
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Precision polymers containing main-chain-amino acids: ADMET polymerization and crystallization†
Jan Freudenberg a,
Silvio Poppeb and
Wolfgang H. Binder*a
a,
Silvio Poppeb and
Wolfgang H. Binder*a
aMartin Luther University Halle-Wittenberg, Faculty of Natural Sciences II, Chair of Macromolecular Chemistry, D-06120 Halle, Germany
bMartin Luther University Halle-Wittenberg, Faculty of Natural Sciences II, Chair of Organic Chemistry, D-06120 Halle, Germany
First published on 9th October 2017
Abstract
The synthesis of new poly(alkyl)-precision oligomers and polymers displaying different types of amino acids (chiral/achiral, polar/non-polar) placed at every 19th carbon atom are presented. Placing ω-alkenyl moieties onto appropriately N- and C-terminus functionalized amino acids successfully enabled ADMET-polymerization of these monomers. Grubbs 1st catalyst under melt-polycondensation-conditions was found to be the most effective with respect to the obtained molecular weights of the polymers, the isomerization of the olefins and their final yield, yielding molecular weights up to ∼22 kDa. The obtained polymers and the subsequent hydrogenation of the double bonds with p-toluenesulfonhydrazide (TsNHNH2) was proven by NMR, GPC, MALDI-ToF-MS and IR measurements. Investigation of the thermal behavior of the monomers and polymers via DSC measurements displays amorphous structures for monomers and polymers with unpolar amino acid side chains, whereas for polymers bearing the polar glutamic- and aspartic acid moieties crystalline morphologies are observed. An ordered lamellar crystal phase is observed where the amino acid branches are either incorporated or excluded from the unit cell, as proven by WAXS data.
Introduction
Understanding the ordering within specific domains of a polymer, based on regular arrangements of (sub-)units is among the fundamental principles in biological and synthetic macromolecules. In contrast to synthetic macromolecules, in which structure formation is based on only a few, often constrained hierarchical levels,1–3 intra- and intermolecular interaction via supramolecular bonds of exactly placed amino acids, combined with rotational constrains caused by rigid bonds in a polypeptide chain are the basis for the formation of regular ordering phenomena in biological macromolecules.4 Such inter- and intrachain macromolecular ordering is often kinetically driven by nucleation dependent processes,5,6 in which the formation of higher structure levels e.g. crystal lamellae,7 an (oligomeric- or peptidic-) gel,8 or an fibrillar aggregate9 strongly depends on the initial formation of primary nuclei.Introducing amino acids as structuring and folding elements into synthetic polymers thus is an important element of molecular design, enabling to determine the molecular shape in the solid state. Different mechanical and thermal properties as well as the crystallization behaviour of such polymers can be engineered by repetitively placing monomers at specific positions within a polymer chain of defined origin.10–15 Acyclic diene metathesis polymerization (ADMET)16 has been proven to be an excellent tool to achieve a polyethylene (PE)-like chain with functional monomers at precise locations.17,18 Size, chemical structure and the steric structure of such “defects” to a large extent determine the final crystallization behaviour in the solid state: when the precisely placed chemical elements are small (such as methyl19,20-, ethyl21- or halogen22–26-groups) they can be incorporated into the crystalline region of a semicrystalline polymer, whereas larger defects (higher alkyl substituents,19,27 phosphoesters28,29 or triazol-rings30 and sulfonic-moieties31) are excluded from the final crystal-lattice.
