
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recyclable cross-linked hydroxythioether particles with tunable structures via robust and efficient thiol-epoxy dispersion polymerizations†
Jiaojun Tan a,
Chunmei Lia,
Kevin De Bruyckerb,
Guoxian Zhanga,
Junwei Gua and
Qiuyu Zhang*a
a,
Chunmei Lia,
Kevin De Bruyckerb,
Guoxian Zhanga,
Junwei Gua and
Qiuyu Zhang*a
aKey Laboratory of Applied Physics and Chemistry in Space of Ministry of Education, School of Science, Northwestern Polytechnical University, Xi'an, 710072, China. E-mail: qyzhang1803@gmail.com
bDepartment of Organic and Macromolecular Chemistry, Polymer Chemistry Research Group, Ghent University, Krijgslaan, 281, S4-bis, B-9000 Ghent, Belgium
First published on 8th November 2017
Abstract
The highly efficient base-catalyzed thiol-epoxy reactions were exploited in dispersion polymerizations as a simple method for the preparation of uniform and cross-linked particles with a tunable size, glass transition temperature (Tg) and network structure. Particles with sizes ranging from 1 μm to 3 μm could be obtained by varying monomer concentration and reaction medium, while the Tgs could be increased from −10 °C to 65 °C by adjusting the monomer functionality and structure. In order to further demonstrate the potential of these particles, the thioether bonds were simply oxidized to sulfoxides and sulfones to increase the Tgs and stiffness of the particles significantly. Moreover, the cross-linked particles proved to be reprocessable to a polymer material when a proper catalyst was incorporated. We believe that this simple and efficient method will become a powerful tool for particle preparation, and paves the way for polymer particle reinforcement and recycling.
Introduction
The preparation and design of polymer particles have drawn significant attention because of their wide applicability in the fields of coatings, separation, photonic crystals and catalyst immobilization, resulting from their tunable size, functionality and surface area.1–4 In the last decades, many methods were developed for particle preparation, such as suspension, emulsion, precipitation and dispersion polymerization.5 However, the chemistry for particle preparation was mainly limited to a radical chain growth of styrenic and acrylic monomers, which is prone to inhibition by oxygen and usually requires a high energy input (e.g. heat or UV-radiation) during initiation and polymerization.5 Furthermore, a long reaction time is always required for this kind of particle preparation. Thus, developing fast and energy-efficient reactions for the preparation of polymer particles is an important goal for both academia and industry.In order to reach this goal, great efforts have been devoted to efficient step-growth poly-additions, such as the production of polyurethane particles via the reaction of a diisocyanate and a diol.6 Another example involves the introduction of azide–alkyne click chemistry to interfacial polymerizations by Landfester and co-workers to prepare nano-capsules efficiently.7,8 While oxygen inhibition is successfully eliminated in these systems, they are still restricted by possible side reactions, lose control of polymerization and limited commercially available monomers. Therefore, the focus has recently shifted to thiol-X reactions for the preparation of polymer particles and capsules, which also tolerate oxygen while they typically proceed efficiently under mild conditions.9 Thiol-ene reactions, for example, gradually revealed their great potential in polymer synthesis and heterogeneous polymerizations.10,11 Since the first report of Du Prez and co-workers on the preparation of functional polymer beads via thiol-ene/yne chemistry in a microfluidic setup,12 both the nucleophilic thiol-Michael and radical-mediated thiol-ene additions were applied to fabricate polymer particles via suspensions,13–18 emulsions,19,20 mini-emulsions,21–25 Pickering emulsion,26 dispersions,27–32 microfluidics.12,33,34 Very recently, thiol-isocyanate reactions have also been implemented to prepare particles and capsules by our group as well as by Ethirajan and co-workers.35–38 The main advantage of using thiol-Michael and thiol-isocyanate reactions over the radical hydrothiolation is the possibility to introduce functionalities that are not compatible with a radical environment, as well as a lower energy input because no UV-irradiation is required.
Similarly to thiol-Michael and thiol-isocyanate reactions, the thiol-epoxy reaction is a simple nucleophilic ring-opening reaction to yield β-hydroxythio-ethers. This addition is catalyzed by a base, which generates a thiolate anion that opens the epoxide ring via an SN2-type mechanism.39,40 Although the thiol-epoxy reaction has been extensively studied and many reports discuss the broad applicability in polymer science,41–54 the use of this reaction in heterogeneous system has not drawn attention of academia or industry yet. Nevertheless, multiple reasons suggest that the thiol-epoxy reactions is an excellent candidate for particle preparation in heterogeneous system. Firstly, the aforementioned benign characteristics make the preparation process simple, efficient and easy to handle. Secondly, the reaction is catalyzed by a strong base rather than initiated by UV-irradiation or heat, leading to a simple control of the reaction. Furthermore, a wide range of organic and inorganic bases were proven to be efficient catalysts, which can be easily dispersed in various reaction media. Finally, readily available starting materials allow large-scale fabrication at low cost.
This work reports on the first application of thiol-epoxy reactions for the fabrication of polymeric particles via dispersion polymerization. Uniform, functional and cross-linked particles can be obtained in a one-pot dispersion polymerization, while the size, morphology and Tg can be easily adjusted by varying the type or amount of monomer, or the reaction medium. Furthermore, we demonstrated that the polymer backbone can be modified by quantitative and selective oxidation of the generated thioethers to sulfoxides or sulfones, which addressed the main disadvantages of thioethers: insufficient rigidity and low Tg. Finally, the cross-linked particles, containing both β-hydroxythioethers and ester groups, are able to be recycled and reprocessed when a suitable catalyst is added.
