
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiocarbamoyl in synthesis of the absorbent bis-mono and dimethine cyanine dyes incorporating porphyrin, thiazoles, thiazolones, thiadiazoles, pyrazoles and pyrazolones†
Rasha E. El-Mekawy‡
 a and
A. A. Fadda*b
a and
A. A. Fadda*b
aDepartment of Petrochemicals, Egyptian Petroleum Research Institute, Nasr City, Cairo, Egypt
bDepartment of Chemistry, Faculty of Science, Mansoura University, Mansoura, Egypt. E-mail: afadda50@yahoo.com
First published on 29th November 2017
Abstract
In this study, we have introduced an outlook dealing with synthetic heteroarenium-substituted pyridines 1 that are suitable as starting materials for the synthesis of a potential new thiocarbamoyl derivative 3 that may be of value in preparative organic and biological chemistry. Herein, a new series of twelve bis-mono and dimethine cyanine dyes incorporating different heterocyclic compounds were designed by efficient and simple reaction of compound 3 with various organic reagents to overcome the obstacles of energetic reactions, because they took long times. Moreover, the electronic behaviors of these compounds were studied because of their significant activity in many medical and other fields. The photosensitizers were elucidated by spectral and analytical analyses.
Introduction
Over the historical development of chemistry, it has been found that sulfur-containing compounds have often exhibited numerous biotechnologies and served as significant enforcement in the pharmaceutical industry.1 A set of compounds containing sulfur atoms are widely used in drugs and natural products. Therefore, sulfur atoms and other heterogeneous atoms must be linked together to increase the efficiency of the compounds; thus, sulfur-nitrogen heterocyclic compounds have attracted the interest from organic and inorganic researchers. Thiocarbamoyls also play a decisive role in the evolution of heterocyclic chemistry and are extensively used as beneficial synthons incorporating –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CS moiety in organic synthesis.2–5 Additionally, for the last several years, there has been considerable progress6,7 in the synthesis and applications of methine cyanine dyes, which are among the most influential functional organic dyes. They have been used as photographic sensitizers,8 optical recording materials in laser disks9 and sensitizers in photovoltaic cells.10 With the growth of relative technologies, methine cyanine dyes have been screened as tools in photodynamic treatment11 and as fluorescent labels, micelles, organelles and probes for cells.12–15 Usually, mono and dimethine cyanines are synthesized by heating a mixture of aromatic aldehydes, a quaternary ammonium salt with an active methyl group and a catalyst agent in an organic environment under reflux.16 This synthesis has intrinsic obstacles as relatively the reaction requires relatively energetic conditions, namely heating with reflux reactants for diverse hours in solvents, many organic solvents inimical to environment and complexity in product isolation. Here, we record our feedback about some efficacious steps for the synthesis of novel hemimethine, bis-hemimethine and bismethine cyanine dyes incorporating porphyrin, substituted thiazoles, thiazolones, thiadiazoles, pyrazoles and pyrazolones.
CS moiety in organic synthesis.2–5 Additionally, for the last several years, there has been considerable progress6,7 in the synthesis and applications of methine cyanine dyes, which are among the most influential functional organic dyes. They have been used as photographic sensitizers,8 optical recording materials in laser disks9 and sensitizers in photovoltaic cells.10 With the growth of relative technologies, methine cyanine dyes have been screened as tools in photodynamic treatment11 and as fluorescent labels, micelles, organelles and probes for cells.12–15 Usually, mono and dimethine cyanines are synthesized by heating a mixture of aromatic aldehydes, a quaternary ammonium salt with an active methyl group and a catalyst agent in an organic environment under reflux.16 This synthesis has intrinsic obstacles as relatively the reaction requires relatively energetic conditions, namely heating with reflux reactants for diverse hours in solvents, many organic solvents inimical to environment and complexity in product isolation. Here, we record our feedback about some efficacious steps for the synthesis of novel hemimethine, bis-hemimethine and bismethine cyanine dyes incorporating porphyrin, substituted thiazoles, thiazolones, thiadiazoles, pyrazoles and pyrazolones.
Results and discussion
We were heedful in constructing heterocycles established on thiocarbamoyl moieties such as N-substituted thiazole, thiadiazoles, pyrazole, thiophene and thiadiazoles. Even heterocyclic compounds incorporating thiocarbamoyl moieties have rarely been stated in the research field of drug construction through synthesis of solid phase.17–22 In the light of the above facts, we have developed a straightforward and efficient mode for the production of bis-thiocarbamoyl intermediate 3. The methods are operationally simple and do not require strict conditions. The base-promoted reaction of two moles of phenyl isothiocyanate and active methylene of 2-benzyl-1-(4-bromocyclohexy)pyridine-1-ium salt (1) was synthesized by heating benzylpyridine with an appropriate amount of 1,4-dibromocyclohexane in refluxing toluene in 85% yield (Scheme 1). The latter reaction was performed in the existence of a catalytic quantity of potassium hydroxide in DMF and yielded the non-separated intermediate 2, which underwent in situ neutralization with dilute HCl, resulting in the formation of a red acyclic intermediate 3. On the other hand, 1 was refluxed with two moles of phenyl isothiocyanate for 4 h in absolute ethanol and fresh pellets of potassium hydroxide afforded a selfsame compound in all elemental and spectral behaviours (IR, 1H-NMR, 13C-NMR, mass spectroscopy and m.p.) with the acyclic intermediate 3 (Scheme 2). Assignment of compound 3 is based on its analytical and spectral data. Infrared spectrum showed absorption bands at υ = 3321 and 2231 cm−1, attributable to two NH and two SH function groups. Moreover, IR spectrum displayed no absorption bands at 1284 cm−1 related to the absence of two SO function groups. This is evidence of the formation of tautomeric structure 3 and not 4 and/or 5. Also, the structure of 3 was inferred by 1H-NMR spectroscopy, which revealed a chemical shift (δ) at 2.27 ppm as pentet signals corresponding to two methine protons of 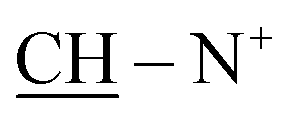 of the cyclohexyl ring and singlet signal at δ 2.17 ppm for two SH protons; this further confirms the formation of tautomeric structure 3. Also, the spectrum showed doublet signals at δ 1.47 ppm, attributable to
of the cyclohexyl ring and singlet signal at δ 2.17 ppm for two SH protons; this further confirms the formation of tautomeric structure 3. Also, the spectrum showed doublet signals at δ 1.47 ppm, attributable to 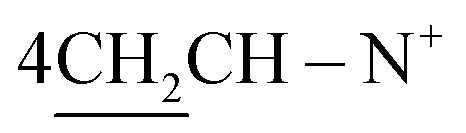 protons, in addition to D2O exchangeable protons as singlet signal at δ 11.2 ppm due to two NH protons. The mass spectrum of structure 3 agreed with the molecular structure at m/z = 690 (M+, −2Br, 92%) with respect to the molecular formula C44H42N4S2Br2.
protons, in addition to D2O exchangeable protons as singlet signal at δ 11.2 ppm due to two NH protons. The mass spectrum of structure 3 agreed with the molecular structure at m/z = 690 (M+, −2Br, 92%) with respect to the molecular formula C44H42N4S2Br2.
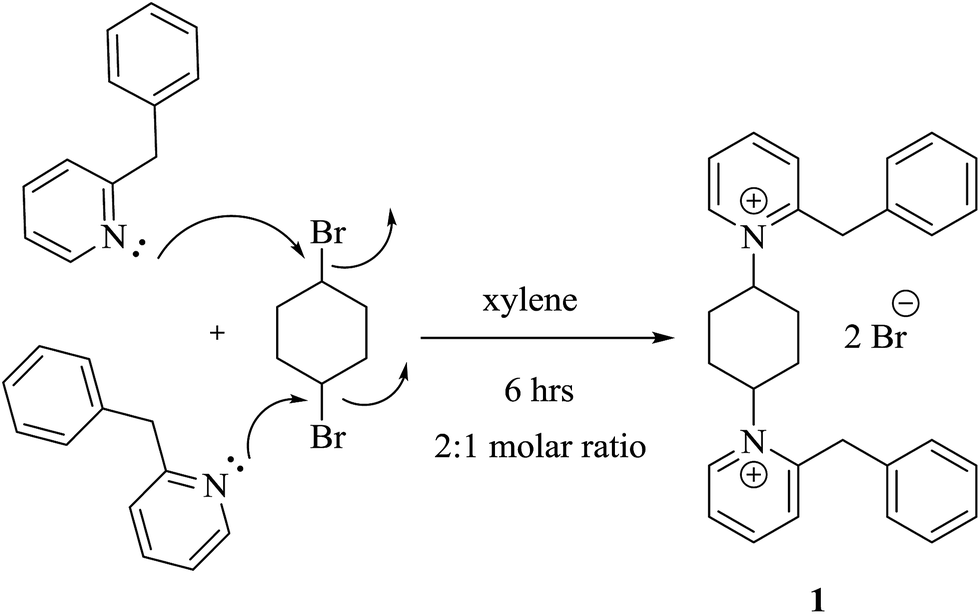 | ||
| Scheme 1 New features of bis-pyridinium salt 1. | ||
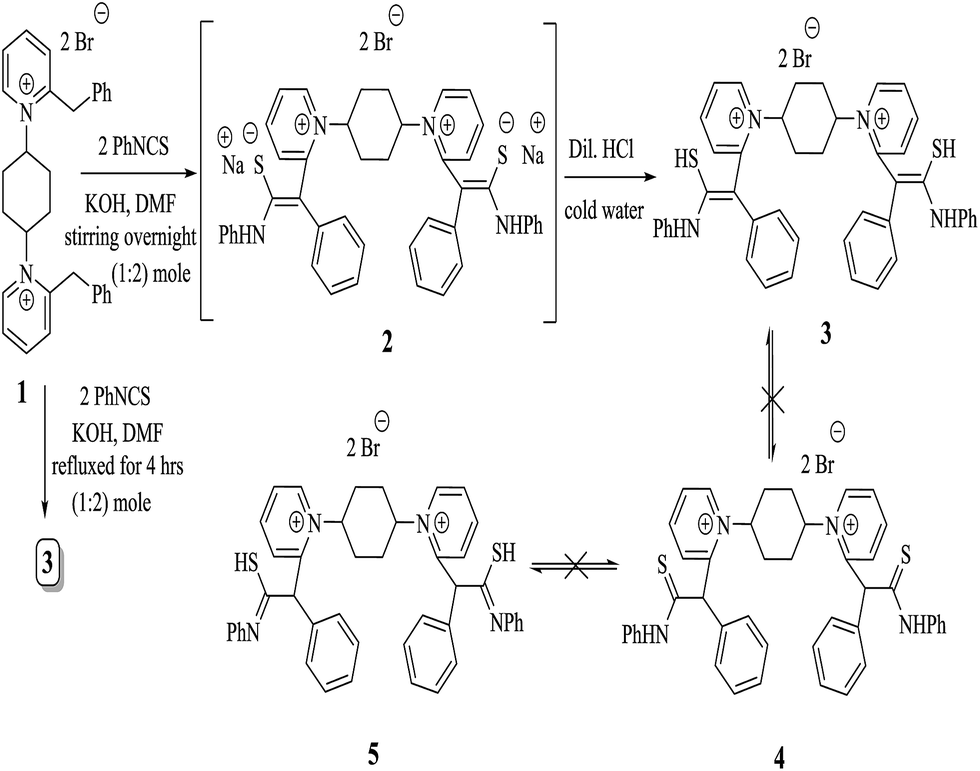 | ||
| Scheme 2 Synthesis of 1,1′-[(cyclohexan-1,4-diyl)bis(2-(Z)-2-mercapto-1-phenyl-2-(phenylamino)vinyl)]bis-pyridinium dibromide (3). | ||
There has been particular emphasis on the synthesis of equipotent five and six heterocycles incorporating sulfur and nitrogen atoms in recent decades due to their virtual possibilities for architectural design of significant methine cyanine dyes. Consequently, the non-isolable intermediate 2 and acyclic intermediate 3 act as liaison to prepare bis-methine cyanine dye 7 bearing two pyridinium salts binding to two thiazolidinone rings via methine bridge. Furthermore, ethyl bromoacetate was added dropwise with in situ stirring to non-isolable intermediate 2 to afford 1,1′(cyclohexane-1,4-diyl)bis(2-((Z)-4-oxo-1-(phenyl-3-phenylthiazolidin-2-ylidene)methyl)pyridine-1-ium)dibromide (7). Alternatively, the isolated compound 3 reacted with ethyl bromoacetate in N,N-dimethylformamide and in the presence of freshly fused potassium carbonate in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio to form compound 7. Thus, a new bis-dimethine cyanine dye 6 was synthesized via stirring of non-isolable thiocarbamoyl 2 and/or isolable thiocarbamoyl 3 with two moles of ethyl bromoacetate overnight; then the reaction mixture was acidified, giving the same chemical structure as compound 6 (Scheme 3). Bis-dimethine cyanine dye 6 was refluxed in the presence of a few drops of triethylamine in N,N-dimethylformamide and then cyclized to thiazolidinone derivative 7, which was identical in all respects with compound 7 (IR, 1H-NMR, 13C-NMR, mass spectroscopy and m.p.).
:2 molar ratio to form compound 7. Thus, a new bis-dimethine cyanine dye 6 was synthesized via stirring of non-isolable thiocarbamoyl 2 and/or isolable thiocarbamoyl 3 with two moles of ethyl bromoacetate overnight; then the reaction mixture was acidified, giving the same chemical structure as compound 6 (Scheme 3). Bis-dimethine cyanine dye 6 was refluxed in the presence of a few drops of triethylamine in N,N-dimethylformamide and then cyclized to thiazolidinone derivative 7, which was identical in all respects with compound 7 (IR, 1H-NMR, 13C-NMR, mass spectroscopy and m.p.).
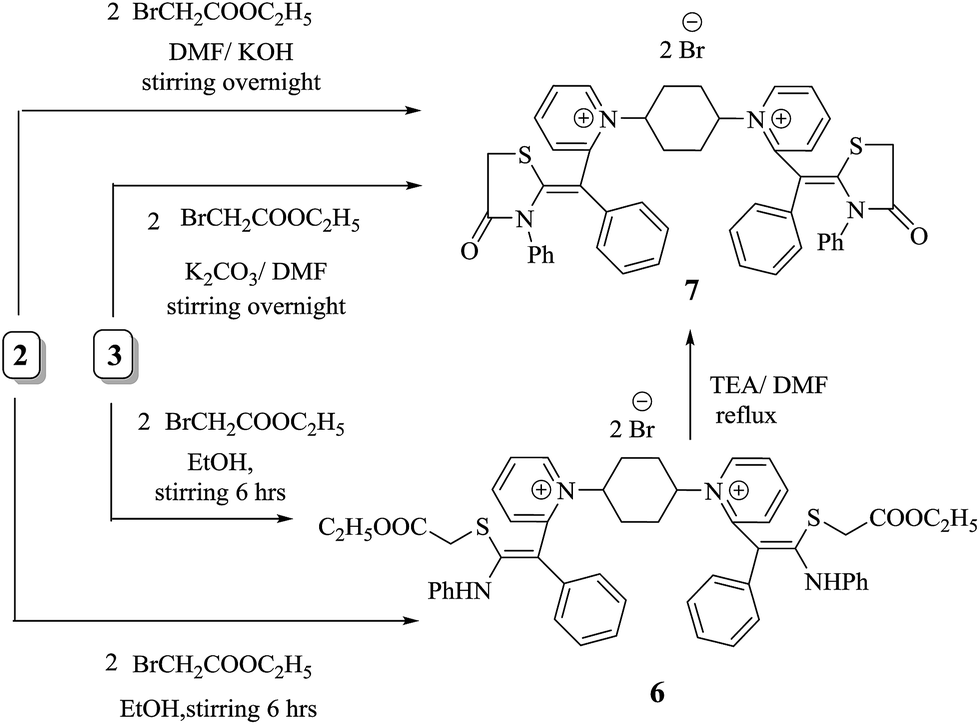 | ||
| Scheme 3 Alternative synthesis of 1,1′-[(cyclohexan-1,4-diyl)bis(2-((Z)-4-oxo-3-phenylthiazolidin-2-ylidene)(phenyl)methyl)]bis-pyridinium dibromide (7). | ||
The skeleton of chemical structure 6 was promoted by its spectral and analytical analyses. Noticeably, the IR spectrum of the synthesized compound revealed absorption bands at υ = 1730 and 3322 cm−1, owing to two –CO and two NH functional groups, respectively. Its 1H-NMR spectrum exhibited distinctive triplet signals at δ = 1.30 ppm, a singlet signal at 3.23 ppm and quartet signals at 4.30 ppm, corresponding to two 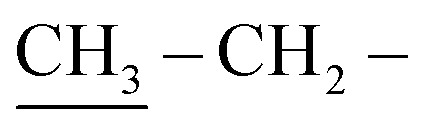 , two
, two 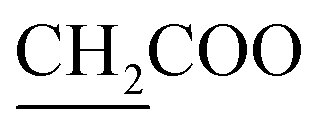 and two
and two 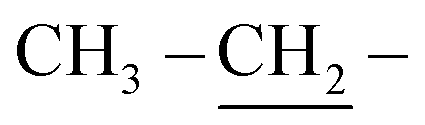 , respectively. On the other hand, a broad singlet band was located at δ = 12.90 ppm, assignable for two NH protons. Structure of 7 was further confirmed by 1H-NMR, which displayed no triplet, quartet, and singlet signals at δ = 1.30, 4.30 and 12.90 ppm because of two CH3, two CH2 of ethoxy groups and two NH groups, which were involved in the reaction forming cyclized thiazolidinone ring. Thus, a singlet signal appeared at δ = 3.12 ppm on account of two protons related to the CH2 group. In addition, the UV-vis spectra showed λmax at 524 nm and λem at 530 nm.
