even after reduction at 600 °C (see Fig. 2), further inhibiting the metal sintering during the cycle experiment (see Fig. S4†). On the other hand, this SMSI effect usually decreases the exposure degree of Pt sites, since it produces Pt atoms partially embedded by oxides patches. Analogous phenomenon has been discussed in other studies on the Pt doped ceria surfaces.35,44As introduced into the Pt/LOC, the tin species can be randomly dispersed on the support surface or located in the vicinity of Pt atoms. TEM images of tin-promoted samples in Fig. 2 show that no obvious changes in the metal particle size were observed upon the tin addition, hence we can discuss the composition-dependent properties of the PtSn catalysts while excluding the particle size effect. A decreasing Pt2+/Pt0 values and the reducing CN values of Pt–O contribution for tin-modified catalysts reveal that the reducibility of platinum is greatly improved possibly owing to the crippling Pt–lanthanum interfaces after the decoration of tin, which is validated by an increase in metal particle size of tin-dopped catalysts during cycle experiment. As indicated by XRD analyses in Fig. 1, introducing a limited quantity of tin results into an increase in the phase composition of La2O3, while an opposite trend is observed when Sn/Pt ratio ≥ 0.94. These results suggest two different modification effects of tin on the Pt–lanthanum interfaces depending on the Sn/Pt ratios. At low Sn content, the dilution effect of Pt by tin predominates and produces individual Pt atoms segregated from lanthanum surfaces; whereas at high Sn content, more Pt atoms are interacted with the tin species and the passivation effect of tin on the catalyst surface prevails.
4.2 Bimetallic particles
The interaction between Pt and the tin species is estimated by XPS, CO-DRIFT and XAS techniques. The Pt–Sn contributions to the Pt LIII-edge oscillation confirm the dilution of Pt atoms by PtSn alloys or SnOx species. XPS characterization manifests that the number of the O-coordinated Pt atoms is greatly decreased accompanied by a limited quantity of metallic Sn0 when Sn/Pt ratio ≥ 0.94, suggesting the generation of Pt–SnOx entities at high Sn loading.In the case of small particles (particle size below 50 Å), the composition-dependent structural changes are reflected in varying CNs of the scattering paths from central atom.9,18 The Pt–O, Pt–Pt and Pt–Sn contributions in the curve fitting of Pt LIII-edge provide further information on the bimetallic structure. The average CN of the Pt–Sn shell for PtSn0.69/LOC is 1.1, and this value is decreased to 0.3 for PtSn0.94/LOC and 0.2 for PtSn1.33/LOC, most likely resulting from the phase segregation of PtSn alloys.41 Meanwhile, the shorter distances of Pt–Pt bond for PtSn0.94/LOC and PtSn1.33/LOC than that for Pt/LOC and PtSn0.69/LOC, suggests a contraction of the interatomic distance due to the formation of small Pt clusters possibly segregated from PtSn alloys. These EXAFS fitting results give a clear illustration on the possible structure evolution of bimetallic NPs from SnOx-patched PtSn alloys to SnOx-patched Pt clusters with increasing Sn/Pt ratios. Besides, the CN value of Pt–O contribution to the PtSn1.33/LOC is decreased to 1.0 with respect to the Pt/LOC (1.5). In addition to the decreased O/La ratio from XPS analyses, it is suggested that surface oxygenic species are partially consumed at high Sn loading. One plausible reason is that surface oxygen vacancy sites are generated, presumably due to the reduction of Pt–SnOx sites through electron capture and transfer upon hydrogen spillover.41,45
From these results we can conclude that bimetallic samples are composed of O-coordinated Pt2+ atoms and Pt0 clusters, which are diluted by the PtSn alloys and SnOx species. Although most of tin cannot be reduced to metallic form and incorporated into an alloy, it affects the geometric structure of platinum and helps to stabilize the Pt atoms. Furthermore, the composition-dependent changes in bimetallic structure are proposed to account for the distinct catalytic behavior.
4.3 The correlations between structure evolution and catalytic behavior
The above results reflect an interaction between Pt and Sn, which is related to the formed PtSn alloys and Pt–SnOx species. Such an interaction exerts a great influence on the reaction route. The product distribution demonstrates that the reaction proceeds through preferential hydrogenation of CO bond for bimetallic system. In particular, over the PtSn0.94/LOC, the selectivity and yield to CROL achieves a 5-fold and 7-fold improvement over the Pt/LOC. Additionally, PtSn0.94/LOC retains its selectivity at the initial level (around 70%), irrespective of the reaction time and hydrogen pressure.