Experiment and characterizations
Materials
Pentaerythritol tetra (3-mercaptopropionate) (PETMP, >90%), trimethylolpropane tris (3-mercaptopropionate) (TMMP, CP), trimethylolpropane triglycidyl ether (TMTGE, RG), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 98%), propargyl acrylate (PA, 98%) were purchased from Sigma Aldrich. 4,4′-Methylene bis (N,N-diglycidylaniline) (TGMDA, 98%) was obtained from J&K. Fluorescein isothiocyanate isomer (FITC isomer, 95%), 1,8-diazabicyclo[5.4.0]-7-undecene (DBU, 98%), 1,1,2,2-tetrahydroperfluorooctyl acrylate (97%), 2-propanol (iPA, 99.9%), 1,3-benzenedimethanethiol (BDT, 98%+) were purchased from Adamas. Tetrabutylammonium fluoride (TBAF, 75 wt% solution in H2O) was obtained from Alfa. All the chemicals were used as received and their structures are shown in Fig. 1. | ||
| Fig. 1 Chemical structures and acronyms for monomers used in this work. | ||
Preparation of the thiol-epoxy-based particles
The particles were prepared via one-step dispersion polymerization via the typical procedure. A stoichiometric amount of thiol and epoxide-containing monomers (2.0 g TMTGE and 2.7 g PETMP), PVP (3.0 g) and 2-propanol (100 g) were added to a 250 mL round bottom flask under shaking or gentle stirring to form a clear and homogeneous solution. DBU (0.3 mL) was added to the solution, and dissolved immediately. Then, the mixture turned milky after several minutes, and the reaction was kept overnight to obtain a full conversion. Finally, the particles were collected by centrifuging the dispersion and washing subsequently with ethanol and water to remove the PVP and unreacted monomers.For the preparation of alkyne functionalized particles, PA (0.11 g), TMTGE (0.90 g), PETMP(1.33 g), PVP (2.0 g) and 2-propanol (50 g) were added to a 100 mL round bottom flask and polymerized using the same conditions and procedure as described above. As for fluorescent labeled particles, FITC (5 mg), TMTGE (1.0 g), PETMP(1.33 g), PVP (2.0 g) and 2-propanol (50 g) were added to a 100 mL round bottom flask and polymerized using the same conditions and procedure as described above.
Characterizations
SEM (scanning electron microscope): SEM images and EDS (energy dispersive spectrometer) spectra were recorded on a Zeiss Merlin Compact. The dispersion of particles was cast on a silicon wafer, air dried and coated with a layer of Pt before imaging. The average size and C.V. (coefficient of variation) of particles were obtained by counting at least 100 particles from the SEM images.

FTIR (Fourier transform infrared spectroscopy): FTIR spectra of particles and monomers were obtained from Bruker Tensor 27 through a transmission model, and the powder sample for FTIR were just washed and air-dried.
DSC (differential scanning calorimetry) was performed on a Mettler Toledo DSC1 calorimeter under a nitrogen flow at a heating rate of 10 °C min−1.
The fluorescence images of FITC-labelled particles were visualized with a fluorescence microscope (Nikon 80i).
XPS (X-ray photoelectron spectroscopy) spectra were measured on a Kratos AXIS Ultra DLD XPS (Kratos, UK) with a monochromatic Al Kα X-ray source to detect the elemental composition of the particles. The shifts of binding energy of XPS curves were corrected by calibration on the lowest C 1s peak at 284.6 eV as a reference.
Results and discussions
Preparation of cross-linked particles
In order to obtain uniform particles, catalyst type and reaction media were optimized in advance. Because extensive studies have demonstrated that LiOH, TBAF, DBU and TBD could all be applied as catalysts for thiol-epoxy reactions,41–43 their catalytic effect in dispersion polymerizations was investigated in this work. LiOH dissolved very little in organic solvents, and a significant amount of water (>10%) was usually required. However, aggregates instead of particles were obtained when water was added to the system, and the possible reasons were as follows: the limited solubility of thiol and epoxy monomers in alcohols solvent, and water made the solubility even worse. The poor solubility leaded to inhomogeneous solution at the initial stage and unstable nucleation. As for TBAF, a high catalyst content (>20 mol%) was necessary for quantitative conversion, while TBAF was commericially available as a 75% aqueous solution. Therefore, TBAF was not a good candidate either for thiol-epoxy dispersion polymerizations. Interestingly, when TBD was adopted as catalyst, the solution became initially turbid and milky, but turned clear eventually. The reason for this phenomenon will be discussed below. Nevertheless, TBD was a solid and needed some seconds to dissolve in the medium, while the catalytic efficiency was too high for thiol-epoxy reactions, so it could not to be dispersed uniformly before turning turbid and aggregates were formed. DBU acted as an ideal catalyst for thiol-epoxy dispersion polymerization, both for its high efficiency and from a practical point of view, and was thus was selected as the prefered catalyst in this work. As for reaction media, methanol and ethanol were the most commonly used solvents for dispersion polymerizations, so they are firstly adopted in this work. However, the limit solubility of thiol and epoxy monomer as well as the polymer chains in methanol or ethanol resulted in inhomogeneous solution at initial stage and unstable nucleation in polymerization stage. As a result, no regular particles were obtained. Fortunately, uniform particles were fabricated when iPA was selected as model medium.Polymer particles were prepared via thiol-epoxy dispersion polymerization, as shown in Fig. 2. Initially, multivalent thiol and epoxy monomers were dissolved together with poly(N-vinylpyrrolidone) (PVP) in isopropyl alcohol (iPA) to form a clear solution, where PVP and iPA served as stabilizer and reaction medium, respectively. As mentioned above, a strong base is required as the catalyst for the thiol-epoxy reaction. Therefore, DBU was added to the solution to deprotonate the thiol monomers, which could subsequently react with the epoxides via an SN2 type mechanism, i.e. at the less hindered side of the epoxide ring, finally generating hydroxythioether linkages through proton transfer. As the polymerization proceeded, the growing hydroxythioether chains could not dissolve in iPA and started to precipitate, which served as the primary nuclei of polymer particles. After the continues attachment and deposition of propagating polymer chains on the surface of nuclei, uniform particles were finally obtained when a high/full conversion reached. The complete fabrication process is triggered by the addition of the amine catalyst and occurred at room temperature without any other energy input such as UV-irradiation or even stirring.