, respectively. On the other hand, a broad singlet band was located at δ = 12.90 ppm, assignable for two NH protons. Structure of 7 was further confirmed by 1H-NMR, which displayed no triplet, quartet, and singlet signals at δ = 1.30, 4.30 and 12.90 ppm because of two CH3, two CH2 of ethoxy groups and two NH groups, which were involved in the reaction forming cyclized thiazolidinone ring. Thus, a singlet signal appeared at δ = 3.12 ppm on account of two protons related to the CH2 group. In addition, the UV-vis spectra showed λmax at 524 nm and λem at 530 nm.
Moreover, chalcones have played a definitive role in the evolution of heterocyclic compound theory. Also, they are applied extensively in organic composition.23,24 A lot of efforts have been devoted to synthesize bis-porphyrinodimethine cyanine dye 8 by reaction of compound 7 with two moles of 1-formyl porphyrin in N,N-dimethylformamide in the presence of a catalytic quantity of piperidine under nitrogen gas for 24 h, affording a good yield. Structure of 8 was based on it spectral and analytical analyses. The IR spectrum showed no characteristic absorption band at υ = 1732 cm−1, attributable to formyl group of porphyrin structure, and this gave an evidence that the functional group was consumed in the reaction, forming arylidene as bis-dimethine cyanine dye 8. 1H-NMR spectrum disclosed doublet signals at δ = 1.41 ppm due to  molecules of cyclohexyl ring, six singlet signals at δ = 4.30, 6.98, 6.46, 6.66, 5.62, and 12.89 ppm corresponding to
molecules of cyclohexyl ring, six singlet signals at δ = 4.30, 6.98, 6.46, 6.66, 5.62, and 12.89 ppm corresponding to 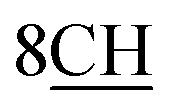 groups of methine bridge of porphyrin skeleton protons,
groups of methine bridge of porphyrin skeleton protons, 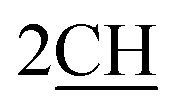 methine protons of pyrrole ring of porphyrin, arylidine moiety (porphyrin
methine protons of pyrrole ring of porphyrin, arylidine moiety (porphyrin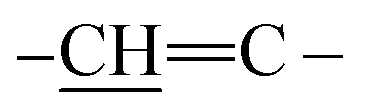 ) and two D2O exchangeable protons of four NH groups of pyrrole rings of porphyrin. Furthermore, mass spectrometry showed molecular ion peak at m/z = 1413 (M+ +2, −2Br, 67%) related to the molecular formula C90H66N12O5S2Br2. 13C-NMR spectrum showed additional confirmation of bis-porphyrinodimethine cyanine dye 8. The UV-vis spectra displayed two interesting and strong absorption bands for λmax at 422 nm and λem at 429 nm with respect to a soret band (B band) of porphyrin structure and λmax at 576 nm and λem at 582 nm corresponding to dimethinecyanine dye (Scheme 4).
) and two D2O exchangeable protons of four NH groups of pyrrole rings of porphyrin. Furthermore, mass spectrometry showed molecular ion peak at m/z = 1413 (M+ +2, −2Br, 67%) related to the molecular formula C90H66N12O5S2Br2. 13C-NMR spectrum showed additional confirmation of bis-porphyrinodimethine cyanine dye 8. The UV-vis spectra displayed two interesting and strong absorption bands for λmax at 422 nm and λem at 429 nm with respect to a soret band (B band) of porphyrin structure and λmax at 576 nm and λem at 582 nm corresponding to dimethinecyanine dye (Scheme 4).
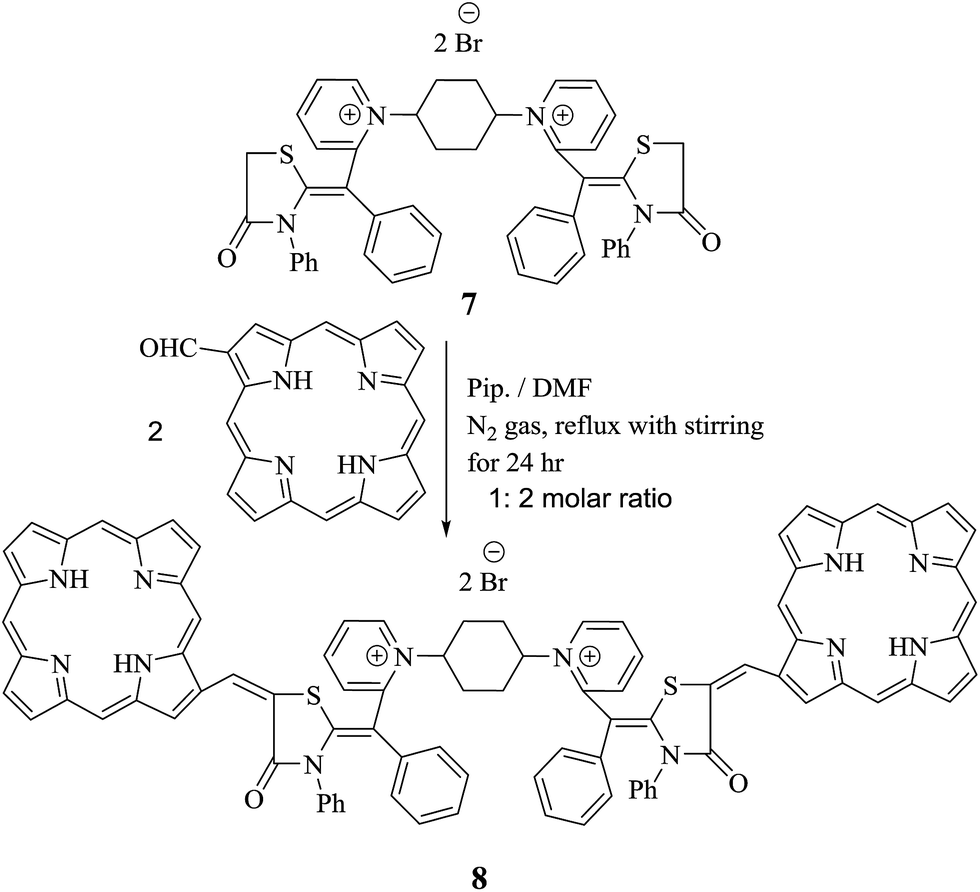 | ||
| Scheme 4 Synthesis of bis-dimethine cyanine dye incorporating porphyrin moiety 8. | ||
In a similar manner, treating the key intermediate 3 with phenacyl bromide in DMF and in the presence of a catalytic quantity of freshly fused potassium carbonate gave a single product that has been analyzed correctly for C60H50N4S2Br2 (10). An alternative synthesis of compound 10 readily occurred by treatment of non-isolable intermediate 2 with phenacyl bromide forming, a compound identical to compound 10 in all respects (IR, 1H-NMR, 13C-NMR, mass spectroscopy and m.p.). During our approach, the formation of novel dimethine cyanine dye 9 was achieved by stirring non-isolable intermediate 2 in basic medium KOH/DMF with alcoholic solution of two moles of phenacyl bromide at room temperature for about 6 h. Subsequently, alkylation of compound 3 with two moles of phenacyl bromide readily led to the formation of a chemical structure identical to compound 9, which underwent cyclization with a strong base such as triethylamine in DMF at 150 °C to provide 1,1′-(cyclohexane-1,4-diyl)bis(2-(Z)-(3,4-diphenylthiazol-2(3H)ylidene)(phenyl)methyl)-bis-pyridin-1-ium dibromide (10) in 75% yield via loss of H2O molecule (Scheme 5).
 | ||
| Scheme 5 Novel 1,1′-[(cyclohexane-1,4-diyl)bis(2-(Z)-(3,4-diphenylthiazol-2(3H)ylidene)(phenyl)methyl)]-bis-pyridinium dibromide (10). | ||
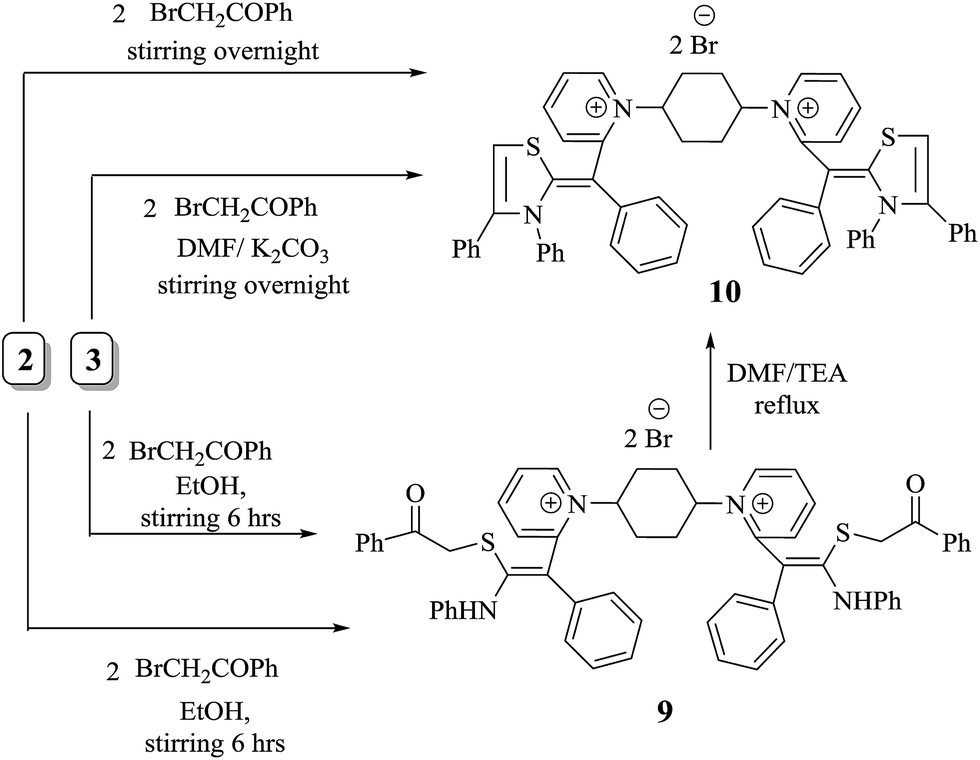
Structure 9 is proposed for the reaction output based on both atomic and spectral analyses. The IR spectroscopy showed significant absorption bands at υ = 3323 and 1728 cm−1, attributable to two NH groups and carbonyl group of 2 Ph–CO, respectively. On the other hand, structure 9 was assigned on the basis of 1H-NMR spectra, which showed a chemical shift (δ) at 2.51 and 11.92 ppm as two singlet signals due to two 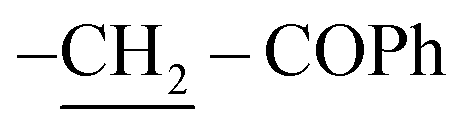 and two NH protons, respectively. The mass spectroscopy gave additional information on compound 9, which showed a molecular ion peak at m/z = 927 (M+, −2Br, 39%) attributed to the molecular formula C60H54N2O2S2Br2.
and two NH protons, respectively. The mass spectroscopy gave additional information on compound 9, which showed a molecular ion peak at m/z = 927 (M+, −2Br, 39%) attributed to the molecular formula C60H54N2O2S2Br2.
Treatment of thiocarbamoyl derivative 3 with anhydrous potassium carbonate in DMF at 150 °C followed by addition of two moles of three different aryl hydrazones resulted in the formation of N-substituted thiadiazole derivatives 12a–c in moderate yields. The latter compounds were alternatively obtained via treatment of non-isolable intermediate 2 with two moles of three different aryl hydrazones, forming compounds with chemical behaviour identical to bis-monomethine cyanine dyes incorporating N-substituted thiadiazole derivatives 12a–c. However, compounds 11a–c were prepared by stirring key non-isolable intermediate 2 with two moles of three different aryl hydrazones for 6 h at room temperature in excellent yields. Also, stirring an appropriate amount of compound 3 with different aryl hydrazones in molar ratio of 1:2 resulted in the formation of compounds 11a–c. Otherwise, the title compounds 11a–c were subjected to facile intramolecular cyclization by loss of aniline molecule in a basic medium Et3N/DMF to give monomethine cyanine dyes 12a–c in reasonable yields. It has been found that bis-dimethine cyanine dyes 11a–c were established on the principle of their spectral and analytical analyses. Generally, the IR showed absorption bands at υ = 1695–1732 cm−1 for two carbonyl groups of two benzoyl groups in each derivative, respectively. Moreover, IR exhibited characteristic absorption bands at υ = 3350–3312 cm−1 on account of four NH functional groups in each compound. However, the 1H-NMR spectra of compound 11a showed significant singlet signals at δ = 3.8, 10.74 and 11.61 ppm corresponding to two –OCH3 in addition to four NH protons (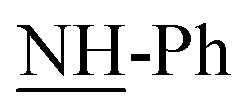 and
and  ). Interestingly, the 1H-NMR spectra of compound 12a assigned the disappearance of singlet signals at δ = 10.73 and 11.60 ppm as well as multiplet signals at 6.73–7.34 ppm to aromatic hydrogens (
). Interestingly, the 1H-NMR spectra of compound 12a assigned the disappearance of singlet signals at δ = 10.73 and 11.60 ppm as well as multiplet signals at 6.73–7.34 ppm to aromatic hydrogens (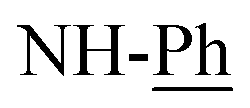 ). This is an excellent fact for cyclization of 11a to 12a. The mass spectroscopy of 12a readily displayed a molecular ion peak at m/z =1009 (M+, −2Br, 42%) with respect to the molecular formula C62H52N6O4S2Br2 (Scheme 6).
). This is an excellent fact for cyclization of 11a to 12a. The mass spectroscopy of 12a readily displayed a molecular ion peak at m/z =1009 (M+, −2Br, 42%) with respect to the molecular formula C62H52N6O4S2Br2 (Scheme 6).