It seems reasonable to accept that the enhanced carbonyl selectivity of bimetallic system is originated from the intrinsic nature of Pt, as our tests show that tin oxides are inactive for CRAL hydrogenation and participate only in the isolation of Pt atoms. It is proposed that the chemisorption behaviors of H2 and CO on metal sites originate from the interacting surfaces of Pt and Sn at the atomic level.41 The H2-TPD and CO-DRIFT measurements are employed to extract the correlation between the bimetallic structure and the catalytic behavior. It is found that incorporating a small amount of tin (Sn/Pt ratio equals to 0.69) substantially increases the number of active Pt sites for hydrogen, presumably due to the prevailing effect of site isolation generated by tin. Meanwhile, this dilution effect favors the reversible adsorption of linear Pt–CO, which is accounted for the improvement in hydrogenation rate for PtSn0.69/LOC compared to the Pt/LOC. However, when Sn/Pt ratio ≥ 0.94, excessive tin results into the passivation effect of the catalyst surface and greatly reduce the exposure of Pt sites for H2 and CO adsorption. Besides, the interaction between CO and Pt is greatly enhanced with increasing Sn loadings, as evidenced by the red-shift of linear Pt–CO band and the desorption behavior from CO-DRIFTS spectra. This increased interaction plays a decisive role in the competitive adsorption and hydrogenation of carbonyl groups, whereas lowers the desorption rate of the products and hinders the recovery of active sites. Consequently, the hydrogenation rate of CRAL is sharply decreased at high Sn loadings.
It is known that the hydrogenation product of unsaturated aldehyde is strongly dependent on the adsorption mode of reactant molecule on the catalyst surface.37,46 According to the reactive performance over the prepared catalysts, two possible adsorption models of CRAL over the prepared Pt and PtSn catalysts are proposed. On the Pt/LOC, the support effect is the primary factor for the selective hydrogenation. The carbonyl oxygen is suggested to interact with the positively charged Pt2+, and the Pt0 site is the adsorption center of CC bond. In this model, the hydrogenation of CC and CO bonds is competitive, which dominates the selectivities to BUAL and CROL, respectively. While for tin-promoted catalysts, additional Pt–SnOx sites are generated onto the catalyst surface. The carbonyl group could chemisorb on the Pt–SnOx entities through the carbonyl carbon with the Pt0 site and carbonyl oxygen with electropositive Snn+ species to form a di-σCO adsorption mode, which is proposed to facilitate the polarization and activation of the CO bond.21,38 Evidently, the activation of the CO bond on the Pt–SnOx entity is much easier than that on the Pt0–Pt2+ couple, which accounts for the additional activity attained on the tin-modified catalysts compared to the Pt/LOC. In addition, oxygen vacancies related to the generation of Pt–SnOx entities is evidenced by the XPS and EXAFS-fitting results. This improved Pt–SnOx couple is likely to further enhance the selective activation and hydrogenation of CO bond.
By analyzing the activity of PtSn/LOC catalysts as a function of the Sn/Pt ratios, there seems to be a dilution effect of Pt sites, yielding higher hydrogenation rates for reaction, and a promoting effect of ionic Sn, favoring the preferential hydrogenation for carbonyl groups.21,34 However, excessive tin loading in the bimetallic catalysts drastically reduces the conversion rate of CRAL. Therefore, the synergy between Pt sites and tin species leads to an optimum Sn/Pt ratio.
5. Conclusion
In summary, liquid-phase CRAL hydrogenation is used as a model reaction to test the tin-promoted Pt/LOC catalysts, which are highly selective to the desired unsaturated alcohol compared to the Pt/LOC. The strong metal–support interaction in the Pt/LOC plays an important role in retaining high dispersion of Pt, giving rise to a good reproducibility during cycle experiment. For bimetallic systems, the distinct reactive performance can be tentatively attributed to the composition-dependent structure of bimetallic particles. The varying Sn/Pt ratios determine the atomic arrangement of bimetallic NPs and the geometric characteristics of Pt, which dominates the adsorption mode of CRAL molecules and is proposed to be accounted for the additional activity towards carbonyl hydrogenation. However, the PtSn0.94/LOC easily suffers from the catalyst deactivation due to the phase segregation of PtSn alloys and the leaching effect of SnOx patches. This work demonstrates that high performing catalysts with both metals can be achieved by adjustment of the bimetallic structure, and provides a further understanding on the fabrication of bimetallic catalysts for specific heterogeneous reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Sincere acknowledge is made to the financial support from the National Science Foundation of China (21401204), Innovation Promotion Association CAS (2017460), the Western Light Program of Chinese Academy of Sciences (2015), and Science and Technology Project of Suzhou City (SYG201627). The X-ray absorption spectroscopy is granted by 4W1B station of Beijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences. The staff members of 4W1B are acknowledged for their support in measurements and data reduction.References
- P. Gallezot and D. Richard, Catal. Rev., 1998, 40, 81–126 CAS.