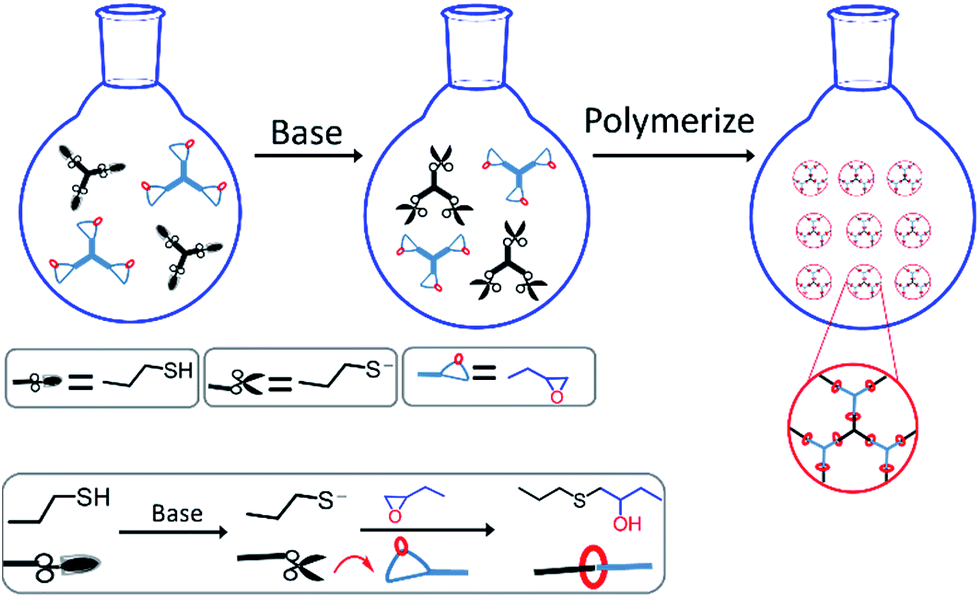 | ||
| Fig. 2 General scheme of thiol-epoxy reactions in dispersion polymerization. | ||
The morphology, size distribution, FTIR spectra and DSC trace of the particles obtained via a typical experiment, as described in the experimental section, are shown in Fig. 3. The particles show a well-defined spherical morphology (Fig. 3A), without the formation of aggregates and with a relatively narrow size distribution around an average diameter of 4.18 μm and C.V. of 8.5% (Fig. 3B). Meanwhile, the structure and high conversion could be verified by FTIR (Fig. 3C) because both the peaks at 2570 cm−1 and 908 cm−1 of monomer mixture, corresponding to the thiol and epoxide, respectively, were absent in the spectrum of particles. As seen in the DSC trace (Fig. 3D), the particles had a relatively low Tg (∼−10 °C) as a result of the flexible hydroxythioether linkages. However, they could only be dispersed and swelled in many organic solvents without dissolving or coagulating, while retaining a good spherical shape.
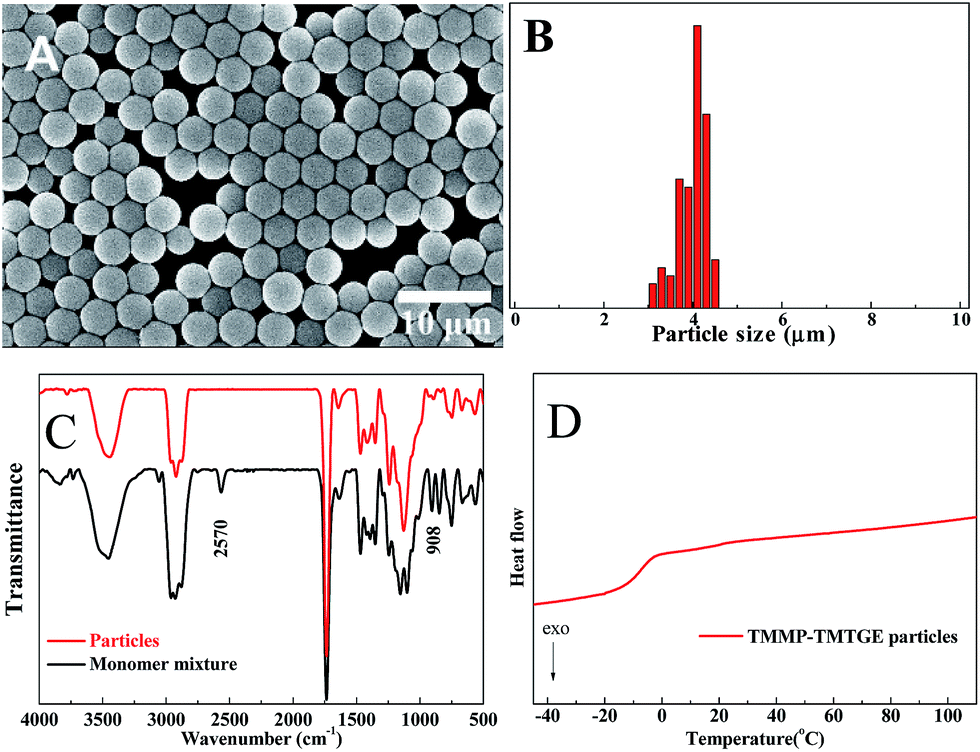 | ||
| Fig. 3 (A) SEM image, (B) size distribution, (C) FTIR spectra, (D) DSC trace of particles prepared by stoichiometric TMMP–TMTGE dispersion polymerization. Polymerization conditions: TMMP (2.7 g), TMTGE (2.0 g), PVP (3.0 g), iPA (100 g) and DBU (0.5 mL). The reaction lasted overnight without stirring. | ||
Effect of stabilizer, monomer, catalyst and solvent on the particle size, morphology and thermal properties
Particles prepared by dispersion polymerization generally undergo a “nucleation stage” and a “particle growing stage”, while the boundary between the two stages is the phase transition from a homogeneous system to a turbid solution. It is quite difficult to prepare functional cross-linked particles via a chain-growth dispersion polymerization, because even the preparation of particles with a low cross-linking density (cross-linker < 5%) or functional particles (functional monomers < 5%) typically requires several steps. The reason is that cross-linkers or functional monomers make the nucleation stage unstable, and aggregates are formed instead. Nevertheless, cross-linked particles were easily obtained via the thiol-epoxy dispersion polymerization as a result of its step-growth nature. In order to get an in-depth knowledge of this method, the effects of many factors including monomer concentration, stabilizer concentration, catalyst loading and monomer type on the size, morphology and thermal properties of the particles were systemically investigated.Firstly, particles were prepared in a constant amount of iPA (100 g), PVP (3.0 g) and DBU (0.5 mL), to which different amounts of monomers were added. As shown in Fig. 4 and Table S1,† the average diameter of particles increased from 0.94 μm to 3.02 μm, while a tendency of high yields and excellent spherical morphology was observed, when the monomer concentration increased from 1.15 wt% to 11.5 wt%. This phenomenon might be explained by the critical chain length and number of particles in the nucleation stage, i.e. (i) solubility of the polymer chain increases with increasing the monomer–solvent ratio, (ii) higher monomer concentrations will make the critical chain length become longer and decrease number of particles in the nucleation stage. Consequently, higher monomer concentrations resulted in a larger particle size. This hypothesis was further verified by the particles prepared with different concentrations of catalyst (DBU) or stabilizer (PVP), as depicted in Fig. S1 and S2 in ESI,† respectively, which show a fairly constant particle size and similar yields as a function of the DBU or PVP concentration. Nevertheless, a minimal amount of DBU was required for an efficient polymerization, because a low concentration of catalyst resulted in some adhesion among particles. As soon as the dilution of the monomers is approaching the solubility limit of the polyhydroxythioether, a vague boundary and low yield was observed between the particles (Fig. 4A), while a further dilution prevented the nucleation and thereby did not yield any particles.