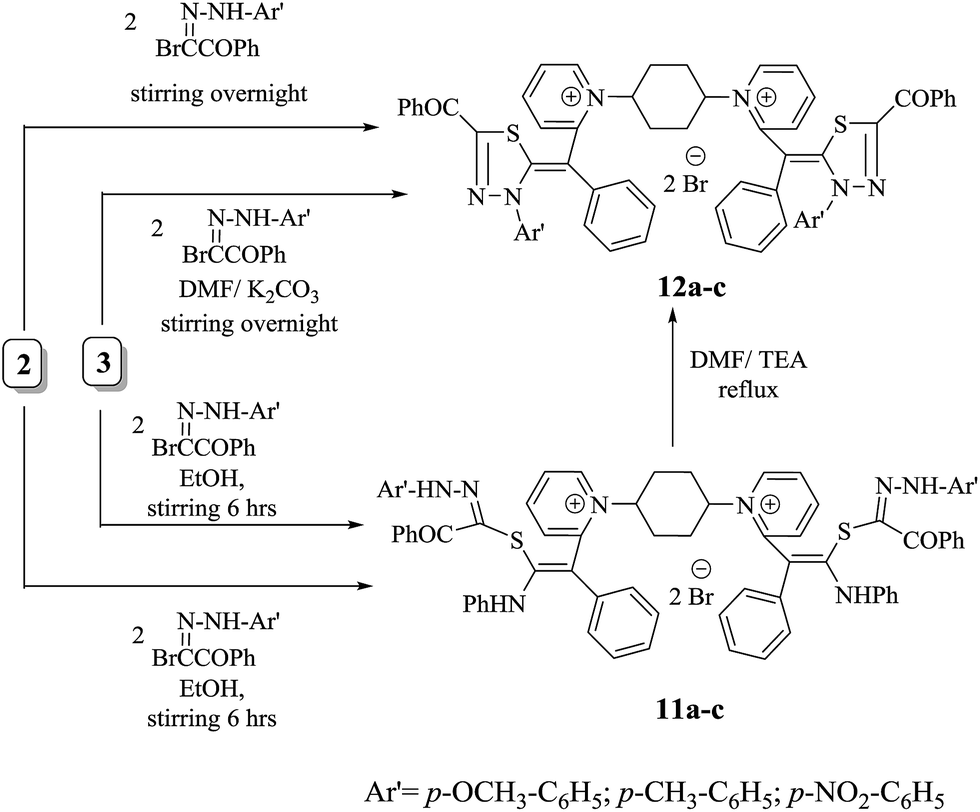 | ||
| Scheme 6 Novel series of monomethine cyanine dyes 12a–c. | ||
The finding of this category of drugs supplies a distinguished historical status of renewed drug evolution and also refers to the unpredictability of biological efficiency because of structural modulation of a prototype of pharmaceutical molecule. The broad area of curative prominence of thiocarbamoyl heterocyclic ring systems assists us to outline the synthesis of a range of new thiocarbamoyl heterocyclic derivatives incorporated in two adjacent dimethine cyanine dyes. Refluxing thiocarbamoyl derivative 3 with thiocarbohydrazide in N,N-dimethylformamide and in the presence of few drops of triethylamine produced bis-monomethine cyanine dye incorporating dithiadiazole derivative 14 instead of bis-monomethine cyanine dye incorporating thiadiazole derivative 13. Transformation of compound 3 into 1,1′-(cyclohexane-1,4-diyl)bis(2-((Z)-2-(dihydrazinylmethyl)disulfanyl)-1-phenyl-2-(phenylamino)vinyl)pyridine-1-ium)dibromide (15) and not into 16 in good yield was achieved upon its heating with thiocarbohydrazide in absolute ethanol and in the presence of freshly fused potassium carbonate. The reaction advancement was tracked by TLC. Furthermore, treatment of 15 with DMF and triethylamine resulted in the cyclization of 15 and furnished the corresponding 1,1′-(cyclohexane-1,4-diyl)bis(2-(Z)-(5-dihydrazinyl-3-phenyl-1,2,3-thiadiazol-2(3H)-ylidene(phenyl)methyl)pyridine-1-ium dibromide (14) in moderate yield, as shown in Scheme 7. The elucidation of compound 14 was concentrated on its spectral and analytical data. The IR spectrum presented strong absorption bands at υ = 3325–3174 cm −1 for two  groups. However, 1H-NMR spectra revealed two singlet signals at 3.8 and 8.8 ppm, corresponding to D2O exchangeable protons of two
groups. However, 1H-NMR spectra revealed two singlet signals at 3.8 and 8.8 ppm, corresponding to D2O exchangeable protons of two 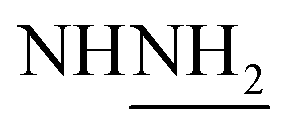 and
and 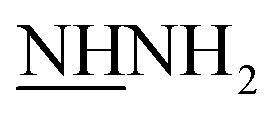 groups, respectively.
groups, respectively.
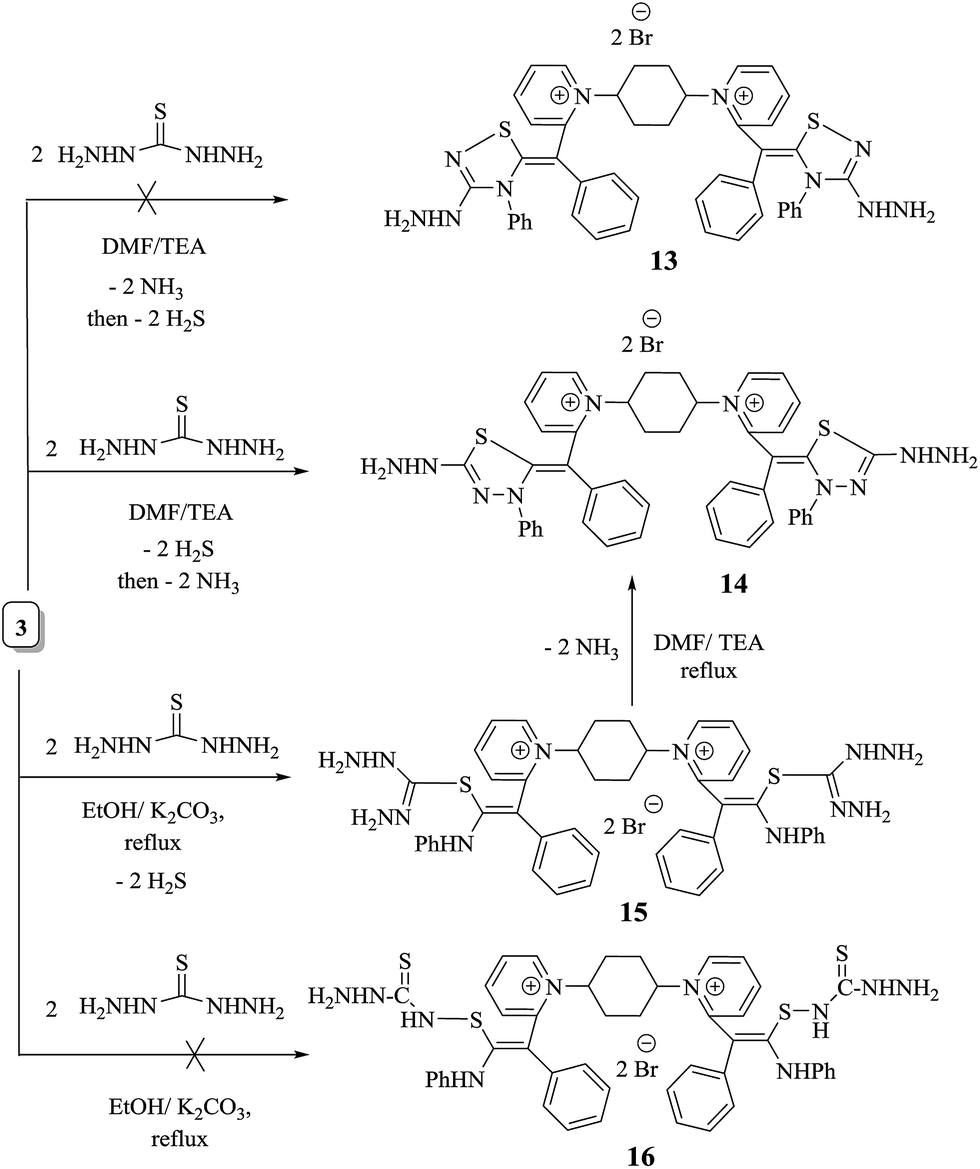 | ||
| Scheme 7 Synthesis of 1,1′-[(cyclohexane-1,4-diyl)bis(2-(Z)-(5-dihydrazinyl-3-phenyl-1,2,3-thiadiazol-2(3H)-ylidene(phenyl)methyl)]bis-pyridinium)dibromide (14). | ||
When compound 14 was subjected to heating in absolute ethanol at reflux and in the presence of triethylamine with ethyl acetoacetate and acetylacetone, single products 17 and 18 were for formed, respectively. Also, the reaction mixture was analyzed by TLC (Scheme 8). The elemental analyses and mass spectrum of the obtained products were in accordance with the molecular formula C54H48N10S4Br2 and C56H52N10S4Br2. Structure of 17 was established on the principle of its correct spectral and analytical analyses. The IR spectrum presented absorption bands at υ = 1652 cm−1 for two amidic carbonyl groups. The 1H-NMR spectrum showed two characteristic singlet signals at δ = 1.94 and 3.02 ppm, corresponding to two 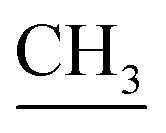 and two
and two 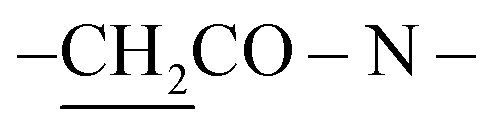 protons, respectively, while mass spectroscopy displayed a molecular ion peak at m/z = 997 (M+, −2Br, 97%) based on the molecular formula C54H48N10S4Br2. Moreover, the UV-vis spectra presented absorption bands at λmax= 574 nm and λem= 586 nm, as presented in the experimental section.
protons, respectively, while mass spectroscopy displayed a molecular ion peak at m/z = 997 (M+, −2Br, 97%) based on the molecular formula C54H48N10S4Br2. Moreover, the UV-vis spectra presented absorption bands at λmax= 574 nm and λem= 586 nm, as presented in the experimental section.
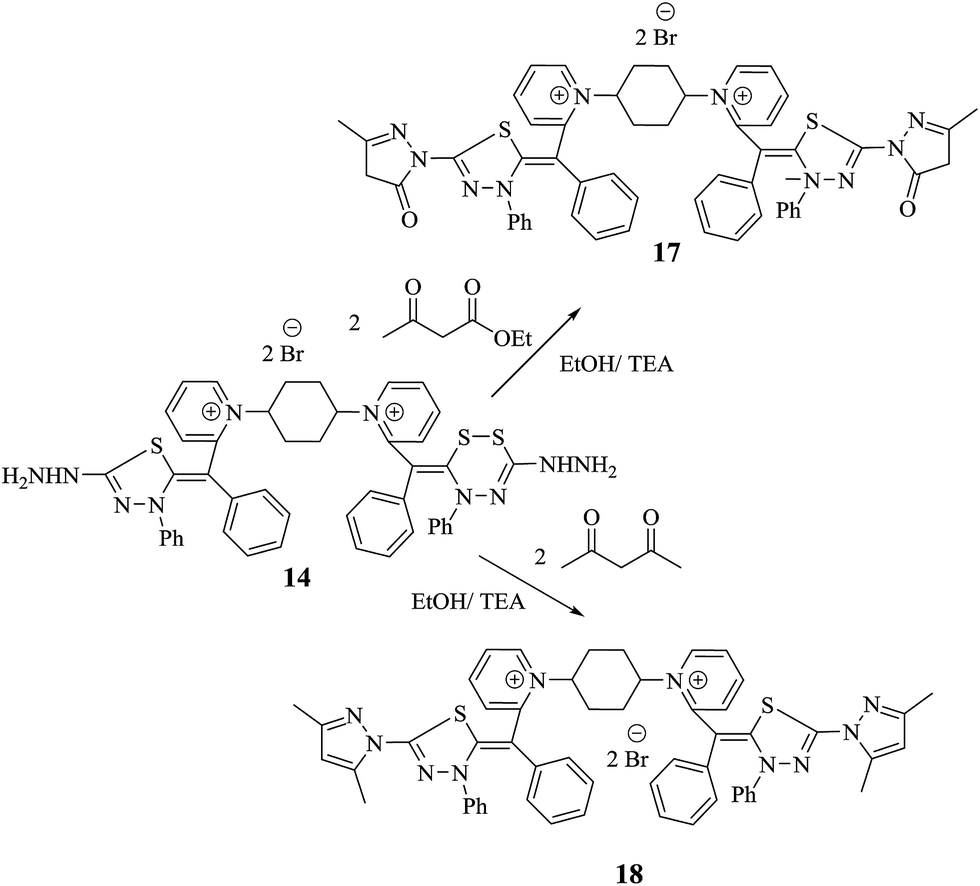 | ||
| Scheme 8 New features of bis-dimethine cyanine dyes incorporated pyrazolone and pyrazole derivatives 17 and 18. | ||
Studies on electronic absorption and emission spectra of synthesized photosensitizers
Chemical modifications produce dramatic shifts in the chromophore absorption spectra, and with a longer methine chain, the fluorescence quantum yield of the dyes is reduced as compared to that with the conjugation and substitution of methine dyes. Synthetic cyanines25–28 are known to absorb between the visible and infrared regions of the electromagnetic spectrum. In addition, cyanines exhibit wide absorption bands and high. Due to these properties, cyanine dyes have been extensively employed in various applications. The electronic absorption spectra of the synthesized carbocyanine dyes exhibited absorption and emission bands at λmax = 468 (490), 502 (522), 447 (462), 478 (508), 558. 430 (583) (439), 562, 436 (58) (440), 820, 438 (845) (442), 596, 428 (615) (438), 622, and 425 (642) (436). These absorption and emission bands are influenced by the nature of quaternary ammonium salt residues of heterocycles and substituent that undergo bathochromic shift and/or hypsochromic shift. Photosensitization increases as π-conjugation increases and vice versa. This nature is attributable to π-delocalization of positive charge of quaternary ammonium salt on nitrogen atoms. As conjugation increases, resonating structure and wavelength increase and excitation energy decreases.The prolonged wavelength absorption of all studied compounds was cited between 461–572 and 525 nm for absorption and between 487 and 597 nm for emission. Also, their molar absorptivity values were between 4.46 and 4.56 L M−1 cm−1 (Table 1). The latter values are linked to the bathochromic and hypsochromic shifts, which affect the longest wavelength absorption band and indicate that the dye is accommodated into the potholes of the double helix.29 Notably, the absorption bands increase according to increasing π-delocalization of positive charge of heteroarenium nitrogen atoms due to increasing resonating structure, leading to more stable and low energy molecules, as shown in Table 1. It is well known that the conjugated system undergoes photosensitization by enough quanta, which leads to excitation of bonding electrons and unpaired electrons, giving possible electronic transitions such as σ–σ*, n–π* and π–π*. This may confirm the later mentioned facts. Compound 8 revealed two characteristic bands at λmax = 422 and 582 nm related to soret bands of two highly extended conjugated porphyrin systems and absorption bands of bis-dimethine cyanine dyes. This means that structure 8 shows a larger bathochromic shift (red shift) than other compounds and high intensity due to high resonating positive charge across the system. It was observed that cyanine dyes linked with heterocyclic rings incorporating more heteroatoms revealed bathochromic shift as follows: by comparing all synthesized compounds: 7 > 6, 8 > 7, 10 > 9, 12a–c > 11a–c, 14 > 15 and 17 > 18. All of these correspond to π-delocalization of the chromophore bis-methine cyanine dyes; as conjugation increases, resonating structure and wavelength increase and excitation energy decreases. In conclusion, we have developed an unprecedented, original two-component, one-pot approach for the synthesis of highly substituted methine cyanine dyes 3, 6, 7, 8, 9, 10, 11, 12, 14, 15, 17 and 18 via a non-isolable intermediate 2 and/or a key thiocarbamoyl intermediate 3 in moderate to high yield. The chemical structures of the obtained potent and selective drugs are analysed by IR, 1H-NMR, 13C-NMR, mass spectroscopy, UV-vis spectroscopy and elemental analyses for C H N element.