The synthetic preparation of biosimilar oligomers and polymers with repetitive amino acid units in the main chains has stimulated a plethora of different methods.32,33 Thus e.g. the synthesis of optically active linear and branched poly(meth)acrylamides bearing L-amino acid moieties by radical polymerization34–37 or the periodically incorporation of amino acid units via ROMP using Grubbs catalysts has been accomplished.38–40 Especially ROMP was found to be an excellent tool for synthesizing polyolefin-graft-oligopeptide polyelectrolytes which can form extended, pearl-like, or multi-molecular structures.41 A periodic arrangement of amino acid branches in precision polymers via ADMET polymerization has been previously achieved allowing to prepare polyolefines bearing ester-terminated and amide-terminated amino acids in the backbone.42 Currently, only lysine as a side-chain moiety and cysteine (within the main chain) have been placed within a poly(alkyl)-chain43,44 resulting in a semi-crystalline morphology of such polymers.
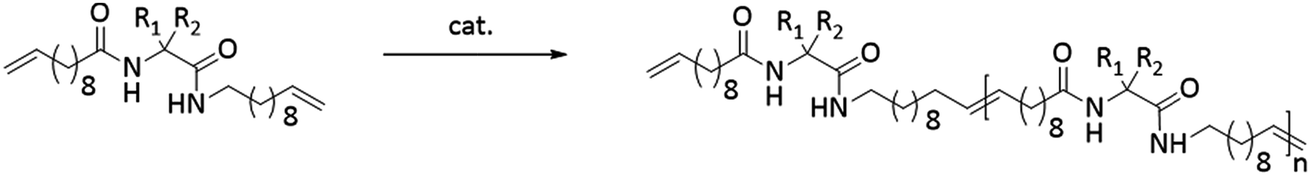
We here report on the introduction of five different amino-acids (chiral/achiral, polar/non-polar) as basic bio-building blocks into the main chain of PE-polymers, placed precisely at every 19th carbon within the main chain by functionalizing the N- and C-terminus of the amino acid (see Fig. 1). Our main interest concerned the synthetic/preparative issue, most of all how ADMET will comply with different amino acids to furnish – after hydrogenation – the final polymers with precisely placed amino acids. On the other hand we were interested in the structure formation of such polymers in the solid (semicrystalline) state: which amino acid will allow crystallization of the poly(alkyl)-chain, which one will inhibit the crystal-formation, and what will happen to the crystal structure if formed.