 | ||
| Fig. 4 SEM images of stoichiometric PETMP–TMTGE particles with different monomer concentrations: (A) (1.15 g) (B) (2.3 g) (C) (4.7 g) (D) (6.9 g) (E) (9.3 g) (F) (11.5 g). Condition used: PVP (3.0 g), 2-propanol (100 g), DBU (0.5 mL) and the reaction lasted overnight without stirring. The size distribution was inserted. | ||
In the next step, TGMDA was introduced as a more rigid epoxy monomer compared to the previously applied TMTGE. In fact, addition of N,N-dimethylformamide (DMF) was required to increase the solubility of TGMDA in iPA. Therefore, the effect of the DMF content on particles size was also evaluated, and Fig. 5 displays the SEM images and size variation of particles prepared via TMMP–TGMDA reactions with different volume ratios of DMF. When the ratio of DMF increased from 3% to 30%, the co-solvent provided a better solubility for monomer and the growing polymer chians, which resulted in a delay of the nucleation stage. As a consequence, the number of nuclei decreased, and the average diameter naturally increased from 1.6 μm to 2.9 μm. At the same time, the C.V. decreased significantly with the ratio of DMF, and only aggregation was obtained when the ratio of DMF exceeded 40%.
 | ||
| Fig. 5 SEM images (A–E) and size variation (F) of particles prepared via TMMP–TGMDA reactions with different volume ratio of DMF: (A) (3%) (B) (5%) (C) (10%) (D) (20%) (E) (30%). Condition used: TMMP (2.4 g), TGMDA (2.2 g), PVP (3.0 g), total solvent (100 g), DBU (0.5 mL) and the reaction lasted overnight without stirring. The size distribution was inserted. | ||
As shown in Fig. 6, the Tg of the polymeric particles could be adjusted by utilizing different thiol and epoxide monomers. As a result of the many thioether linkages, as well as the flexible multivalent epoxide, particles prepared with TMTGE had a low Tg of approximately −10 °C (vide supra). By using the more rigid multivalent epoxides that introduced aromatic and aliphatic rings, on the other hand, particles could be obtained with a Tg of up to 65 °C.
 | ||
| Fig. 6 DSC traces of particles prepared with different monomer types. | ||
Feasibility of selective oxidization of hydroxythioether to sulfoxides/sulfones
While the Tg of the obtained particles can be improved by increasing the rigidity in the applied monomers, as discussed above, the flexible hydroxythioethers will always have a negative influence, thereby limiting the applications. Therefore, a post-polymerisation modification to achieve materials with a high Tg is highly desired. Recently, Tew, Bowman and co-workers have demonstrated that thioethers can be easily oxidized to sulfoxides or sulfones, which dramatically change the polymer properties, including polarity, rigidity and mechanical properties.55,56 Inspired by this concept, we explored the feasibility of oxidation of hydroxythioether to sulfoxides/sulfones and further address the critical limitation of (hydroxy)thioether particles, i.e. insufficient rigidity and flexibility.As shown in Fig. 7, thioethers can be efficiently oxidized to sulfoxides with only a slow (further) oxidation to sulfones when H2O2 was used as oxidant. On the other hand, m-chloroperbenzoic acid (mCPBA) quickly oxidizes thioethers to their highest oxidative state.55,56 First, particles (0.2 g) prepared by TMMP and ERL 4221 were immersed in a solution containing an excess of H2O2 (6 mL) and 6 mL ethanol. After stirring the reaction overnight, the oxidized particles were harvested by repeating three cycles of centrifugation and washing with ethanol. For the oxidation with mCPBA, the same procedure as for H2O2 was used, except that the amounts of mCPBA and ethanol were 0.8 g and 20 mL, respectively. The properties and structures of particles after the oxidation were investigated by DSC, FTIR and XPS and depicted in Fig. 8. As anticipated, the Tgs of the oxidized particles significantly increased by the oxidative treatment, i.e. from 32 °C to 54 °C and 81 °C when H2O2 and mCPBA were used as oxidants, respectively (Fig. 8A), while FTIR analysis confirmed the presence of three new peaks after the oxidation (Fig. 8B). Two of them (1300 and 1128 cm−1) were ascribed to the sulfones, while the third at 1010 cm−1 is characteristic for sulfoxides.56,57 In order to compare the intensities, the absorption of the esters at 1730 cm−1 were set as internal reference to eliminate errors of sample concentrations. While a significant amount of over-oxidation is observed in the case of H2O2, the FTIR spectra reflect the higher oxidative reactivity of mCPBA, yielding predominantly sulfone groups. As depicted in Fig. S3,† the morphology of oxidized particles kept almost the same as before, because oxidation would increase stiffness and dimensional stability of the particles.