| Compound no. | Absorption bands λmax (nm) | Emission bands λem (nm) | Molar absorptivity logε (L mol−1 cm−1) |
Stoke shift λmax–λem | Quantum yield (Q) |
|---|---|---|---|---|---|
| 3 | 461 | 487 | 4.46 | 26 | 1.06 |
| 6 | 485 | 492 | 4.48 | 7 | 1.01 |
| 7 | 524 | 530 | 4.51 | 6 | 1.01 |
| 8 | 422(576) | 429(582) | 4.52(4.73) | 7(6) | 1.02(1.010) |
| 9 | 486 | 495 | 4.48 | 9 | 1.06 |
| 10 | 525 | 533 | 4.52 | 9 | 1.02 |
| 11a | 490 | 501 | 4.48 | 11 | 1.02 |
| 11b | 491 | 506 | 4.49 | 15 | 1.03 |
| 11c | 496 | 508 | 4.49 | 12 | 1.02 |
| 12a | 526 | 534 | 4.51 | 8 | 1.02 |
| 12b | 528 | 536 | 4.52 | 8 | 1.01 |
| 12c | 532 | 536 | 4.52 | 4 | 1.01 |
| 14 | 546 | 557 | 4.53 | 11 | 1.02 |
| 15 | 513 | 532 | 4.50 | 19 | 1.03 |
| 17 | 570 | 597 | 4.55 | 27 | 1.04 |
| 18 | 572 | 589 | 4.56 | 17 | 1.03 |
Experimental
Melting degrees were uncorrected in degree centigrade and tested on a Gallenkamp electric melting point instrument. The infrared (IR) spectra were detected (KBr disk) on a Mattson 5000 FTIR spectrometer, the 1H-NMR and 13C-NMR spectra were obtained on a Bruker WPSY 200 MHz spectrometer with TMS as an interior standard and the chemical shifts were in δ ppm using (DMSO-d6) and/or CDCl3 as solvents and recorder at the Faculty of Science, Mansoura University, Egypt. Mass spectroscopy was performed at 70 eV using Varian MAT 311 and elemental analyses (C, H and N) were conducted at the Microanalytical Center, Faculty of Science, Cairo University. The outcomes were got in a kindly approval (±0.03) with the studied rates.Synthesis
Yield 85%; white needle crystals; m.p. = 163 °C; IR (KBr): υ/cm−1 = 3081 (Ar-CH), 2920 (CH2), 1622 (CN); 1H-NMR (DMSO-d6) δ/ppm = 1.39 (d, J = 5.3 Hz, 8H, 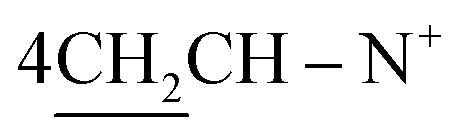 ), 2.26 (m, J = 7.2 Hz, 2H,
), 2.26 (m, J = 7.2 Hz, 2H, 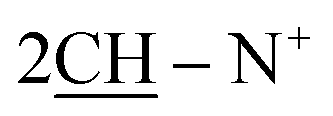 of cyclohexyl ring), 3.81 (s, J = 6.8 Hz, 4H,
of cyclohexyl ring), 3.81 (s, J = 6.8 Hz, 4H, 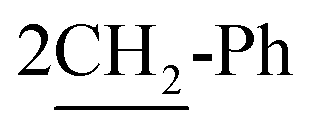 ), 7.20–7.28 (m, 10H, 2Ar-H), 8.04 (d.d, J = 7.0 Hz, 2H, 2CH of pyridinium ring), 8.52 (d, J = 6.2 Hz, 2H, 2CH of pyridinium ring), 8.56 (d.d, J = 5.8 Hz, J = 4.9 Hz, 2H, 2CH of pyridinium ring), 8.91 (d, J = 7.0 Hz, 2H, 2CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 24.4, 39.4, 57.6, 125.4, 125.7, 126.4, 128.6, 129.0, 136.2, 145.6, 151.3; MS: (m/z, %) 422 (M+ +2, −2Br, 100%), 356 (42%), 332 (34%), 255 (20%), 241 (57%), 213 (62%), 185 (72%), 158 (55%), 117 (70%), 76 (23%). Anal. calcd for C30H32N2Br2 (580): C, 85.67; H, 7.67; N, 6.66%. Found: C, 85.70; H, 7.79; N, 6.65%.
), 7.20–7.28 (m, 10H, 2Ar-H), 8.04 (d.d, J = 7.0 Hz, 2H, 2CH of pyridinium ring), 8.52 (d, J = 6.2 Hz, 2H, 2CH of pyridinium ring), 8.56 (d.d, J = 5.8 Hz, J = 4.9 Hz, 2H, 2CH of pyridinium ring), 8.91 (d, J = 7.0 Hz, 2H, 2CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 24.4, 39.4, 57.6, 125.4, 125.7, 126.4, 128.6, 129.0, 136.2, 145.6, 151.3; MS: (m/z, %) 422 (M+ +2, −2Br, 100%), 356 (42%), 332 (34%), 255 (20%), 241 (57%), 213 (62%), 185 (72%), 158 (55%), 117 (70%), 76 (23%). Anal. calcd for C30H32N2Br2 (580): C, 85.67; H, 7.67; N, 6.66%. Found: C, 85.70; H, 7.79; N, 6.65%.
Yield 96%; yellow crystals; m.p. = 196–197 °C; IR (KBr): υ/cm−1 = 3321 (2NH), 3009 (Ar-CH), 2921 (CH2), 2231 (2SH), 1598 (CN); 1H-NMR (DMSO-d6) δ/ppm = 1.40 (d, J = 7.2 Hz, 8H, 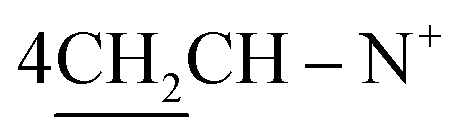 ), 2.27 (m, J = 6.1 Hz, 2H,
), 2.27 (m, J = 6.1 Hz, 2H, 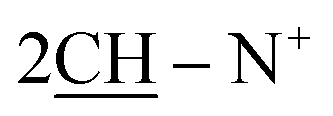 of cyclohexyl ring), 2.17 (s, J = 5.0 Hz, 2H, 2SH), 7.20–7.37 (m, J = 7.5 Hz, 20H, 4Ar-H), 6.73 (d, J = 6.8 Hz, 2H, 2CH of pyridinium ring), 6.69 (d.d, J = 7.5 Hz, 2H, 2CH of pyridinium ring), 7.07 (d.d, J = 8.0 Hz, 2H, 2CH of pyridinium ring), 8.01 (d, J = 5.2 Hz, 2H, 2CH of pyridinium ring), 11.20 (s, 2H, 2NH); 13C-NMR (DMSO-d6) δ/ppm = 24.5, 58.6, 125.4, 114.6, 119.5, 122.1, 125.7, 126.4, 128.6, 129.0, 136.2, 140.9, 145.6, 147.6, 152.3; MS: (m/z, %) 690 (M+, −2Br, 92%), 657 (40%), 624 (14%), 533 (20%), 443 (57%), 366 (62%), 290 (72%), 278 (15%), 266 (100%), 188 (23%), 110 (53%), 82 (74%), 54 (61%). Anal. calcd for C44H42N4S2Br2 (850): C, 76.48; H, 6.13; N, 8.11%. Found: C, 76.45; H, 6.10; N, 8.09%.
of cyclohexyl ring), 2.17 (s, J = 5.0 Hz, 2H, 2SH), 7.20–7.37 (m, J = 7.5 Hz, 20H, 4Ar-H), 6.73 (d, J = 6.8 Hz, 2H, 2CH of pyridinium ring), 6.69 (d.d, J = 7.5 Hz, 2H, 2CH of pyridinium ring), 7.07 (d.d, J = 8.0 Hz, 2H, 2CH of pyridinium ring), 8.01 (d, J = 5.2 Hz, 2H, 2CH of pyridinium ring), 11.20 (s, 2H, 2NH); 13C-NMR (DMSO-d6) δ/ppm = 24.5, 58.6, 125.4, 114.6, 119.5, 122.1, 125.7, 126.4, 128.6, 129.0, 136.2, 140.9, 145.6, 147.6, 152.3; MS: (m/z, %) 690 (M+, −2Br, 92%), 657 (40%), 624 (14%), 533 (20%), 443 (57%), 366 (62%), 290 (72%), 278 (15%), 266 (100%), 188 (23%), 110 (53%), 82 (74%), 54 (61%). Anal. calcd for C44H42N4S2Br2 (850): C, 76.48; H, 6.13; N, 8.11%. Found: C, 76.45; H, 6.10; N, 8.09%.
General procedure for the generation of bis-dimethine cyanine dyes 7, 10 and 12a–c
:2 of absolute ethanol:DMF to give compounds 7, 10 and 12a–c.1,1′-[(Cyclohexan-1,4-diyl)bis(2-((Z)-4-oxo-3-phenylthiazolidin-2-ylidene)(phenyl)methyl)]bis-pyridinium dibromide (7). Yield 52%; red crystals; m.p. = 210–211 °C; IR (KBr): υ/cm−1 = 3009 (Ar-CH), 2922 (CH2), 1690 (2C
O), 1598 (CN); 1H-NMR (DMSO-d6) δ/ppm = 1.42 (d, J = 6.1 Hz, 8H, 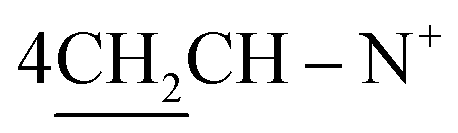 ), 2.28 (m, J = 7.3 Hz, 2H,
), 2.28 (m, J = 7.3 Hz, 2H, 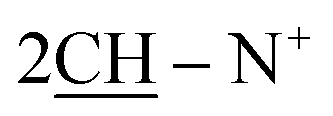 of cyclohexyl ring), 3.123 (s, J = 5.3 Hz, 4H, 2CH2 of thiazolidinone ring), 7.21–7.47 (m, 20H, 4Ar-H), 7.79 (d, J = 5.9 Hz, 2H, 2CH of pyridinium ring), 8.01 (d.d, J = 5.7 Hz, 2H, 2CH of pyridinium ring), 8.14 (d.d, J = 6.9 Hz, 2H, 2CH of pyridinium ring), 8.56 (d, J = 7.2 Hz, 2H, 2CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 22.9, 24.5, 59.0, 109.5, 122.9, 125.4, 116.6, 118.5, 123.2, 125.7, 126.4, 128.6, 129.0, 136.4, 140.9, 145.6, 147.6, 153.3. 178.9; MS: (m/z, %) 771 (M+, −2Br−, 39%), 727 (69%), 683 (51%), 607 (27%), 531 (59%), 397 (62%), 263 (64%), 251 (45%), 239 (100%), 161 (23%), 83 (68%), 55 (64%). Anal. calcd for C48H42N4O2S2Br2 (931): C, 74.78; H, 5.49; N, 7.27%. Found: C, 74.80; H, 5.52; N, 7.29%.
of cyclohexyl ring), 3.123 (s, J = 5.3 Hz, 4H, 2CH2 of thiazolidinone ring), 7.21–7.47 (m, 20H, 4Ar-H), 7.79 (d, J = 5.9 Hz, 2H, 2CH of pyridinium ring), 8.01 (d.d, J = 5.7 Hz, 2H, 2CH of pyridinium ring), 8.14 (d.d, J = 6.9 Hz, 2H, 2CH of pyridinium ring), 8.56 (d, J = 7.2 Hz, 2H, 2CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 22.9, 24.5, 59.0, 109.5, 122.9, 125.4, 116.6, 118.5, 123.2, 125.7, 126.4, 128.6, 129.0, 136.4, 140.9, 145.6, 147.6, 153.3. 178.9; MS: (m/z, %) 771 (M+, −2Br−, 39%), 727 (69%), 683 (51%), 607 (27%), 531 (59%), 397 (62%), 263 (64%), 251 (45%), 239 (100%), 161 (23%), 83 (68%), 55 (64%). Anal. calcd for C48H42N4O2S2Br2 (931): C, 74.78; H, 5.49; N, 7.27%. Found: C, 74.80; H, 5.52; N, 7.29%.
1,1′-[(Cyclohexane-1,4-diyl)bis(2-(Z)-(3,4-di-phenylthiazol-2(3H)-ylidene)(phenyl)methyl)]bis-pyridinium dibromide (10). Yield 62%; red crystals; m.p. = 240 °C; IR (KBr): υ/cm−1 = 3023 (Ar-CH), 2920 (CH2), 1565 (C
N); 1H-NMR (CHCl3) δ/ppm = 1.41 (d, J = 7.0 Hz, 8H, 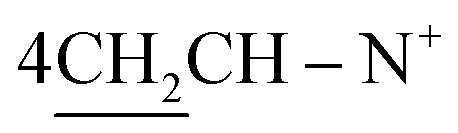 ), 2.27 (m, J = 7.2 Hz, 2H,
), 2.27 (m, J = 7.2 Hz, 2H, 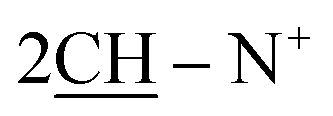 of cyclohexyl ring), 6.73 (s, J = 5.9 Hz, 2H, 2CH of thiazole ring), 7.20–7.45 (m, 20H, 4Ar-H), 7.29 (d, J = 8.0 Hz, 2H, 2CH of pyridinium ring), 7.52–7.71 (m, 10H, 2Ar-H), 8.11 (d.d, J = 7.0 Hz, 2H, 2CH of pyridinium ring), 8.13 (d.d, J = 5.6 Hz, 2H, 2CH of pyridinium ring), 8.93 (d, J = 6.7 Hz, 2H, 2CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 24.0, 29.5, 58.0, 106.5, 110.9, 122.4, 127.9, 128.3, 128.6, 129.5, 132.5, 133.4, 137.6, 140.9, 145.6, 147.6, 147.9; MS: (m/z, %) 892 (M+ +1, −2Br−, 25%), 816 (32%), 740 (11%), 664 (64%), 588 (19%), 505 (62%), 422 (100%), 410 (35%), 346 (10%), 270 (73%), 258 (68%), 246 (64%), 181 (25%), 116 (89%), 76 (68%). Anal. calcd for C60H50N4S2Br2 (1051): C, 80.86; H, 5.66; N, 6.89%. Found: C, 80.87; H, 5.68; N, 6.92%.
of cyclohexyl ring), 6.73 (s, J = 5.9 Hz, 2H, 2CH of thiazole ring), 7.20–7.45 (m, 20H, 4Ar-H), 7.29 (d, J = 8.0 Hz, 2H, 2CH of pyridinium ring), 7.52–7.71 (m, 10H, 2Ar-H), 8.11 (d.d, J = 7.0 Hz, 2H, 2CH of pyridinium ring), 8.13 (d.d, J = 5.6 Hz, 2H, 2CH of pyridinium ring), 8.93 (d, J = 6.7 Hz, 2H, 2CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 24.0, 29.5, 58.0, 106.5, 110.9, 122.4, 127.9, 128.3, 128.6, 129.5, 132.5, 133.4, 137.6, 140.9, 145.6, 147.6, 147.9; MS: (m/z, %) 892 (M+ +1, −2Br−, 25%), 816 (32%), 740 (11%), 664 (64%), 588 (19%), 505 (62%), 422 (100%), 410 (35%), 346 (10%), 270 (73%), 258 (68%), 246 (64%), 181 (25%), 116 (89%), 76 (68%). Anal. calcd for C60H50N4S2Br2 (1051): C, 80.86; H, 5.66; N, 6.89%. Found: C, 80.87; H, 5.68; N, 6.92%.