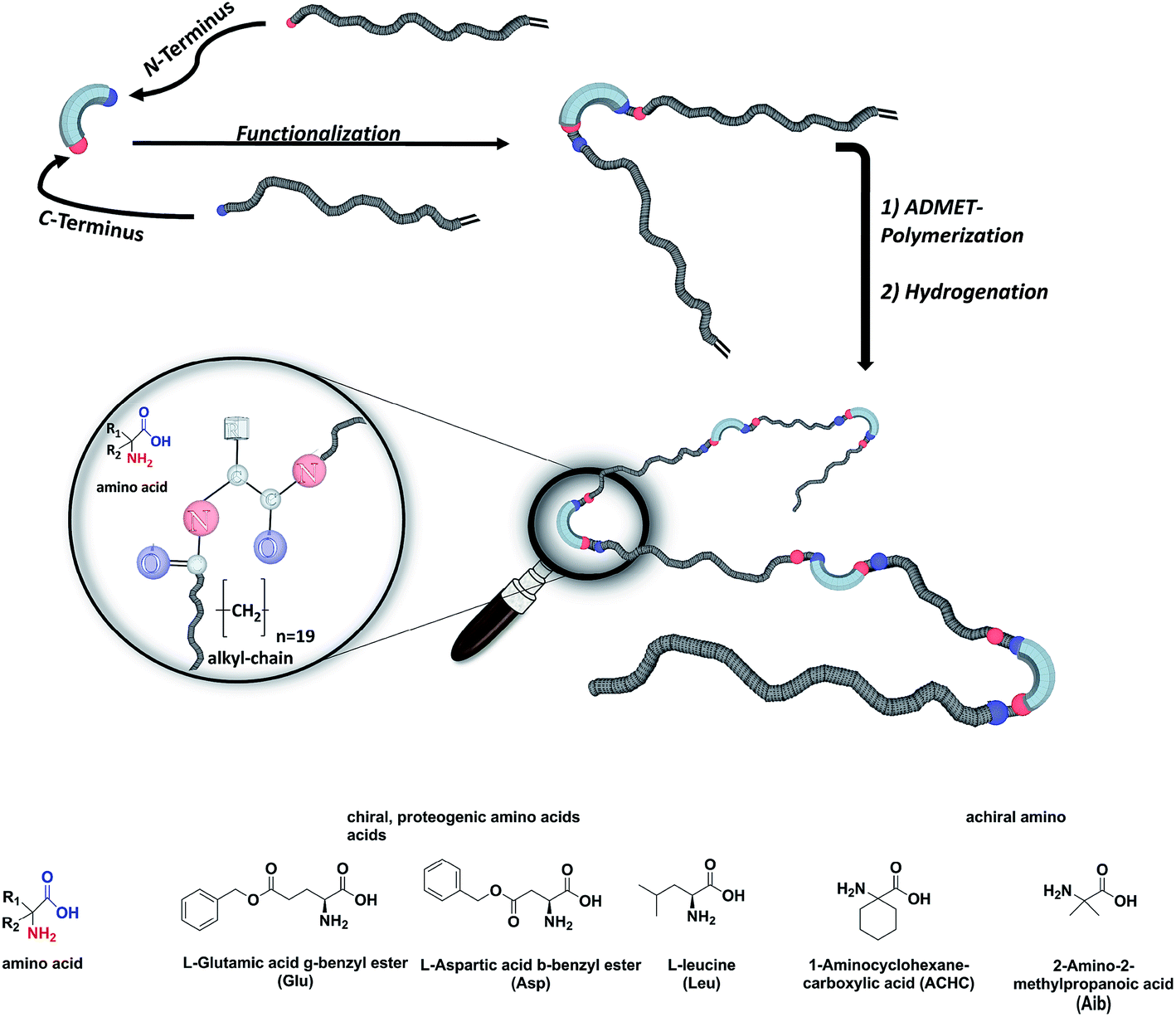 | ||
| Fig. 1 Schematic representation of the synthesis of PE precision polymers with amino acid defects. | ||
Experimental section
Materials
Amino acids L-glutamic acid γ-benzyl ester and L-aspartic acid β-benzyl ester were purchased from carbolution-chemicals, 1-aminocyclohexanecarboxylic acid, 2-aminoisobutyric acid and LiAlH4 were purchased from ABCR and 10-undecenoyl chloride was purchased from Acros Organics. All other chemicals were purchased from Sigma-Aldrich. All chemicals were used without further preparations or purification steps. The solvents which were used for reactions were dried and freshly distilled before using. THF was dried and distilled over sodium and benzophenone under N2 atmosphere, for drying of dichloromethane (DCM) and dimethyl formamide (DMF) CaH2 was used. For the reactions under protective gas atmosphere nitrogen (N2) was used, which was purchased from LINDE.Instrument and measurements
Nuclear magnetic resonance (NMR) spectra were recorded at 27 °C on a NMR spectrometer of VARIAN. The 1H-NMR spectroscopy were performed at 400 MHz and 13C-NMR spectroscopy at 100 MHz. For all samples CDCl3 or DMSO-d6 were used as solvents, in case of the hydrogenated polymers 4a–4e 15 vol% TFA were added. The splitting patterns were subdivided in singuletts (s), dubletts (d), triplets (t) and multiplett (m) during analysis. Chemical shifts (δ) were given in ppm referred to the solvent residue peak and tetramethylsilane were used as internal standard.Infrared-spectroscopy (IR) was measured by VERTEX 70 IR spectrometer of Bruker by using a single reflex-diamond-ATR-unit. The absorption bands are reported in cm−1 in an area of 400–4000 cm−1. The software OPUS was used for interpretation of the obtained bands.