 | ||
| Fig. 7 Oxidation of thioether linkage to sulfoxide/sulfone via H2O2 and mCPBA (3-chloroperbenzoic acid). | ||
 | ||
| Fig. 8 (A) DSC traces, (B) FTIR, (C) XPS spectra of pristine, H2O2 oxidized, mCPBA oxidized TMMP-ERL 4221 particles. | ||
The sulfur (2p) XPS spectra (Fig. 8C) gave a deeper information on the oxidative state of the sulfur atoms in the materials. In this work, there were three possible sulfur species (thioether, sulfoxide and sulfone) with their respective binding energies located at 163.0 eV (thioether), 165.2 eV (sulfoxide) and 167.2 eV (sulfone).58 It was clear that the peak of particles oxidized by mCPBA is mainly located at the region of sulfones, and only a small part can be ascribed to sulfoxides. At the same time, the peak of particles oxidized by H2O2, ranging from 163.4 eV to 168.5 eV, indicated the presence of both sulfoxide and sulfone, which confirms the FTIR analysis discussed above. In summary, the flexible thioether links in the produced particles can be easily oxidized to more rigid sulfoxides or sulfones using cheap and commercially available oxidants, which significantly enhances the accompanying material properties.
Feasibility of remolding and reprocessing of particles based on transesterification
As mentioned above, the solution of TBD-catalyzed TMMP–TMTGE dispersion polymerization in ethanol became initially turbid but eventually turned clear again, as shown in Fig. S4.† To our surprise, both the thiol and epoxy groups were almost quantitatively consumed, as judged from FTIR analysis (Fig. S5†). Nevertheless, TBD is a well-known and highly effective catalyst of transesterification, while the obtained polymer contains a large amount of ester functionalities, introduced from TMMP, and secondary hydroxyl groups as a result of the epoxide ring opening. Moreover, the polymer is suspended in a medium of primary alcohols. Very recently, the group of Bowman demonstrated that TBD promotes the transesterification between secondary hydroxyl and ester groups at elevated temperatures, resulting in a network with vitrimer properties.59 Therefore, a plausible explanation for the apparent solubility of our network, is the TBD-catalyzed alcoholysis of the TMMP–TMTGE particles into low molecular weight compounds, as shown in Fig. 9. | ||
| Fig. 9 Scheme of TBD-catalyzed TMMP–TMTGE polymerization and transesterification in EtOH. | ||
The hypothesis of a TBD-catalyzed transesterification, as described above, was verified by attempting to either redissolve or remold ethanol-free TMMP–TMTGE particles. To this end, particles (1.0 g) and TBD (0.1 g) were dispersed in ethanol (15 mL) to obtain a turbid suspension (Fig. 10A). After 3 hours at room temperature, the particles were completely dissolved, forming a clear solution (Fig. 10B and C), which indicates that ethanol was indeed involved in transesterifications, which cuts the cross-linked structures into small and soluble pieces in presence of TBD. The control sample remained unchanged (Fig. 10E), and the same condition was adopted except ethanol was replaced by acetone. Next, the solution in Fig. 10C was heated in open air to evaporate the ethanol, enabling the dissolved molecules to reform a bulk solid material (Fig. 10D). Finally, this bulk material could dissolve in ethanol again, which served as a typical solvent-assisted pressure-free method for reprocessing polymers.
 | ||
| Fig. 10 The reprocessing procedure of TMMP–TMTGE particles: (A) the initial stage (B) 2 hour (C) 3 hour after dispersing TMMP–TMTGE particles and TBD in EtOH. (D) Bulk polymer obtained via evaporating ethanol. (E) Control samples, dispersion of TMMP–TMTGE particles and TBD in acetone. | ||
Next to the alcoholysis of the network into soluble molecules, the remolding of TMMP–TMTGE particles was also targeted. Therefore, TBD was incorporated into particles via a swelling method, in which particles (4.0 g) were dispersed in acetone (30 mL) containing TBD (0.4 g), followed by evaporation of the acetone in vacuo. The particles containing TBD (Fig. 11A) were then pressed for 1 hour in a mold at 180 °C to obtain a rectangular plate (Fig. 11C). Finally, the Tgs of both the bulk polymer obtained by the solvent-assisted pressure-free method and the particles after hot pressing were investigated by DSC. Only a small increase in Tg is observed in both cases (Fig. S6†), which might be explained by a further curing or some oxidation, for example during the heating.60 In summary, these particles were shown to be reprocessable to bulk materials with similar thermal properties to the pristine network via either a solvent-assisted approach or pressing at elevated temperatures.
 | ||
| Fig. 11 (A) The pristine particles containing TBD, (B) particles in the mold, (C) polymer plate after hot press. | ||
Functionalization of thioether particles
A significant feature of the step-growth mechanism that was applied for the synthesis of particles via a thiol-Michael- or thiol-isocyanate-based heterophase polymerizations, was the facile functionalization via an off-stoichiometric amount of reagents. However, in the case of the investigated thiol-epoxy dispersions, an off-stoichiometric reaction did not generate functional particles at all, even with a thiol or epoxy excess of 30%, as evidenced by FTIR analysis (Fig. S7†). The reason for this phenomenon can be found in the strong base that is used for the polymerizations, which can convert the excess of thiols to disulfides when no degassing or inert atmosphere is applied,61 while the excess of epoxides can also react with the dispersion medium (iPA) in an alkaline environment. Therefore, we attempted to add the functional monomers together with thiol and epoxy monomers at the initial stage. As shown in Fig. 12A, the peak at 2120 cm−1 proved