1,1′-[(Cyclohexane-1,4-diyl)bis(2-(Z)-(5-benzoyl-3-(4-methoxyphenyl)-1,3,4-thidiazol-2(3H)-ylidene)(phenyl)methyl)]bis-pyridinium dibromide (12a). Yield 39%; brown crystals; m.p. = 230–232 °C; IR (KBr): υ/cm−1 = 3002 (Ar-CH), 2922 (CH2), 1737 (C
O), 1568 (CN); 1H-NMR (CHCl3) δ/ppm = 1.42 (d, J = 7.3 Hz, 8H, 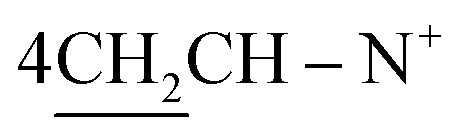 ), 2.27 (m, J = 5.3 Hz, 2H,
), 2.27 (m, J = 5.3 Hz, 2H, 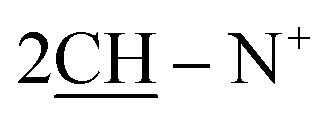 of cyclohexyl ring), 3.81 (s, J = 7.5 Hz, 6H, 2OCH3), 7.13 (d, J = 7.1 Hz, 4H, C–H of C6H4–OCH3-p), 7.29 (d, J = 7.9 Hz, 2H, 2CH of pyridinium ring), 7.34–7.50 (m, J = 5.4 Hz, 10H, 2Ar-H), 7.52 (d, J = 5.6 Hz, 4H, C–H of C6H4–OCH3-p), 7.49–7.81 (m, 10H, 2Ar-H), 8.12 (d.d, J = 7.3 Hz, 2H, 2CH of pyridinium ring), 8.14 (d.d, J = 7.0 Hz, 2H, 2CH of pyridinium ring), 8.90 (d, J = 7.5 Hz, 2H, 2CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 24.0, 31.2, 55.8, 68.3, 115.1, 120.7, 122.7, 127.9, 128.3, 128.5, 137.0, 137.9, 139.0, 140.0, 148.3, 149.6, 155.3, 179.3; MS: (m/z, %) 1009 (M+, −2Br−, 75%), 933 (47%), 857 (18%),
of cyclohexyl ring), 3.81 (s, J = 7.5 Hz, 6H, 2OCH3), 7.13 (d, J = 7.1 Hz, 4H, C–H of C6H4–OCH3-p), 7.29 (d, J = 7.9 Hz, 2H, 2CH of pyridinium ring), 7.34–7.50 (m, J = 5.4 Hz, 10H, 2Ar-H), 7.52 (d, J = 5.6 Hz, 4H, C–H of C6H4–OCH3-p), 7.49–7.81 (m, 10H, 2Ar-H), 8.12 (d.d, J = 7.3 Hz, 2H, 2CH of pyridinium ring), 8.14 (d.d, J = 7.0 Hz, 2H, 2CH of pyridinium ring), 8.90 (d, J = 7.5 Hz, 2H, 2CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 24.0, 31.2, 55.8, 68.3, 115.1, 120.7, 122.7, 127.9, 128.3, 128.5, 137.0, 137.9, 139.0, 140.0, 148.3, 149.6, 155.3, 179.3; MS: (m/z, %) 1009 (M+, −2Br−, 75%), 933 (47%), 857 (18%), 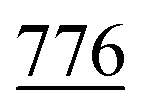 (64%), 669 (59%), 641 (32%), 613 (17%), 529 (65%), 445 (100%), 369 (23%), 293 (46%), 281 (16%), 269 (81%), 258 (68%), 246 (64%), 181 (25%), 116 (89%), 76 (68%). Anal. calcd for C62H52N6O4S2Br2 (1169): C, 73.79; H, 5.19; N, 8.33%. Found: C, 73.79; H, 5.19; N, 8.33%.
(64%), 669 (59%), 641 (32%), 613 (17%), 529 (65%), 445 (100%), 369 (23%), 293 (46%), 281 (16%), 269 (81%), 258 (68%), 246 (64%), 181 (25%), 116 (89%), 76 (68%). Anal. calcd for C62H52N6O4S2Br2 (1169): C, 73.79; H, 5.19; N, 8.33%. Found: C, 73.79; H, 5.19; N, 8.33%.
1,1′-[(Cyclohexane-1,4-diyl)bis(2-(Z)-(5-benzoyl-3-(4-methylphenyl)-1,3,4-thidiazol-2(3H)-ylidene) (phenyl)methyl)]bis-pyridinium dibromide (12b). Yield 39%; brown crystals; m.p. = 231–234 °C; IR (KBr): υ/cm−1 = 3002 (Ar-CH), 2922 (CH2), 1737 (C
O), 1568 (CN); 1H-NMR (CHCl3) δ/ppm = 1.42 (d, J = 6.1 Hz 8H, 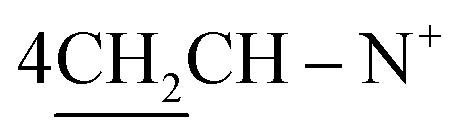 ), 2.27 (m, J = 7.3 Hz, J = 7.0 Hz, 2H,
), 2.27 (m, J = 7.3 Hz, J = 7.0 Hz, 2H, 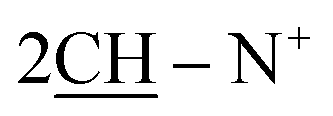 of cyclohexyl ring), 2.31 (s, J = 7.1 Hz, 6H), 2CH36.96 (d, J = 6.4 Hz, 4H, C–H of C6H4–OCH3-p), 7.13 (d, J = 7.3 Hz, 4H, C–H of C6H4–OCH3-p), 7.17–7.35 (m, 10H, 2CO–Ar-H), 7.49–7.81 (m, J = 5.9 Hz, 10H, 2Ar-H), 8.05 (d, J = 5.6 Hz, 2H, 2CH of pyridinium ring), 8.12 (d.d, J = 6.4 Hz, 2H, 2CH of pyridinium ring), 8.15(d.d, J = 7.1 Hz, 2H, 2CH of pyridinium ring), 8.92 (d, J = 7.3 Hz, 2H, 2CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 24.4, 58.0, 99.4, 114.5, 116.2, 124.5, 127.9, 128.5, 128.6, 128.9, 130.2, 131.2, 132.5, 134.5, 143.2, 143.3, 145.8, 147.1, 148.3, 188.7.; MS: (m/z, %) 977 (M+, −2Br−, 93%), 901 (47%), 825 (29%), 760 (34%), 669 (59%), 695 (81%), 667 (57%), 639 (55%), 555 (100%), 471 (23%), 395 (76%), 319 (16%), 269 (81%), 258 (68%), 246 (64%), 181 (25%), 116 (89%), 76 (68%). Anal. calcd for C62H52N6O2S2Br2 (1137): C, 76.20; H, 5.36; N, 8.60%. Found: C, 76.23; H, 5.36; N, 8.60%.
of cyclohexyl ring), 2.31 (s, J = 7.1 Hz, 6H), 2CH36.96 (d, J = 6.4 Hz, 4H, C–H of C6H4–OCH3-p), 7.13 (d, J = 7.3 Hz, 4H, C–H of C6H4–OCH3-p), 7.17–7.35 (m, 10H, 2CO–Ar-H), 7.49–7.81 (m, J = 5.9 Hz, 10H, 2Ar-H), 8.05 (d, J = 5.6 Hz, 2H, 2CH of pyridinium ring), 8.12 (d.d, J = 6.4 Hz, 2H, 2CH of pyridinium ring), 8.15(d.d, J = 7.1 Hz, 2H, 2CH of pyridinium ring), 8.92 (d, J = 7.3 Hz, 2H, 2CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 24.4, 58.0, 99.4, 114.5, 116.2, 124.5, 127.9, 128.5, 128.6, 128.9, 130.2, 131.2, 132.5, 134.5, 143.2, 143.3, 145.8, 147.1, 148.3, 188.7.; MS: (m/z, %) 977 (M+, −2Br−, 93%), 901 (47%), 825 (29%), 760 (34%), 669 (59%), 695 (81%), 667 (57%), 639 (55%), 555 (100%), 471 (23%), 395 (76%), 319 (16%), 269 (81%), 258 (68%), 246 (64%), 181 (25%), 116 (89%), 76 (68%). Anal. calcd for C62H52N6O2S2Br2 (1137): C, 76.20; H, 5.36; N, 8.60%. Found: C, 76.23; H, 5.36; N, 8.60%.
1,1′-[(Cyclohexane-1,4-diyl)bis(2-(Z)-(5-benzoyl-3-(4-nitrophenyl)-1,3,4-thidiazol-2(3H)-ylidene)(phenyl)methyl)]bis-pyridinium dibromide (12c). Yield 87%; reddish brown crystals; m.p. = 253–254 °C; IR (KBr): υ/cm−1 = 3002 (Ar-CH), 2922 (CH2), 1737 (C
O), 1568 (CN); 1H-NMR (CHCl3) δ/ppm = 1.42 (d, J = 7.3 Hz, 8H, 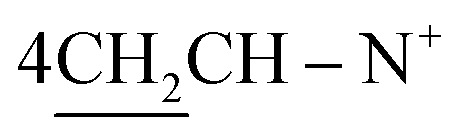 ), 2.26 (m, J = 7.6 Hz, 2H,
), 2.26 (m, J = 7.6 Hz, 2H, 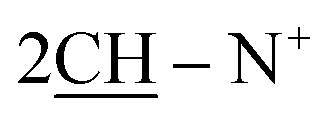 of cyclohexyl ring), 2.31 (s, J = 6.4 Hz, 6H, 2CH3), 7.33–7.91 (m, 20H, 4Ar-H), 7.91 (d, J = 6.9 Hz, 4H, C–H of C6H4–NO2-p), 8.05 (d, J = 6.8 Hz, 2H, 2CH of pyridinium ring), 8.12 (d.d, J = 6.5 Hz, 2H, 2CH of pyridinium ring), 8.15 (d.d, J = 6.8 Hz, 2H, 2CH of pyridinium ring), 8.28 (d, J = 6.5 Hz, 4H, C–H of C6H4–NO2-p), 8.92 (d, J = 7.3 Hz, 2H, 2CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 24.5, 58.2, 99.4, 115.5, 116.4, 124.5, 127.9, 128.7, 128.9, 129.9, 131.2, 132.2, 132.5, 134.5, 143.2, 143.5, 145.8, 147.2, 148.4, 188.8; MS: (m/z, %) 1039 (M+, −2Br−, 43%), 993 (87%), 947 (39%), 871 (57%), 795 (89%), 691 (81%), 587 (57%), 503 (55%), 421 (100%), 333 (53%), 245 (16%), 167 (26%), 89 (89%), 49 (68%). Anal. calcd for C60H46N8O6S2Br2 (1199): C, 69.35; H, 4.46; N, 10.78%. Found: C, 69.37; H, 4.48; N, 10.80%.
of cyclohexyl ring), 2.31 (s, J = 6.4 Hz, 6H, 2CH3), 7.33–7.91 (m, 20H, 4Ar-H), 7.91 (d, J = 6.9 Hz, 4H, C–H of C6H4–NO2-p), 8.05 (d, J = 6.8 Hz, 2H, 2CH of pyridinium ring), 8.12 (d.d, J = 6.5 Hz, 2H, 2CH of pyridinium ring), 8.15 (d.d, J = 6.8 Hz, 2H, 2CH of pyridinium ring), 8.28 (d, J = 6.5 Hz, 4H, C–H of C6H4–NO2-p), 8.92 (d, J = 7.3 Hz, 2H, 2CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 24.5, 58.2, 99.4, 115.5, 116.4, 124.5, 127.9, 128.7, 128.9, 129.9, 131.2, 132.2, 132.5, 134.5, 143.2, 143.5, 145.8, 147.2, 148.4, 188.8; MS: (m/z, %) 1039 (M+, −2Br−, 43%), 993 (87%), 947 (39%), 871 (57%), 795 (89%), 691 (81%), 587 (57%), 503 (55%), 421 (100%), 333 (53%), 245 (16%), 167 (26%), 89 (89%), 49 (68%). Anal. calcd for C60H46N8O6S2Br2 (1199): C, 69.35; H, 4.46; N, 10.78%. Found: C, 69.37; H, 4.48; N, 10.80%.
General procedure for generation of bis-dimethine cyanine dyes 6, 9 and 11a–c
1,1′-[(Cyclohexan-1,4-diyl)bis(2-(Z)-2((2-ethoxy-2-oxoethyl)thio)-1-phenyl-2-(phenylamino)vinyl)]bis-pyridinium dibromide (6). Yield 47%; yellow powder; m.p. = 222–223 °C; IR (KBr): υ/cm−1 = 3323 (NH), 3009 (Ar-CH), 2922 (CH2), 1730 (C
O), 1569 (CN); 1H-NMR (CHCl3) δ/ppm = 1.31 (t, J = 7.0 Hz, 6H, 2CH3), 1.40 (d, J = 5.8 Hz, 8H, 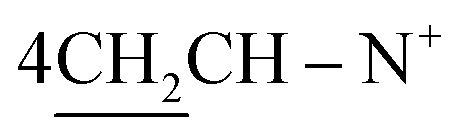 ), 2.26 (m, J = 6.3 Hz, 2H,
), 2.26 (m, J = 6.3 Hz, 2H, 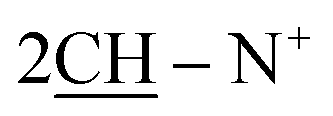 of cyclohexyl ring), 3.23 (s, J = 5.7 Hz, 4H,
of cyclohexyl ring), 3.23 (s, J = 5.7 Hz, 4H, 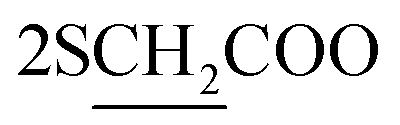 ), 4.30 (q, J = 5.4 Hz, 4H, 2CH2), 7.32–7.81 (m, 20H, 4Ar-H), 8.02 (d, J = 5.7 Hz, 2H, 2CH of pyridinium ring), 8.10 (d.d, J = 7.2 Hz, 2H, 2CH of pyridinium ring), 8.13 (d.d, J = 7.0 Hz, 2H, 2CH of pyridinium ring), 8.82 (d, J = 8.0 Hz, 2H, 2CH of pyridinium ring), 12.90 (s, 2H, 2NH); 13C-NMR (DMSO-d6) δ/ppm = 14.1, 24.5, 26.5, 58.2, 60.6, 115.6, 116.9, 124.5, 127.9, 128.7, 128.9, 129.9, 132.2, 132.8, 132.9, 134.6, 143.5, 143.7, 145.8, 147.2, 148.4, 170.8; MS: (m/z, %) 865 (M+ +2, −2Br−, 76%), 820 (27%), 775 (29%), 747 (87%), 719 (89%), 673 (31%), 627 (57%), 551 (45%), 475 (12%), 449 (53%), 423 (16%), 335 (26%), 247 (100%), 169 (34%), 91 (53%), 51 (63%). Anal. calcd for C60H46N8O6S2Br2 (1023): C, 72.36; H, 3.61; N, 6.49%. Found: C, 72.37; H, 3.64; N, 6.52%.
), 4.30 (q, J = 5.4 Hz, 4H, 2CH2), 7.32–7.81 (m, 20H, 4Ar-H), 8.02 (d, J = 5.7 Hz, 2H, 2CH of pyridinium ring), 8.10 (d.d, J = 7.2 Hz, 2H, 2CH of pyridinium ring), 8.13 (d.d, J = 7.0 Hz, 2H, 2CH of pyridinium ring), 8.82 (d, J = 8.0 Hz, 2H, 2CH of pyridinium ring), 12.90 (s, 2H, 2NH); 13C-NMR (DMSO-d6) δ/ppm = 14.1, 24.5, 26.5, 58.2, 60.6, 115.6, 116.9, 124.5, 127.9, 128.7, 128.9, 129.9, 132.2, 132.8, 132.9, 134.6, 143.5, 143.7, 145.8, 147.2, 148.4, 170.8; MS: (m/z, %) 865 (M+ +2, −2Br−, 76%), 820 (27%), 775 (29%), 747 (87%), 719 (89%), 673 (31%), 627 (57%), 551 (45%), 475 (12%), 449 (53%), 423 (16%), 335 (26%), 247 (100%), 169 (34%), 91 (53%), 51 (63%). Anal. calcd for C60H46N8O6S2Br2 (1023): C, 72.36; H, 3.61; N, 6.49%. Found: C, 72.37; H, 3.64; N, 6.52%.