- X. Yang, A. Wang, X. Wang, T. Zhang, K. Han and J. Li, J. Phys. Chem. C, 2009, 113, 20918–20926 CAS.
- M. Abid, V. Paul-Boncour and R. Touroude, Appl. Catal., A, 2006, 297, 48–59 CrossRef CAS.
- A. Dandekar and M. A. Vannice, J. Catal., 1999, 183, 344–354 CrossRef CAS.
- H. Yoshitake and N. Saito, Microporous Mesoporous Mater., 2013, 168, 51–56 CrossRef CAS.
- H.-Y. Chen, C.-T. Chang, S.-J. Chiang, B.-J. Liaw and Y.-Z. Chen, Appl. Catal., A, 2010, 381, 209–215 CrossRef CAS.
- P. Reyes, H. Rojas and J. L. G. Fierro, Appl. Catal., A, 2003, 248, 59–65 CrossRef CAS.
- X. Yang, Y. Mueanngern, Q. A. Baker and L. R. Baker, Catal. Sci. Technol., 2016, 6, 6824–6835 CAS.
- A. Siani, O. S. Alexeev, G. Lafaye and M. D. Amiridis, J. Catal., 2009, 266, 26–38 CrossRef CAS.
- I. M. J. Vilella, S. R. de Miguel and O. A. Scelza, J. Mol. Catal. A: Chem., 2008, 284, 161–171 CrossRef CAS.
- I. M. Vilella, I. Borbáth, J. L. Margitfalvi, K. Lázár, S. R. de Miguel and O. A. Scelza, Appl. Catal., A, 2007, 326, 37–47 CrossRef CAS.
- F. Coloma, J. Llorca, N. Homs, P. R. de la Piscina, F. Rodriguez-Reinoso and A. Sepulveda-Escribano, Phys. Chem. Chem. Phys., 2000, 2, 3063–3069 RSC.
- J. Shi, M. Zhang, W. Du, W. Ning and Z. Hou, Catal. Sci. Technol., 2015, 5, 3108–3112 CAS.
- Z. Tian, C. Liu, Q. Li, J. Hou, Y. Li and S. Ai, Appl. Catal., A, 2015, 506, 134–142 CrossRef CAS.
- W. O. Oduro, N. Cailuo, K. M. K. Yu, H. Yang and S. C. Tsang, Phys. Chem. Chem. Phys., 2011, 13, 2590–2602 RSC.
- H. Rong, Z. Niu, Y. Zhao, H. Cheng, Z. Li, L. Ma, J. Li, S. Wei and Y. Li, Chem.–Eur. J., 2015, 21, 12034–12041 CrossRef CAS PubMed.
- S. T. Christensen, H. Feng, J. L. Libera, N. Guo, J. T. Miller, P. C. Stair and J. W. Elam, Nano Lett., 2010, 10, 3047–3051 CrossRef CAS PubMed.
- G. J. Siri, J. M. Ramallo-López, M. L. Casella, J. L. G. Fierro, F. G. Requejo and O. A. Ferretti, Appl. Catal., A, 2005, 278, 239–249 CrossRef CAS.
- F. Coloma, A. Sepúlveda-Escribano, J. L. G. Fierro and F. Rodríguez-Reinoso, Appl. Catal., A, 1996, 136, 231–248 CrossRef CAS.
- J. P. Stassi, P. D. Zgolicz, V. I. Rodríguez, S. R. de Miguel and O. A. Scelza, Appl. Catal., A, 2015, 497, 58–71 CrossRef CAS.
- J. P. Stassi, P. D. Zgolicz, S. R. de Miguel and O. A. Scelza, J. Catal., 2013, 306, 11–29 CrossRef CAS.