the successful incorporation of an alkyne in the particles via the reaction of propargyl acrylate (PA) with the thiol under base catalysis. On the other hand, a fluorescent dye was also successfully linked to the particles using a fluorescein isothiocyanate (FITC) isomer, as evidenced by the fluorescence microscopy image in Fig. 12B. Nevertheless, this method could not be generally applied for a broad range of functionalities, since the nucleation stage was easily disturbed by functional monomers. For example, only floccule and aggregates were obtained in an attempt to attach a fluorinated acrylate (THPFA) to particles. This problem might be solved in the future by applying a one-pot multi-step dispersion, a technique that is currently under development in our labs, in which functional monomers are added after the nucleation stage to avoid the aforementioned nucleation issues.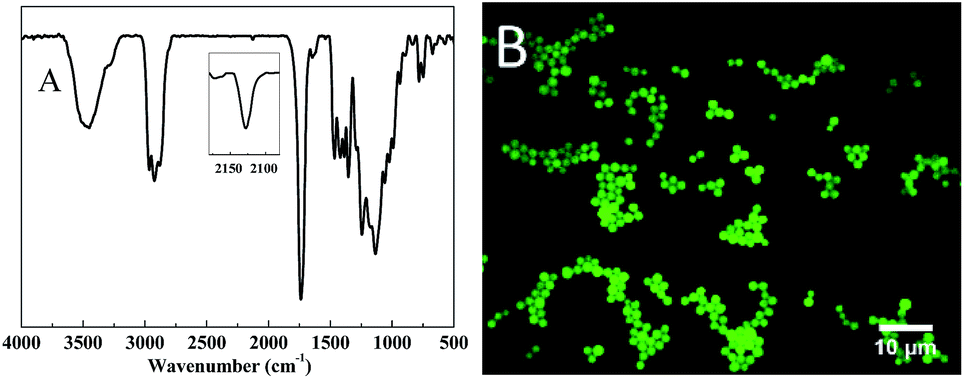 | ||
| Fig. 12 (A) FTIR spectrum of alkyne functionalized particles, (B) fluorescence image of fluorescent labeled particles by fluorescein isothiocyanate isomer. The conditions are provided in the experimental section. | ||
Conclusion
In summary, uniform hydroxythioether particles have been prepared via a base-catalyzed thiol-epoxy dispersion polymerization for the first time. The step-growth nature of this process makes the preparation quite efficient and easy to operate with a low energy input. The influences of stabilizer and monomer concentration, as well as the type of catalyst and solvent on the particle preparation were systematically investigated. Next, the critical limitations of hydroxythioether-based particles including insufficient rigidity and low Tgs could be significantly improved by oxidation of the thioether bonds to sulfoxides and sulfones with simple methods and cheap oxidants. Moreover, the ester and secondary hydroxyl groups in the particles allow the recycling of the cross-linked polymer when a proper catalyst is incorporated, which greatly reduces environmental pressure and increases the utilization efficiency. Finally, functionalities could be introduced in the particles by simply adding a functional monomer to the mixture of multivalent thiol and epoxide. This work provides a simple, efficient and promising method for the preparation, reinforcement and recycling of polymer particles, and is expected to become a strong tool for further research and industrial applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the financial support provided by the National Natural Science of China (51433008, 21704084), the International Cooperation and Exchanges NSFC (51711530233). K. D. B. acknowledges the Research Foundation-Flanders (FWO) for the funding of his PhD fellowship.Notes and references
- F. S. Macintyre, D. C. Sherrington and L. Tetley, Synthesis of ultrahigh surface area monodisperse porous polymer nanospheres, Macromolecules, 2006, 39, 5381–5384 CrossRef CAS.
- L. Chen, Y. Honsho, S. Seki and D. Jiang, Light-harvesting conjugated microporous polymers: rapid and highly efficient flow of light energy with a porous polyphenylene framework as antenna, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS PubMed.
- Y. Xia, B. Gates, Y. Yin and Y. Lu, Monodispersed colloidal spheres: old materials with new applications, Adv. Mater., 2000, 12, 693–713 CrossRef CAS.
- J.-S. Song, F. Tronc and M. A. Winnik, Two-stage dispersion polymerization toward monodisperse, controlled micrometer-sized copolymer particles, J. Am. Chem. Soc., 2004, 126, 6562–6563 CrossRef CAS PubMed.
- M. T. Gokmen and F. E. Du Prez, Porous Polymer Particles—A Comprehensive Guide To Synthesis, Characterization, Functionalization And Applications, Prog. Polym. Sci., 2012, 37, 365–405 CrossRef CAS.
- F. Tiarks, K. Landfester and M. Antonietti, One-step preparation of polyurethane dispersions by miniemulsion polyaddition, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2520–2524 CrossRef CAS.
- J. M. Siebert, G. Baier, A. Musyanovych and K. Landfester, Towards copper-free nanocapsules obtained by orthogonal interfacial “click” polymerization in miniemulsion, Chem. Commun., 2012, 48, 5470–5472 RSC.
- R. m. Roux, L. Sallet, P. Alcouffe, S. p. Chambert, N. Sintes-Zydowicz, E. Fleury and J. Bernard, Facile and rapid access to glyconanocapsules by CuAAC interfacial polyaddition in miniemulsion conditions, ACS Macro Lett., 2012, 1, 1074–1078 CrossRef CAS.
- L.-T. T. Nguyen, M. T. Gokmen and F. E. Du Prez, Kinetic comparison of 13 homogeneous thiol–X reactions, Polym. Chem., 2013, 4, 5527–5536 RSC.
- A. B. Lowe, Thiol-ene “click” reactions and recent applications in polymer and materials synthesis, Polym. Chem., 2010, 1, 17–36 RSC.
- A. B. Lowe, Thiol-ene “click” reactions and recent applications in polymer and materials synthesis: a first update, Polym. Chem., 2014, 5, 4820–4870 RSC.
- R. A. Prasath, M. T. Gokmen, P. Espeel and F. E. Du Prez, Thiol-ene and thiol-yne chemistry in microfluidics: a straightforward method towards macroporous and nonporous functional polymer beads, Polym. Chem., 2010, 1, 685–692 RSC.
- O. Z. Durham, S. Krishnan and D. A. Shipp, Polymer Microspheres Prepared by Water-Borne Thiol–Ene Suspension Photopolymerization, ACS Macro Lett., 2012, 1, 1134–1137 CrossRef CAS.
- O. Z. Durham and D. A. Shipp, Suspension thiol-ene photopolymerization: Effect of stabilizing agents on particle size and stability, Polymer, 2014, 55, 1674–1680 CrossRef CAS.
- O. Z. Durham, H. R. Norton and D. A. Shipp, Functional polymer particles via thiol–ene and thiol–yne suspension “click” polymerization, RSC Adv., 2015, 5, 66757–66766 RSC.
- O. Z. Durham and D. A. Shipp, Suspension “click” polymerizations: thiol-ene polymer particles prepared with natural gum stabilizers, Colloid Polym. Sci., 2015, 293, 2385–2394 CAS.