1,1′-[(Cyclohexane-1,4-diyl)bis(2-(Z)-2((2-oxo-2-phenylethyl)thio)-1-phenyl-2-(phenylamine)vinyl)]bis-pyridinium dibromide (9). Yield 75%; yellow powder; m.p. = 206 °C; IR (KBr): υ/cm−1 = 3322 (NH), 3002 (Ar-CH), 2922 (CH2), 1728 (C
O), 1569 (CN); 1H-NMR (DMSO-d6) δ/ppm = 1.42 (d, J = 7.1 Hz, 8H, 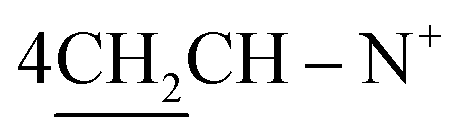 ), 2.26 (m, J = 7.6 Hz, 2H,
), 2.26 (m, J = 7.6 Hz, 2H, 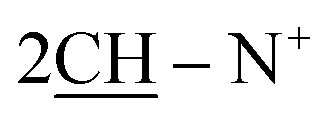 of cyclohexyl ring), 2.51 (s, J = 7.2 Hz, 4H,
of cyclohexyl ring), 2.51 (s, J = 7.2 Hz, 4H, 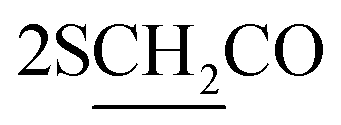 ), 7.22–7.34 (m, 20H, 4Ar-H), 7.43–74 (m, 10H, 2Ar-H), 8.05 (d, J = 7.4 Hz, 2H, 2CH of pyridinium ring), 8.12 (d.d, J = 7.1 Hz, 2H, 2CH of pyridinium ring), 8.14 (d.d, J = 7.3 Hz, 2H, 2CH of pyridinium ring), 8.83 (d, J = 5.8 Hz, 2H, 2CH of pyridinium ring), 11.92 (s, J = 6.4 Hz, 2H, 2NH); 13C-NMR (CHCl3) δ/ppm = 24.2, 26.8, 58.2, 61.6, 116.9, 117.3, 124.5, 128.0, 128.7, 128.9, 129.9, 131.4, 132.8, 132.9, 134.6, 143.7, 143.5, 145.8, 147.3, 148.5, 177.2.; MS: (m/z, %) 927 (M+, −2Br, 39%), 852 (27%), 775 (29%), 747 (87%), 718 (89%), 675 (31%), 627 (57%), 552 (45%), 475 (12%), 449 (53%), 423 (16%), 335 (26%), 247 (100%), 170 (34%), 90 (53%), 50 (63%). Anal. calcd for C60H46N8O6S2Br2 (1087): C, 77.72; H, 5.87; N, 6.04%. Found: C, 77.72; H, 5.87; N, 6.04%.
), 7.22–7.34 (m, 20H, 4Ar-H), 7.43–74 (m, 10H, 2Ar-H), 8.05 (d, J = 7.4 Hz, 2H, 2CH of pyridinium ring), 8.12 (d.d, J = 7.1 Hz, 2H, 2CH of pyridinium ring), 8.14 (d.d, J = 7.3 Hz, 2H, 2CH of pyridinium ring), 8.83 (d, J = 5.8 Hz, 2H, 2CH of pyridinium ring), 11.92 (s, J = 6.4 Hz, 2H, 2NH); 13C-NMR (CHCl3) δ/ppm = 24.2, 26.8, 58.2, 61.6, 116.9, 117.3, 124.5, 128.0, 128.7, 128.9, 129.9, 131.4, 132.8, 132.9, 134.6, 143.7, 143.5, 145.8, 147.3, 148.5, 177.2.; MS: (m/z, %) 927 (M+, −2Br, 39%), 852 (27%), 775 (29%), 747 (87%), 718 (89%), 675 (31%), 627 (57%), 552 (45%), 475 (12%), 449 (53%), 423 (16%), 335 (26%), 247 (100%), 170 (34%), 90 (53%), 50 (63%). Anal. calcd for C60H46N8O6S2Br2 (1087): C, 77.72; H, 5.87; N, 6.04%. Found: C, 77.72; H, 5.87; N, 6.04%.
2-((Z)-2-(E)-1-(2-(4-methoxyphenyl)hydrazono)-2-oxo-2-phenylethylthio)-1-phenyl-2-(phenylamino)vinyl)-1-(4-(2-((Z)-2-((Z)-pyridin-1-ium)cyclohexyl)bis-pyridinium)dibromide (11a). Yield 54%; red powder; m.p. = 235 °C; IR (KBr): υ/cm−1 = 3312–3324 (2NH), 3004 (Ar-CH), 2925 (CH2), 1695 (C
O), 1570 (CN); 1H-NMR (CHCl3) δ/ppm = 1.71 (d, J = 7.3 Hz, 8H, 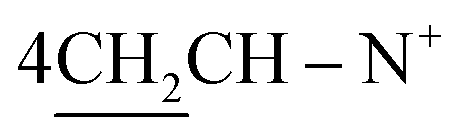 ), 2.26 (m, J = 7.1 Hz, 2H,
), 2.26 (m, J = 7.1 Hz, 2H, 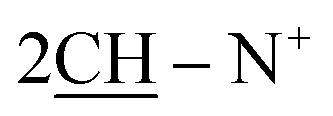 of cyclohexyl ring), 3.80 (s, J = 6.7 Hz, 3H,
of cyclohexyl ring), 3.80 (s, J = 6.7 Hz, 3H, 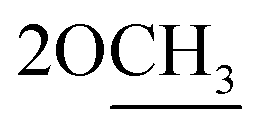 ), 6.73–7.10 (m, 8H,
), 6.73–7.10 (m, 8H, 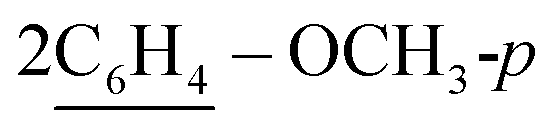 ), 7.17–7.52 (m, 20H, 4Ar-H), 7.53–74 (m, 10H, 2Ar-H), 8.05 (d, J = 5.8 Hz, 2H, 2CH of pyridinium ring), 8.12 (d.d, J = 6.1 Hz, 2H, 2CH of pyridinium ring), 8.14 (d.d, J = 8.0 Hz, 2H, 2CH of pyridinium ring), 8.83 (d, J = 7.8 Hz, 2H, 2CH of pyridinium ring), 10.74 (s, J = 7.0 Hz, 2H, 2NH), 11.61 (s, J = 7.1 Hz, 2H, 2NH); 13C-NMR (CHCl3) δ/ppm = 33.5, 27.0, 54.2, 115.1, 117.3, 122.4, 125.4, 127.1, 127.2, 127.9, 128.9, 128.3, 128.6, 129.2, 129.5, 133.6, 134.5, 136.4, 147.1, 158.3, 188.7; MS: (m/z, %) 1195 (M+, −2Br−, 74%), 1164 (27%), 1133 (29%), 1058 (87%), 983 (89%), 908 (31%), 880 (57%), 852 (45%), 748 (12%), 644 (53%), 632 (16%), 620 (26%), 588 (100%), 556 (34%), 465 (53%), 374 (63%), 362 (21%), 286 (81%), 210 (63%), 132 (34%), 54 (36%). Anal. calcd for C74H66N8O4S2Br2 (1355): C, 74.35; H, 5.56; N, 9.37%. Found: C, 74.38; H, 5.57; N, 9.40%.
), 7.17–7.52 (m, 20H, 4Ar-H), 7.53–74 (m, 10H, 2Ar-H), 8.05 (d, J = 5.8 Hz, 2H, 2CH of pyridinium ring), 8.12 (d.d, J = 6.1 Hz, 2H, 2CH of pyridinium ring), 8.14 (d.d, J = 8.0 Hz, 2H, 2CH of pyridinium ring), 8.83 (d, J = 7.8 Hz, 2H, 2CH of pyridinium ring), 10.74 (s, J = 7.0 Hz, 2H, 2NH), 11.61 (s, J = 7.1 Hz, 2H, 2NH); 13C-NMR (CHCl3) δ/ppm = 33.5, 27.0, 54.2, 115.1, 117.3, 122.4, 125.4, 127.1, 127.2, 127.9, 128.9, 128.3, 128.6, 129.2, 129.5, 133.6, 134.5, 136.4, 147.1, 158.3, 188.7; MS: (m/z, %) 1195 (M+, −2Br−, 74%), 1164 (27%), 1133 (29%), 1058 (87%), 983 (89%), 908 (31%), 880 (57%), 852 (45%), 748 (12%), 644 (53%), 632 (16%), 620 (26%), 588 (100%), 556 (34%), 465 (53%), 374 (63%), 362 (21%), 286 (81%), 210 (63%), 132 (34%), 54 (36%). Anal. calcd for C74H66N8O4S2Br2 (1355): C, 74.35; H, 5.56; N, 9.37%. Found: C, 74.38; H, 5.57; N, 9.40%.
2-((Z)-2-(E)-1-(2-(4-methylphenyl)hydrazono)-2-oxo-2-phenylethylthio)-1-phenyl-2-(phenylamino)vinyl)-1-(4-(2-(Z)-2-((Z)-pyridin-1-ium)cyclohexyl)bis-pyridinium dibromide (11b). Yield 65%; green powder; m.p. = 241–242 °C; IR (KBr): υ/cm−1 = 3312–3324 (2NH), 3006 (Ar-CH), 2925 (CH2), 1730 (C
O), 1570 (CN); 1H-NMR (DMSO-d6) δ/ppm = 1.53 (d, J = 7.3 Hz, 8H, 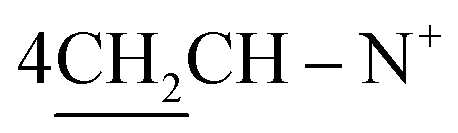 ), 2.26 (m, J = 5.9 Hz, 2H,
), 2.26 (m, J = 5.9 Hz, 2H, 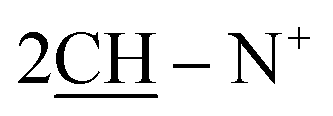 of cyclohexyl ring), 2.30 (s, J = 6.6 Hz, 3H, 2CH3), 7.17–7.32 (m, 20H, 4Ar-H), 7.45 (d, J = 7.5 Hz, 4H,
of cyclohexyl ring), 2.30 (s, J = 6.6 Hz, 3H, 2CH3), 7.17–7.32 (m, 20H, 4Ar-H), 7.45 (d, J = 7.5 Hz, 4H, 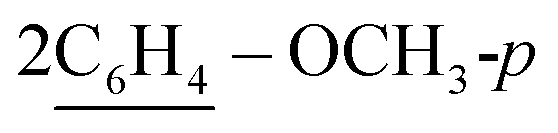 ), 7.49 (d, J = 8.0 Hz, 4H,
), 7.49 (d, J = 8.0 Hz, 4H, 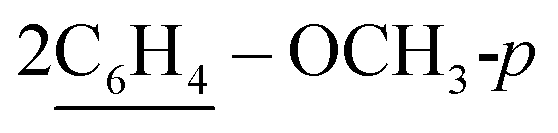 ), 7.53–7.56 (m, 10H, 2Ar-H), 8.02 (d, J = 6.9 Hz, 2H, 2CH of pyridinium ring), 8.11 (d.d, J = 5.9 Hz, 2H, 2CH of pyridinium ring), 8.14 (d.d, J = 5.7 Hz, 2H, 2CH of pyridinium ring), 8.86 (d, J = 5.8 Hz, 2H, 2CH of pyridinium ring), 10.75 (s, J = 5.9 Hz, 2H, 2NH), 11.62 (s, 6.8, 2H, 2NH); 13C-NMR (CHCl3) δ/ppm = 24.5, 58.2, 116.2, 117.2, 122.4, 125.4, 127.9, 128.6, 129.2, 131.2, 133.6, 134.5, 136.6, 140.0, 145.8, 147.1, 158.8, 187.7; MS: (m/z, %) 1163 (M+, −2Br−, 94%), 1148 (37%), 1133 (89%), 1057 (26%), 980 (38%), 910 (69%), 880 (59%), 852 (75%), 748 (42%), 644 (55%), 632 (66%), 620 (90%), 588 (100%), 556 (35%), 465 (24%), 374 (13%), 362 (22%), 286 (86%), 210 (64%), 132 (33%), 54 (29%). Anal. calcd for C74H66N8O2S2Br2 (1323): C, 76.39; H, 5.75; N, 9.63%. Found: C, 76.39; H, 5.75; N, 9.63%.
), 7.53–7.56 (m, 10H, 2Ar-H), 8.02 (d, J = 6.9 Hz, 2H, 2CH of pyridinium ring), 8.11 (d.d, J = 5.9 Hz, 2H, 2CH of pyridinium ring), 8.14 (d.d, J = 5.7 Hz, 2H, 2CH of pyridinium ring), 8.86 (d, J = 5.8 Hz, 2H, 2CH of pyridinium ring), 10.75 (s, J = 5.9 Hz, 2H, 2NH), 11.62 (s, 6.8, 2H, 2NH); 13C-NMR (CHCl3) δ/ppm = 24.5, 58.2, 116.2, 117.2, 122.4, 125.4, 127.9, 128.6, 129.2, 131.2, 133.6, 134.5, 136.6, 140.0, 145.8, 147.1, 158.8, 187.7; MS: (m/z, %) 1163 (M+, −2Br−, 94%), 1148 (37%), 1133 (89%), 1057 (26%), 980 (38%), 910 (69%), 880 (59%), 852 (75%), 748 (42%), 644 (55%), 632 (66%), 620 (90%), 588 (100%), 556 (35%), 465 (24%), 374 (13%), 362 (22%), 286 (86%), 210 (64%), 132 (33%), 54 (29%). Anal. calcd for C74H66N8O2S2Br2 (1323): C, 76.39; H, 5.75; N, 9.63%. Found: C, 76.39; H, 5.75; N, 9.63%.
2-((Z)-2-((E)-1-(2-(4-nitrophenyl)hydrazono)-2-oxo-2-phenylethylthio)-1-phenyl-2-(phenylamino)vinyl)-1-(4-(2-(Z)-2-((Z)-pyridin-1-ium)cyclohexyl)bis-pyridinium)dibromide (11c). Yield 65%; green powder; m.p. = 195 °C; IR (KBr): υ/cm−1 = 3332–3350 (2NH), 3006 (Ar-CH), 2925 (CH2), 1732 (C
O), 1570 (CN); 1H-NMR (DMSO-d6) δ/ppm = 1.72 (d, J = 6.7 Hz, 8H, 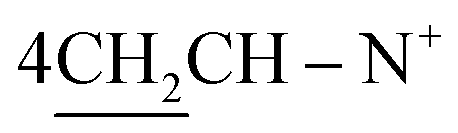 ), 2.28 (m, J = 6.4 Hz, 2H,
), 2.28 (m, J = 6.4 Hz, 2H, 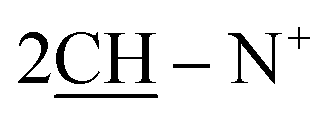 of cyclohexyl ring), 7.17–7.32 (m, 20H, 4Ar-H), 7.47 (d, J = 7.1 Hz, 4H,
of cyclohexyl ring), 7.17–7.32 (m, 20H, 4Ar-H), 7.47 (d, J = 7.1 Hz, 4H, 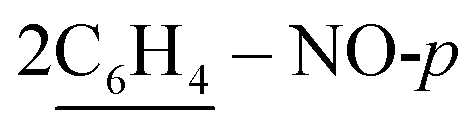 ), 7.52 (d, J = 7.9 Hz, 4H,
), 7.52 (d, J = 7.9 Hz, 4H, 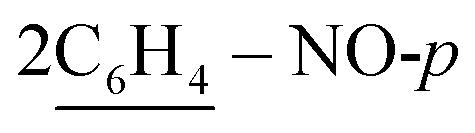 ), 7.55–7.57 (m, 10H, 2Ar-H), 8.03 (d, J = 7.0 Hz, 2H, 2CH of pyridinium ring), 8.12 (d.d, J = 6.8 Hz, 2H, 2CH of pyridinium ring), 8.15 (d.d, J = 6.3 Hz, 2H, 2CH of pyridinium ring), 8.84 (d, J = 5.6 Hz, 2H, 2CH of pyridinium ring), 10.74 (s, J = 6.0 Hz, 2H, 2NH), 11.60 (s, J = 6.3 Hz, 2H, 2NH); 13C-NMR (CHCl3) δ/ppm = 24.4, 58.3, 116.3, 116.3, 117.2, 123.5, 126.4, 127.9, 128.5, 129.3, 132.0, 133.6, 134.3, 136.6, 140.1, 145.7, 147.2, 158.5, 187.3; MS: (m/z, %) 1225 (M+, −2Br−, 41%), 1179 (17%), 1133 (59%), 1057 (66%), 980 (72%), 911 (54%), 881 (60.3%), 851 (74%), 747 (32%), 644 (19%), 632 (36%), 621 (32%), 588 (100%), 556 (95%), 465 (48%), 374 (75%), 362 (12%), 286 (14%), 210 (24%), 132 (36%), 54 (47%). Anal. calcd for C72H60N10O6S2Br2 (1385): C, 70.57; H, 4.94; N, 11.43%. Found: C, 70.58; H, 4.97; N, 11.46%.