- A. Huidobro, A. Sepúlveda-Escribano and F. Rodríguez-Reinoso, J. Catal., 2002, 212, 94–103 CrossRef CAS.
- A. B. da Silva, E. Jordão, M. J. Mendes and P. Fouilloux, Appl. Catal., A, 1997, 148, 253–264 CrossRef CAS.
- Y.-H. Hou, W.-C. Han, W.-S. Xia and H.-L. Wan, ACS Catal., 2015, 5, 1663–1674 CrossRef CAS.
- T. Levan, M. Che, J. M. Tatibouet and M. Kermarec, J. Catal., 1993, 142, 18–26 CrossRef CAS.
- W. Gu, J. Liu, M. Hu, F. Wang and Y. Song, ACS Appl. Mater. Interfaces, 2015, 7, 26914–26922 CAS.
- X. Huang, C. Dang, H. Yu, H. Wang and F. Peng, ACS Catal., 2015, 5, 1155–1163 CrossRef CAS.
- L. Jin, Y. Zhang, J. P. Dombrowski, C.-H. Chen, A. Pravatas, L. Xu, C. Perkins and S. L. Suib, Appl. Catal., B, 2011, 103, 200–205 CrossRef CAS.
- Y. Zhang, L. Jin, K. Sterling, Z. Luo, T. Jiang, R. Miao, C. Guild and S. L. Suib, Green Chem., 2015, 17, 3600–3608 RSC.
- F. Hou, H. Zhao, J. Zhao, J. Yang, L. Yan, H. Song and L. Chou, J. Nanopart. Res., 2016, 18, 1–17 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- S. Irusta, L. M. Cornaglia and E. A. Lombardo, Mater. Chem. Phys., 2004, 86, 440–447 CrossRef CAS.
- K. S. Kim, N. Winograd and R. E. Davis, J. Am. Chem. Soc., 1971, 93, 6296–6297 CrossRef CAS.
- J. Shi, M. Zhang, W. Du, W. Ning and Z. Hou, Catal. Sci. Technol., 2015, 5, 3108–3112 CAS.
- J. Lee, Y. Ryou, X. Chan, T. J. Kim and D. H. Kim, J. Phys. Chem. C, 2016, 120, 25870–25879 CAS.
- Y. Zhou, D. E. Doronkin, M. Chen, S. Wei and J.-D. Grunwaldt, ACS Catal., 2016, 6, 7799–7809 CrossRef CAS.
- J. Haubrich, D. Loffreda, F. Delbecq, P. Sautet, Y. Jugnet, C. Becker and K. Wandelt, J. Phys. Chem. C, 2010, 114, 1073–1084 CAS.
- K. Taniya, H. Jinno, M. Kishida, Y. Ichihashi and S. Nishiyama, J. Catal., 2012, 288, 84–91 CrossRef CAS.
- S.-H. Liu, F.-S. Zheng and J.-R. Wu, Appl. Catal., B, 2011, 108, 81–89 Search PubMed.
- F. Behafarid, L. K. Ono, S. Mostafa, J. R. Croy, G. Shafai, S. Hong, T. S. Rahman, S. R. Bare and B. Roldan Cuenya, Phys. Chem. Chem. Phys., 2012, 14, 11766–11779 RSC.
- H. Rong, Z. Niu, Y. Zhao, H. Cheng, Z. Li, L. Ma, J. Li, S. Wei and Y. Li, Chem.–Eur. J., 2015, 21, 12034–12041 CrossRef CAS PubMed.
- Z. Niu, N. Becknell, Y. Yu, D. Kim, C. Chen, N. Kornienko, G. A. Somorjai and P. Yang, Nat. Mater., 2016, 15, 1188–1194 CrossRef CAS PubMed.
- Y. Uemura, Y. Inada, K. K. Bando, T. Sasaki, N. Kamiuchi, K. Eguchi, A. Yagishita, M. Nomura, M. Tada and Y. Iwasawa, Phys. Chem. Chem. Phys., 2011, 13, 15833 RSC.
- Y. Nagai, T. Hirabayashi, K. Dohmae, N. Takagi, T. Minami, H. Shinjoh and S. i. Matsumoto, J. Catal., 2006, 242, 103–109 CrossRef CAS.
- R. Prins, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS PubMed.
- F. Delbecq and P. Sautet, J. Catal., 2002, 211, 398–406 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10084a |
| This journal is © The Royal Society of Chemistry 2017 |