- J. Tan, C. Li, J. Zhou, C. Yin, B. Zhang, J. Gu and Q. Zhang, Fast and facile fabrication of porous polymer particles via thiol–ene suspension photopolymerization, RSC Adv., 2014, 4, 13334–13339 RSC.
- E. M. Barker and J. P. Buchanan, Thiol-ene polymer microbeads prepared under high-shear and their successful utility as a heterogeneous photocatalyst via C 60-capping, Polymer, 2016, 92, 66–73 CrossRef CAS.
- O. Z. Durham, D. V. Chapman, S. Krishnan and D. A. Shipp, Radical Mediated Thiol-Ene Emulsion Polymerizations, Macromolecules, 2017, 50, 775–783 CrossRef CAS.
- D. Liu, B. Yu, X. Jiang and J. Yin, Responsive hybrid microcapsules by the one-step interfacial thiol–ene photopolymerization, Langmuir, 2013, 29, 5307–5314 CrossRef CAS PubMed.
- F. Jasinski, E. Lobry, B. Tarablsi, A. Chemtob, C. l. Croutxé-Barghorn, D. Le Nouen and A. Criqui, Light-mediated thiol–ene polymerization in miniemulsion: a fast route to semicrystalline polysulfide nanoparticles, ACS Macro Lett., 2014, 3, 958–962 CrossRef CAS.
- D. Amato, D. Amato, A. Flynt and D. Patton, Functional, sub-100 nm polymer nanoparticles via thiol–ene miniemulsion photopolymerization, Polym. Chem., 2015, 6, 5625–5632 RSC.
- C. Wang, S. Chatani, M. Podgórski and C. N. Bowman, Thiol-Michael addition miniemulsion polymerizations: functional nanoparticles and reactive latex films, Polym. Chem., 2015, 6, 3758–3763 RSC.
- F. Jasinski, A. s. Rannée, J. Schweitzer, D. Fischer, E. Lobry, C. l. Croutxé-Barghorn, M. Schmutz, D. Le Nouen, A. Criqui and A. Chemtob, Thiol–ene linear step-growth photopolymerization in miniemulsion: fast rates, redox-responsive particles, and semicrystalline films, Macromolecules, 2016, 49, 1143–1153 CrossRef CAS.
- T. O. Machado, P. B. Cardoso, P. E. Feuser, C. Sayer and P. H. Araújo, Thiol-ene miniemulsion polymerization of a biobased monomer for biomedical applications, Colloids Surf., B, 2017, 159, 509–517 CrossRef CAS PubMed.
- C. Li, J. Tan, J. Gu, L. Qiao, B. Zhang and Q. Zhang, Rapid and efficient synthesis of isocyanate microcapsules via thiol-ene photopolymerization in Pickering emulsion and its application in self-healing coating, Compos. Sci. Technol., 2016, 123, 250–258 CrossRef CAS.
- C. Wang, M. Podgórski and C. N. Bowman, Monodisperse functional microspheres from step-growth “click” polymerizations: preparation, functionalization and implementation, Mater. Horiz., 2014, 1, 535–539 RSC.
- C. Wang, X. Zhang, M. Podgórski, W. Xi, P. Shah, J. Stansbury and C. N. Bowman, Monodispersity/Narrow polydispersity cross-Linked microparticles prepared by step-growth thiol–michael addition dispersion polymerizations, Macromolecules, 2015, 48, 8461–8470 CrossRef CAS.
- K. Jiang, Y. Liu, Y. Yan, S. Wang, L. Liu and W. Yang, Combined chain-and step-growth dispersion polymerization toward PSt particles with soft, clickable patches, Polym. Chem., 2017, 8, 1404–1416 RSC.
- C. Wang, M. M. Zieger, A. Schenzel, M. Wegener, J. Willenbacher, C. Barner-Kowollik and C. N. Bowman, Photoinduced tetrazole-based functionalization of off-stoichiometric clickable microparticles, Adv. Funct. Mater., 2017, 27, 1605317, DOI:10.1002/adfm.201605317.
- F. Alimohammadi, C. Wang, O. Z. Durham, H. R. Norton, C. N. Bowman and D. A. Shipp, Radical mediated thiol-ene/yne dispersion polymerizations, Polymer, 2016, 105, 180–186 CrossRef CAS.
- C. O. Bounds, R. Goetter, J. A. Pojman and M. Vandersall, Preparation and application of microparticles prepared via the primary amine-catalyzed michael addition of a trithiol to a triacrylate, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 409–422 CrossRef CAS.
- D. V. Amato, H. Lee, J. r. G. Werner, D. A. Weitz and D. L. Patton, Functional Microcapsules via Thiol–Ene Photopolymerization in Droplet-Based Microfluidics, ACS Appl. Mater. Interfaces, 2017, 9, 3288–3293 CAS.
- E.-K. Fleischmann, F. R. Forst, K. Köder, N. Kapernaum and R. Zentel, Microactuators from a main-chain liquid crystalline elastomer via thiol–ene “click” chemistry, J. Mater. Chem. C, 2013, 1, 5885–5891 RSC.
- J. Tan, C. Li, H. Li, H. Zhang, J. Gu, B. Zhang, H. Zhang and Q. Zhang, Water-borne thiol–isocyanate click chemistry in microfluidics: rapid and energy-efficient preparation of uniform particles, Polym. Chem., 2015, 6, 4366–4373 RSC.
- J. Tan, C. Li, S. Dan, H. Li, J. Gu, B. Zhang, H. Zhang and Q. Zhang, One-step thiol-isocyanate dispersion polymerization: Preparation of uniform, cross-linked and functional particles, Chem. Eng. J., 2016, 304, 461–468 CrossRef CAS.
- S. Kuypers, S. K. Pramanik, L. D'Olieslaeger, G. Reekmans, M. Peters, J. D'Haen, D. Vanderzande, T. Junkers, P. Adriaensens and A. Ethirajan, Interfacial thiol-isocyanate reactions for functional nanocarriers: A facile route towards tunable morphologies and hydrophilic payload encapsulation, Chem. Commun., 2015, 51, 15858–15861 RSC.