), 7.55–7.57 (m, 10H, 2Ar-H), 8.03 (d, J = 7.0 Hz, 2H, 2CH of pyridinium ring), 8.12 (d.d, J = 6.8 Hz, 2H, 2CH of pyridinium ring), 8.15 (d.d, J = 6.3 Hz, 2H, 2CH of pyridinium ring), 8.84 (d, J = 5.6 Hz, 2H, 2CH of pyridinium ring), 10.74 (s, J = 6.0 Hz, 2H, 2NH), 11.60 (s, J = 6.3 Hz, 2H, 2NH); 13C-NMR (CHCl3) δ/ppm = 24.4, 58.3, 116.3, 116.3, 117.2, 123.5, 126.4, 127.9, 128.5, 129.3, 132.0, 133.6, 134.3, 136.6, 140.1, 145.7, 147.2, 158.5, 187.3; MS: (m/z, %) 1225 (M+, −2Br−, 41%), 1179 (17%), 1133 (59%), 1057 (66%), 980 (72%), 911 (54%), 881 (60.3%), 851 (74%), 747 (32%), 644 (19%), 632 (36%), 621 (32%), 588 (100%), 556 (95%), 465 (48%), 374 (75%), 362 (12%), 286 (14%), 210 (24%), 132 (36%), 54 (47%). Anal. calcd for C72H60N10O6S2Br2 (1385): C, 70.57; H, 4.94; N, 11.43%. Found: C, 70.58; H, 4.97; N, 11.46%.
2-((Z)-(E)-4-oxo-3-phenyl-5-(porphyrin-2-ylmethylene)thiazolidin-2-ylidene)(phenyl)(methyl)-1-(4-(2-((Z)-(E)-4-oxo-3-phenyl-5-(porphyrin-2-ylmethylene)thiazolidin-2-ylidene(phenyl)(methyl)pyridin-1-ium-1-yl)cyclohexyl)bis-pyridinium)dibromide (8). In a round bottom flask fitted with a reflux condenser, compound 7 (1 mmol, 0.93 gm) and porphyrin-2-carbaldehyde (0.002 mmol, 0.676 gm) were dissolved in N,N-dimethylformamide by heating for 24 h with magnetic stirring in presence of piperidine as a catalyst under nitrogen gas. The solution contents were heated gradually to 100 °C. The reaction mixture was maintained at 100 °C for about 12 h and then raise the temperature was increased to 150 °C followed by continuous heating at this temperature for 12 h. The reaction contents were observed using TLC. The contents were cooled and poured it onto ice-cold water. The solid was filtered and washed with cold water. The isolated solid 8 was purified by HPLC (CHCl3
:hexane:NH2OH, 10:5:0.5) as eluent.Yield 43%; violet crystals; m.p. > 300 °C; IR (KBr): υ/cm−1 = 3312–3325 (4NH), 3123–3145 (2Ar-CH), 2930 (CH2), 1692 (CO), 1632–1634 (CN+); 1H-NMR (DMSO-d6) δ/ppm = 1.41 (d, J = 7.3 Hz, 8H, 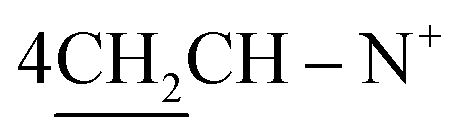 ), 2.28 (m, J = 7.1 Hz, 1H,
), 2.28 (m, J = 7.1 Hz, 1H, 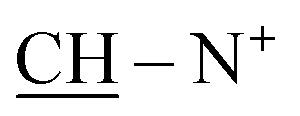 of cyclohexyl ring), 4.30 (s, J = 7.6 Hz, 4H, 4CH methine bridge of porphyrin ring), 5.62 (s, J = 5.5 Hz, 2H, 2NH), 6.24 (d, J = 7.0 Hz, 4H, 4CH methine of pyrrole ring of porphyrin ring), 6.46 (s, J = 7.3 Hz, 2H, 2CH methine of pyrrole ring of porphyrin ring), 6.66 (d, J = 7.1 Hz, 2H,
of cyclohexyl ring), 4.30 (s, J = 7.6 Hz, 4H, 4CH methine bridge of porphyrin ring), 5.62 (s, J = 5.5 Hz, 2H, 2NH), 6.24 (d, J = 7.0 Hz, 4H, 4CH methine of pyrrole ring of porphyrin ring), 6.46 (s, J = 7.3 Hz, 2H, 2CH methine of pyrrole ring of porphyrin ring), 6.66 (d, J = 7.1 Hz, 2H, 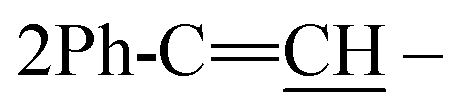 porphyrin), 6.98 (s, J = 5.9 Hz, 4H, 4CH methine bridge of porphyrin ring), 7.84 (d, J = 5.8 Hz, 2H, 2CH methine of pyrrole ring of porphyrin ring), 7.10–7.19 (m, 10H, 2Ar-H), 7.29 (d, J = 6.7 Hz, 1H, CH of pyridinium ring), 7.30–7.43 (m, 4H, Ar-H), 7.84 (d, J = 5.3 Hz, 2H, 2CH methine of pyrrole ring of porphyrin ring), 8.05 (d, J = 6.1 Hz, 1H, CH of pyridinium ring), 8.11 (d.d, J = 7.6 Hz, 1H, CH of pyridinium ring), 8.14 (d.d, J = 7.0 Hz, 1H, CH of pyridinium ring), 8.92 (d, J = 5.7 Hz, 1H, CH of pyridinium ring), 12.89 (s, J = 5.9 Hz, 2H, 2NH); 13C-NMR (DMSO-d6) δ/ppm = 33.5, 58.0, 109.0, 109.8, 112.1, 114.8, 122.9, 127.2, 127.9, 128.0, 128.3, 128.6, 129.8, 130.9, 132.7, 133.5, 135.5, 140.0, 145.8, 146.9, 147.1, 148.4, 180.6; MS: (m/z, %) 1413 (M+ +2, −2Br, 67%), 1102 (73%), 793 (39%), 780 (47%), 767 (81%), 755 (95%), 743 (100%), 715 (17%), 687 (28%), 597 (72%), 507 (62%), 463 (39%), 419 (54%), 343 (71%), 253 (38%), 175 (84%), 97 (17%), 57 (64%). Anal. calcd for C51H44N6Br2 (1571): C, 76.57; H, 4.71; N, 11.91%. Found: C, 76.59; H, 4.73; N, 11.94%.
porphyrin), 6.98 (s, J = 5.9 Hz, 4H, 4CH methine bridge of porphyrin ring), 7.84 (d, J = 5.8 Hz, 2H, 2CH methine of pyrrole ring of porphyrin ring), 7.10–7.19 (m, 10H, 2Ar-H), 7.29 (d, J = 6.7 Hz, 1H, CH of pyridinium ring), 7.30–7.43 (m, 4H, Ar-H), 7.84 (d, J = 5.3 Hz, 2H, 2CH methine of pyrrole ring of porphyrin ring), 8.05 (d, J = 6.1 Hz, 1H, CH of pyridinium ring), 8.11 (d.d, J = 7.6 Hz, 1H, CH of pyridinium ring), 8.14 (d.d, J = 7.0 Hz, 1H, CH of pyridinium ring), 8.92 (d, J = 5.7 Hz, 1H, CH of pyridinium ring), 12.89 (s, J = 5.9 Hz, 2H, 2NH); 13C-NMR (DMSO-d6) δ/ppm = 33.5, 58.0, 109.0, 109.8, 112.1, 114.8, 122.9, 127.2, 127.9, 128.0, 128.3, 128.6, 129.8, 130.9, 132.7, 133.5, 135.5, 140.0, 145.8, 146.9, 147.1, 148.4, 180.6; MS: (m/z, %) 1413 (M+ +2, −2Br, 67%), 1102 (73%), 793 (39%), 780 (47%), 767 (81%), 755 (95%), 743 (100%), 715 (17%), 687 (28%), 597 (72%), 507 (62%), 463 (39%), 419 (54%), 343 (71%), 253 (38%), 175 (84%), 97 (17%), 57 (64%). Anal. calcd for C51H44N6Br2 (1571): C, 76.57; H, 4.71; N, 11.91%. Found: C, 76.59; H, 4.73; N, 11.94%.
1,1′-[(cyclohexane-1,4-diyl)bis(2-(Z)-(5-hydrazinyl-3-pheny-1,2,3-thiadiazol-2(3H)-ylidene) (phenyl)methyl)]bis-pyridinium dibromide (14).
Yield 64%; brown powder; m.p. = 215–216 °C; IR (KBr): υ/cm−1 = 3174–3325 (2NHNH2), 3114–3025 (2Ar-CH), 2931 (CH2), 1574–1578 (CN+); 1H-NMR (DMSO-d6) δ/ppm = 1.42 (d, J = 6.7 Hz, 8H, 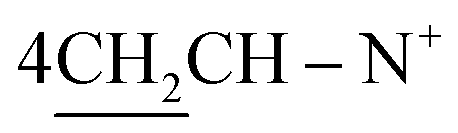 ), 2.27 (m, J = 7.0 Hz, 1H,
), 2.27 (m, J = 7.0 Hz, 1H, 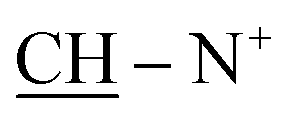 of cyclohexyl ring), 3.81 (s, J = 6.8 Hz, 4H, 2NH2), 6.87–6.94 (m, 10H, 2Ar-H), 7.21–7.39 (m, 10H, 2Ar-H), 78.05 (d, J = 4.8 Hz, 1H, CH of pyridinium ring), 8.00 (d.d, J = 6.2 Hz, 1H, CH of pyridinium ring), 8.13 (d.d, J = 7.3 Hz, 1H, CH of pyridinium ring), 8.78 (d, J = 6.8 Hz, 1H, CH of pyridinium ring), 8.80 (s, J = 6.7 Hz, 2H, 2NH); 13C-NMR (DMSO-d6) δ/ppm = 25.1, 35.5, 58.2, 107.2, 109.7, 112.8, 115.2, 122.1, 127.5, 127.8, 128.1, 128.6, 129.1, 129.8, 131.9, 132.7, 134.5, 135.5, 140.4, 145.4, 146.9, 147.3, 148.5; MS: (m/z, %) 865 (M+, −2Br, 24%), 834 (29%), 803 (62%), 761 (62%), 687 (32%), 602 (15%), 516 (100%), 440 (74%), 365 (92%), 353 (37%), 342 (56%), 263 (81%), 185 (52%), 175 (45%), 97 (37%), 57 (32%). Anal. calcd for C46H44N10S4Br2 (1025): C, 61.44; H, 5.60; N, 18.69%. Found: C, 61.44; H, 5.60; N, 18.69%
of cyclohexyl ring), 3.81 (s, J = 6.8 Hz, 4H, 2NH2), 6.87–6.94 (m, 10H, 2Ar-H), 7.21–7.39 (m, 10H, 2Ar-H), 78.05 (d, J = 4.8 Hz, 1H, CH of pyridinium ring), 8.00 (d.d, J = 6.2 Hz, 1H, CH of pyridinium ring), 8.13 (d.d, J = 7.3 Hz, 1H, CH of pyridinium ring), 8.78 (d, J = 6.8 Hz, 1H, CH of pyridinium ring), 8.80 (s, J = 6.7 Hz, 2H, 2NH); 13C-NMR (DMSO-d6) δ/ppm = 25.1, 35.5, 58.2, 107.2, 109.7, 112.8, 115.2, 122.1, 127.5, 127.8, 128.1, 128.6, 129.1, 129.8, 131.9, 132.7, 134.5, 135.5, 140.4, 145.4, 146.9, 147.3, 148.5; MS: (m/z, %) 865 (M+, −2Br, 24%), 834 (29%), 803 (62%), 761 (62%), 687 (32%), 602 (15%), 516 (100%), 440 (74%), 365 (92%), 353 (37%), 342 (56%), 263 (81%), 185 (52%), 175 (45%), 97 (37%), 57 (32%). Anal. calcd for C46H44N10S4Br2 (1025): C, 61.44; H, 5.60; N, 18.69%. Found: C, 61.44; H, 5.60; N, 18.69%
2-((Z)-2-((E)-hydrazinyl(hydrazono)methyl)thio)-1-phenyl-2-(phenylamino)vinyl)-1-(4-(2-((Z)-2-(((Z)-hydrazinyl(hydrazono)methyl)thio)-1-phenyl-2-(phenylamino)vinyl)-bis-pyridinium)dibromide (15). In a round bottom flask fitted with a reflux water condenser, compound 3 (1 mmol, 0.85 gm) and thiocarbohydrazide (2 mmol, 0.212 gm) were refluxed in 20 ml absolute ethanol in the presence of freshly fused potassium carbonate. The reaction mixture was stirred for 30 min until all materials were dissolved. Then, the reflux was continued for about 3.5 h. The solution was cooled in ice-salt bath until it reached 0 °C. The product was then filtered using a pump, washed with some water, drained well, and then wash with 25 ml ethanol and dried in air. The crude product was recrystallized using methanol. The yield of the formed compound 15 was 75%.
Yield 75%; red powder; m.p. = 256 °C; IR (KBr): υ/cm−1 = 3174–3325 (4NH, 4NH2), 3115–3029 (4Ar-CH), 2932 (CH2), 1575–1579 (CN+); 1H-NMR (DMSO-d6) δ/ppm = 1.43 (d, J = 7.4 Hz, 8H, 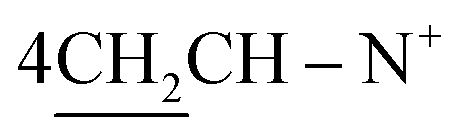 ), 2.25 (m, J = 7.3 Hz, 1H,
), 2.25 (m, J = 7.3 Hz, 1H, 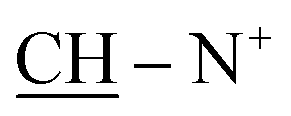 of cyclohexyl ring), 3.81 (s, J = 7.8 Hz, 4H,
of cyclohexyl ring), 3.81 (s, J = 7.8 Hz, 4H,  ), 4.2 (s, J = 7.1 Hz, 4H,
), 4.2 (s, J = 7.1 Hz, 4H, 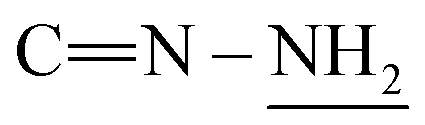 ), 6.87–6.94 (m, 10H, 2Ar-H), 7.21–7.39 (m, 10H, 2Ar-H), 7.85 (d, J = 7.3 Hz, 1H, CH of pyridinium ring), 8.02 (d.d, J = 7.0 Hz, 1H, CH of pyridinium ring), 8.12 (d.d, J = 7.5 Hz, 1H, CH of pyridinium ring), 8.76 (d, J = 7.0 Hz, 1H, CH of pyridinium ring), 8.80 (s, J = 6.8 Hz, 2H,
), 6.87–6.94 (m, 10H, 2Ar-H), 7.21–7.39 (m, 10H, 2Ar-H), 7.85 (d, J = 7.3 Hz, 1H, CH of pyridinium ring), 8.02 (d.d, J = 7.0 Hz, 1H, CH of pyridinium ring), 8.12 (d.d, J = 7.5 Hz, 1H, CH of pyridinium ring), 8.76 (d, J = 7.0 Hz, 1H, CH of pyridinium ring), 8.80 (s, J = 6.8 Hz, 2H,  ), 10.62 (s, J = 6.4 Hz, 2H,
), 10.62 (s, J = 6.4 Hz, 2H, 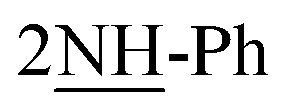 ); 13C-NMR (DMSO-d6) δ/ppm = 25.2, 59.3, 106.2, 109.1, 112.7, 115.3, 122.2, 127.7, 127.5, 128.2, 128.4, 129.2, 129.7, 131.4, 132.7, 134.5, 135.4, 141.8, 145.4, 146.9, 147.3, 151.0; MS: (m/z, %) 899 (M+, −2Br, 56%), 868 (46%), 837 (32%), 761 (62%), 685 (32%), 601 (90%), 517 (100%), 441 (25%), 365 (78%), 353 (47%), 341 (19%), 263 (80%), 185 (51%), 175 (44%), 97 (37%), 57 (64%). Anal. calcd for C46H50N12S4Br2 (1059): C, 55.53; H, 5.07; N, 16.89%. Found: C, 55.50; H, 5.04; N, 16.87%.
); 13C-NMR (DMSO-d6) δ/ppm = 25.2, 59.3, 106.2, 109.1, 112.7, 115.3, 122.2, 127.7, 127.5, 128.2, 128.4, 129.2, 129.7, 131.4, 132.7, 134.5, 135.4, 141.8, 145.4, 146.9, 147.3, 151.0; MS: (m/z, %) 899 (M+, −2Br, 56%), 868 (46%), 837 (32%), 761 (62%), 685 (32%), 601 (90%), 517 (100%), 441 (25%), 365 (78%), 353 (47%), 341 (19%), 263 (80%), 185 (51%), 175 (44%), 97 (37%), 57 (64%). Anal. calcd for C46H50N12S4Br2 (1059): C, 55.53; H, 5.07; N, 16.89%. Found: C, 55.50; H, 5.04; N, 16.87%.
2-((Z)-(3-methyl-5-(3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)-3-phenyl-1,3λ4,4-thiadiazol-2(3H)-ylidene)(phenyl)methyl)-1-(4-2-((Z)-(5-(3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)-3-phenyl-1,3,4-thiadiazol-2(3H)-ylidene(phenyl)methyl)pyridin-1-ium-1-yl)cyclohexyl)-bis-pyridinium dibromide (17). Yield 35%; red powder; m.p. = 205–207 °C; IR (KBr): υ/cm−1 = 3115–3029 (2Ar-CH), 2932 (CH2), 1703 (2C = O), 1575–1579 (C
N+); 1H-NMR (DMSO-d6) δ/ppm = 1.45 (t, J = 7.0 Hz, 8H, 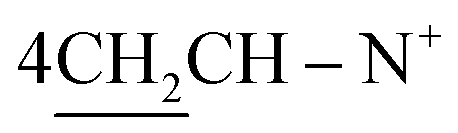 ), 1.93 (s, J = 6.2 Hz, 6H, 2CH3), 2.26 (m, J = 6.7 Hz, 1H,
), 1.93 (s, J = 6.2 Hz, 6H, 2CH3), 2.26 (m, J = 6.7 Hz, 1H, 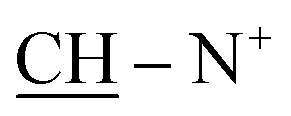 of cyclohexyl ring),3.02 (s, J = 5.6 Hz, 4H, 2CH2), 7.32–7.90 (m, 20H, 2Ar-H), 7.87 (d, J = 5.4 Hz, 1H, CH of pyridinium ring), 8.04 (d.d, J = 5.7 Hz, 1H, CH of pyridinium ring), 8.13 (d.d, J = 5.9 Hz, 1H, CH of pyridinium ring), 8.75 (d, J = 6.4 Hz, 1H, CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 16.4, 26.6, 30.7, 42.3, 68.3, 100.9, 117.9, 119.3, 120.9, 121.4, 122.4, 122.7, 129.5, 129.7, 137.6, 146.3, 147.4, 149.6, 150.3, 150.3, 155.7, 165.0; MS: (m/z, %) 997 (M+, −2Br, 100%), 871 (87%), 795 (42%), 745 (28%), 695 (39%), 645 (84%), 595 (18%), 545 (45%), 495 (61%), 445 (47%), 395 (59%), 345 (80%), 295 (12%), 245 (51%), 195 (74%), 145 (81%), 97 (17%), 57 (60%). Anal. calcd for C54H48N10O2S4Br2 (1157): C, 65.04; H, 4.85; N, 14.05%. Found: C, 65.01; H, 4.81; N, 14.02%.
of cyclohexyl ring),3.02 (s, J = 5.6 Hz, 4H, 2CH2), 7.32–7.90 (m, 20H, 2Ar-H), 7.87 (d, J = 5.4 Hz, 1H, CH of pyridinium ring), 8.04 (d.d, J = 5.7 Hz, 1H, CH of pyridinium ring), 8.13 (d.d, J = 5.9 Hz, 1H, CH of pyridinium ring), 8.75 (d, J = 6.4 Hz, 1H, CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 16.4, 26.6, 30.7, 42.3, 68.3, 100.9, 117.9, 119.3, 120.9, 121.4, 122.4, 122.7, 129.5, 129.7, 137.6, 146.3, 147.4, 149.6, 150.3, 150.3, 155.7, 165.0; MS: (m/z, %) 997 (M+, −2Br, 100%), 871 (87%), 795 (42%), 745 (28%), 695 (39%), 645 (84%), 595 (18%), 545 (45%), 495 (61%), 445 (47%), 395 (59%), 345 (80%), 295 (12%), 245 (51%), 195 (74%), 145 (81%), 97 (17%), 57 (60%). Anal. calcd for C54H48N10O2S4Br2 (1157): C, 65.04; H, 4.85; N, 14.05%. Found: C, 65.01; H, 4.81; N, 14.02%.
1,1′-[(Cyclohexane-1,4-diyl)bis(2-(Z)-(5-(3,5-dimethyl-1H-pyrazol-1-yl)-3-phenyl-1,3,4-thiadiazol-2(3H)ylidene)(phenyl)methyl)]bis-pyridinium dibromide (18). Yield 96%; red crystal; m.p > 300 °C; IR (KBr): υ/cm−1 = 3117–3021 (2Ar-CH), 2931 (CH2), 1575–1579 (C
N+); 1H-NMR (DMSO-d6) δ/ppm =1.40 (d, J = 7.1 Hz, 8H, 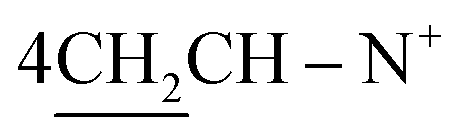 ), 2.20 (s, J = 6.4 Hz, 6H, 2CH3), 2.26 (m, J = 7.3 Hz, 1H,
), 2.20 (s, J = 6.4 Hz, 6H, 2CH3), 2.26 (m, J = 7.3 Hz, 1H, 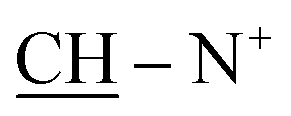 of cyclohexyl ring), 2. 48 (s, J = 7.0 Hz, 6H, 2CH3), 6.22 (s, J = 6.5 Hz, 2H,
of cyclohexyl ring), 2. 48 (s, J = 7.0 Hz, 6H, 2CH3), 6.22 (s, J = 6.5 Hz, 2H, 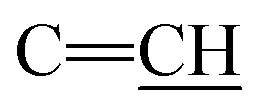 ), 7.21–7.84 (m, 20H, 2Ar-H), 7.92 (d, J = 7.2 Hz, 1H, CH of pyridinium ring), 8.06 (d.d, J = 7.3 Hz, 1H, CH of pyridinium ring), 8.15 (d.d, J = 7.6 Hz, 1H, CH of pyridinium ring), 8.74 (d, J = 5.1 Hz, 1H, CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 13.2, 13.5, 85.2, 16.4, 100.9, 110.0, 112.3, 113.3, 117.2, 120.0, 121.4, 122.7, 124.0, 127.9, 128.6, 128.9. 129.7, 132.5, 136.3, 137.0, 147.3, 155.7, 165.0; MS: (m/z, %) 929 (M+, −2Br, 97%), 853 (67%), 778 (92%), 762 (48%), 747 (84%), 732 (84%), 717 (58%), 652 (89%), 586 (61%), 502 (47%), 417 (59%), 329 (80%), 241 (12%), 163 (51%), 84 (72%), 45 (61%). Anal. calcd for C56H52N10S2Br2 (1157): C, 72.38; H, 5.65; N, 15.07%. Found: C, 72.41; H, 5.68; N, 15.09%.
), 7.21–7.84 (m, 20H, 2Ar-H), 7.92 (d, J = 7.2 Hz, 1H, CH of pyridinium ring), 8.06 (d.d, J = 7.3 Hz, 1H, CH of pyridinium ring), 8.15 (d.d, J = 7.6 Hz, 1H, CH of pyridinium ring), 8.74 (d, J = 5.1 Hz, 1H, CH of pyridinium ring); 13C-NMR (DMSO-d6) δ/ppm = 13.2, 13.5, 85.2, 16.4, 100.9, 110.0, 112.3, 113.3, 117.2, 120.0, 121.4, 122.7, 124.0, 127.9, 128.6, 128.9. 129.7, 132.5, 136.3, 137.0, 147.3, 155.7, 165.0; MS: (m/z, %) 929 (M+, −2Br, 97%), 853 (67%), 778 (92%), 762 (48%), 747 (84%), 732 (84%), 717 (58%), 652 (89%), 586 (61%), 502 (47%), 417 (59%), 329 (80%), 241 (12%), 163 (51%), 84 (72%), 45 (61%). Anal. calcd for C56H52N10S2Br2 (1157): C, 72.38; H, 5.65; N, 15.07%. Found: C, 72.41; H, 5.68; N, 15.09%.
Conclusion
In this letter, we have developed an unprecedented, original two-component, one-pot approach for the synthesis of highly substituted methine cyanine dyes 3, 6, 7, 8, 9, 10, 11, 12, 14, 15, 17 and 18 via a non-isolable intermediate 2 and/or a key thiocarbamoyl intermediate 3 in moderate to high yield. The chemical structures of obtained potent and selective drugs were investigated by IR, 1H-NMR, 13C-NMR, mass spectroscopy, UV-vis and elemental analyses for C H N elements.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors would like to thank their institutions.Notes and references
- N. Matsumoto and M. Takahashi, Tetrahedron, 2002, 58, 10073 CrossRef CAS.
- A. A. Fadda, R. E. El-Mekawy and M. Taha Abdel Aa, Phosphorus, Sulfur, and Silicon and the Related Elements, 2015, 191(8), 1148 CrossRef.
- A. A. Fadda, S. I. El-Desoky, H. A. Etman, S. B. Bondock and M. A. Metwally, Sulfur Lett., 2002, 25, 199 CrossRef.
- A. A. Fadda, S. I. El-Desoky, H. A. Etman, S. B. Bondock and M. A. Metwally, Sulfur Lett., 2003, 26, 127–137 CrossRef.
- A. A. Fadda, M. R. Hala and M. E. A. Zaki, Molecules, 2000, 5, 701 CrossRef CAS.
- A. A. Fadda, M. E. A. Zaki, K. Samir and F. A. Amer, Phosphorus, Sulfur, and Silicon and the Related Elements, 2006, 181, 1815 CrossRef.
- T. G. Deligeorgiev, D. A. Zaneva, H. E. Katerinopoulos and V. N. Kolev, Dyes Pigm., 1999, 41, 49 CrossRef CAS.
- I. M. Warner, S. A. Soper and L. B. McGown, Anal. Chem., 1996, 68, 73R CrossRef.
- T. Karatsu, M. Yanai, S. Yagai, J. Mizukami, T. Urano and A. Kitamura, J. Photochem. Photobiol., A, 2005, 170, 123 CrossRef CAS.
- H. Hoppe and N. S. Sariciftci, J. Mater. Sci., 2004, 19241945 Search PubMed.
- M. Matsui, Y. Hashimoto, K. Funabiki, J. Y. Jin, T. Yoshida and H. Minoura, Synth. Met., 2005, 148, 147 CrossRef CAS.
- E. Delaey, F. van Laar, D. De Vos, A. Kamuhabwa, P. Jacobs and P. de Witte, J. Photochem. Photobiol., B, 2000, 55, 27 CrossRef CAS.
- H. Hilal and J. A. Taylor, Dyes Pigm., 2007, 75, 483 CrossRef CAS.
- H. J. Karlsson, M. H. Bergqvist, P. Lincoln and G. Westman, Bioorg. Med. Chem., 2004, 12, 2369 CrossRef CAS PubMed.
- Q. Li, Y. Kim, J. Namm, A. Kulkarni, G. R. Rosania, Y. H. Ahn and Y. T. Chang, Chem. Biol., 2006, 13, 615 CrossRef CAS PubMed.
- F. M. Hamer, The Cyanine Dyes and Related Compound; Interscience, New York, 1964 Search PubMed.
- A. A. Fadda, E. Abdel-Latif and R. E. El-Mekawy, Phosphorus, Sulfur, and Silicon and the Related Elements, 2008, 183(8), 1940 CrossRef CAS.
- A. A. Fadda, E. Abdel-Latif and R. E. El-Mekawy, Eur. J. Med. Chem., 2009, 44(3), 2223 CrossRef PubMed.
- A. A. Fadda, E. Abdel-Latif and R. E. El-Mekawy, Pharmacol. Pharm., 2012, 3, 148 CrossRef CAS.
- R. E. El-Mekawy, Pharmacology, 2013, 4(11), 590 CAS.
- H. M. Refat, A. A. Fadda, R. E. El-Mekawy and A. M. Sleat, Heterocycles, 2015, 19, 21–26 Search PubMed.
- S. Harusawa, K. Yoshida, C. Kojima, L. Araki and T. Kurihara, Tetrahedron, 2004, 60, 11911 CrossRef CAS.
- Y. B. Vibhute and M. A. Basser, Indian J. Chem., 2003, 42B, 202 CAS.
- N. M. Hamada and E. M. Sharshira, Molecules, 2011, 16, 2304 CrossRef CAS PubMed.
- H. Musso, Tetrahedron, 1979, 35, 2843 CrossRef CAS.
- C. Reichardt, J. Phys. Org. Chem., 1995, 8, 761 CrossRef CAS.
- A. A. Fadda and R. E. El-Mekawy, Dyes Pigm., 2015, 118, 45 CrossRef CAS.
- A. A. Fadda and R. E. El-Mekawy, Dyes Pigm., 2013, 99, 512 CrossRef CAS.
- I. Timtcheva, V. Maximova, T. Deligeorgiev, D. Zanevac and I. Ivanovd, J. Photochem. Photobiol., A, 2000, 130, 7 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10165a |
| ‡ Present address: Department of Chemistry, Faculty of Applied Science, Umm Al-Qura University, Makkah Al Mukarrama, Saudi Arabia. |
| This journal is © The Royal Society of Chemistry 2017 |