- C. Li, J. Tan, H. Li, D. Yin, J. Gu, B. Zhang and Q. Zhang, Thiol–isocyanate click reaction in a Pickering emulsion: a rapid and efficient route to encapsulation of healing agents, Polym. Chem., 2015, 6, 7100–7111 RSC.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Thiol-click chemistry: a multifaceted toolbox for small molecule and polymer synthesis, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- M. C. Stuparu and A. Khan, Thiol-epoxy “click” chemistry: Application in preparation and postpolymerization modification of polymers, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3057–3070 CrossRef CAS.
- S. De and A. Khan, Efficient synthesis of multifunctional polymers via thiol-epoxy “click” chemistry, Chem. Commun., 2012, 48, 3130–3132 RSC.
- I. Gadwal, J. Rao, J. Baettig and A. Khan, Functionalized molecular bottlebrushes, Macromolecules, 2013, 47, 35–40 CrossRef.
- I. Gadwal, M. C. Stuparu and A. Khan, Homopolymer bifunctionalization through sequential thiol–epoxy and esterification reactions: an optimization, quantification, and structural elucidation study, Polym. Chem., 2015, 6, 1393–1404 RSC.
- S. Binder, I. Gadwal, A. Bielmann and A. Khan, Thiol-epoxy polymerization via an AB monomer: Synthetic access to high molecular weight poly (β-hydroxythio-ether) s, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2040–2046 CrossRef CAS.
- A. Saha, S. De, M. C. Stuparu and A. Khan, Facile and General Preparation of Multifunctional Main-Chain Cationic Polymers through Application of Robust, Efficient, and Orthogonal Click Chemistries, J. Am. Chem. Soc., 2012, 134, 17291–17297 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Cu (0)-mediated living radical polymerization: recent highlights and applications; a perspective, Polym. Chem., 2016, 7, 1002–1026 RSC.
- Q. Zhang, A. Anastasaki, G.-Z. Li, A. J. Haddleton, P. Wilson and D. M. Haddleton, Multiblock sequence-controlled glycopolymers via Cu (0)-LRP following efficient thiol–halogen, thiol–epoxy and CuAAC reactions, Polym. Chem., 2014, 5, 3876–3883 RSC.
- J. A. Carioscia, J. W. Stansbury and C. N. Bowman, Evaluation and control of thiol–ene/thiol–epoxy hybrid networks, Polymer, 2007, 48, 1526–1532 CrossRef CAS PubMed.
- K. Jin, N. Wilmot, W. H. Heath and J. M. Torkelson, Phase-Separated Thiol–Epoxy–Acrylate Hybrid Polymer Networks with Controlled Cross-Link Density Synthesized by Simultaneous Thiol–Acrylate and Thiol–Epoxy Click Reactions, Macromolecules, 2016, 49, 4115–4123 CrossRef CAS.
- E. A. Dhulst, W. H. Heath and J. M. Torkelson, Hybrid thiol-acrylate-epoxy polymer networks: Comparison of one-pot synthesis with sequential reactions and shape memory properties, Polymer, 2016, 96, 198–204 CrossRef CAS.
- Y. Jian, Y. He, Y. Sun, H. Yang, W. Yang and J. Nie, Thiol–epoxy/thiol–acrylate hybrid materials synthesized by photopolymerization, J. Mater. Chem. C, 2013, 1, 4481–4489 RSC.
- S. Li, J. Han and C. Gao, High-density and hetero-functional group engineering of segmented hyperbranched polymers via click chemistry, Polym. Chem., 2013, 4, 1774–1787 RSC.
- H. Lin, J. Ou, Z. Liu, H. Wang, J. Dong and H. Zou, Thiol-epoxy click polymerization for preparation of polymeric monoliths with well-defined 3D framework for capillary liquid chromatography, Anal. Chem., 2015, 87, 3476–3483 CrossRef CAS PubMed.
- H. Lin, L. Chen, J. Ou, Z. Liu, H. Wang, J. Dong and H. Zou, Preparation of well-controlled three-dimensional skeletal hybrid monoliths via thiol–epoxy click polymerization for highly efficient separation of small molecules in capillary liquid chromatography, J. Chromatogr. A, 2015, 1416, 74–82 CrossRef CAS PubMed.
- J. M. Sarapas and G. N. Tew, Thiol-Ene Step-Growth as a Versatile Route to Functional Polymers, Angew. Chem., Int. Ed., 2016, 55, 15860–15863 CrossRef CAS PubMed.
- M. Podgórski, C. Wang, Y. Yuan, D. Konetski, I. Smalyukh and C. N. Bowman, Pristine Polysulfone Networks as a Class of Polysulfide-Derived High-Performance Functional Materials, Chem. Mater., 2016, 28, 5102–5109 CrossRef.
- R. Qu, C. Sun, C. Wang, C. Ji, Y. Sun, L. Guan, M. Yu and G. Cheng, Preparation and metal binding behavior of poly (4-vinylbenzyl 2-hydroxyethyl) sulfoxide and sulfone, Eur. Polym. J., 2005, 41, 1525–1530 CrossRef CAS.
- X. Zhao, Q. Zhang, C.-M. Chen, B. Zhang, S. Reiche, A. Wang, T. Zhang, R. Schlögl and D. S. Su, Aromatic sulfide, sulfoxide, and sulfone mediated mesoporous carbon monolith for use in supercapacitor, Nano Energy, 2012, 1, 624–630 CrossRef CAS.
- G. B. Lyon, L. M. Cox, J. T. Goodrich, A. D. Baranek, Y. Ding and C. N. Bowman, Remoldable Thiol–Ene Vitrimers for Photopatterning and Nanoimprint Lithography, Macromolecules, 2016, 49, 8905–8913 CrossRef CAS.
- Q. Shi, K. Yu, M. L. Dunn, T. Wang and H. J. Qi, Solvent Assisted Pressure-Free Surface Welding and Reprocessing of Malleable Epoxy Polymers, Macromolecules, 2016, 49, 5527–5537 CrossRef CAS.
- N. Feillée, M. De Fina, A. Ponche, C. Vaulot, S. Rigolet, L. Jacomine, H. Majjad, C. Ley and A. Chemtob, Step-growth thiol–thiol photopolymerization as radiation curing technology, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 117–128 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional Fig. S1–S6. See DOI: 10.1039/c7ra10481b |
| This journal is © The Royal Society of Chemistry 2017 |