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF-MS) measurements were performed on a Bruker Autoflex III system (Bruker Daltonics) using a nitrogen laser operating at a wave length of λ = 337 nm in reflection or linear modes. The polymer was dissolved in THF with a concentration of 10 mg mL−1. The matrix dithranol was dissolved in THF with a concentration of 10 mg mL−1, for the salt KTFA a concentration of 5 mg mL−1 in THF was used. The volume ratio of the components was matrix![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :analyt:salt was for all measurements 100:10:1. Calibration was done with PEG-standard (Mp = 2000 g mol−1 and 4200 g mol−1). Data evaluation was carried out on flexAnalysis software (3.4).
:analyt:salt was for all measurements 100:10:1. Calibration was done with PEG-standard (Mp = 2000 g mol−1 and 4200 g mol−1). Data evaluation was carried out on flexAnalysis software (3.4).
For gel permeation chromatography (GPC) measurements a Viscotek GPCmax VE 2002 with a HHR-HGuard-17369 and a GMH HR-N-18055 column in tetrahydrofuran (THF) was used. Measurements were done at 22 °C and concentration of the samples were c = 5 mg ml−1. Detection was realized by refractive index with a VE3580 RI detector of Viscotek at a temperature of 35 °C and a flow rate of 1 mL min−1. External calibration was done with polystyrene (Mp = 1000–115000 g mol−1).
For differential scanning calorimetry (DSC) measurements a NETZSCH DC 204F1 Phoenix, which was calibrated with indium, tin, bismuth, and zinc was used. Nitrogen was used as purge gas and all samples (3g–8g) were placed in standard aluminum pans. For analysis of obtained data Netzsch Proteus – Thermal Analysis (version 5.2.1.) and OriginPro 8G was used. For calculation of the crystallinity α equation below was used:
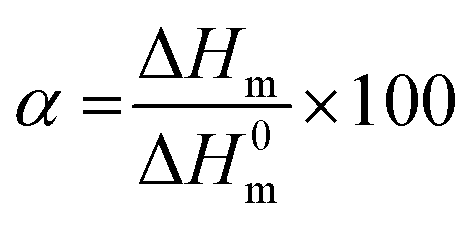
X-ray diffraction (XRD) 2D patterns were detected on Vantec 500 detector (Bruker AXS) with a detector to sample distance of 9.00 cm. Measurements of the samples were done as droplet which was heated up on a glass plate to isotropic state followed by cooling the sample with a define cooling rate of 5 K min−1 on a temperature-controlled heating stage. Measuring temperature for 4a was 60.0 °C and for 4b 70.0 °C, measuring time for both samples were 30 min.
Dialysis tube (MWCO: 1000 g mol−1) was purchased from Carl Roth GmbH.
Monomer synthesis
General procedure for the amino acids bearing a benzyl protection group in the side chain (method A) (1a–1b). N-terminus functionalization was realized by the Einhorn method for the amino acids bearing a benzyl protection group in the side chain. Therefore, the protected amino acid (1 eq.) was dissolved in THF (12 mL) and pyridine (10 mL) was added to the solution. The mixture was cooled to 0 °C and 10-undecenoyl chloride (1.3 eq.) was added dropwise to the solution, which then was stirred for 24 h at room temperature. Then the reaction mixture was poured into cold 1 M HCl-solution (300 mL) and diethyl ether (200 mL) was added. The organic phase were separated, extracted two times with 1 M HCl solution (2 × 100 mL) and dried over Na2SO4. After filtration the mixture was concentrated in vacuum and recrystallized three times in ethyl acetate
:n-hexane (1:40) mixture to yield a white solid 1a–1b (72–76%).N-Glu (1a): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 1.27 (m, 10H, Hd − Hh), 1.60 (m, 2H, Hi), 2.02–2.20 (m, 6H, H4 + Hc + Hj), 2.49 (m, 1H, H3), 2.57 (m, 1H, H3), 4.54 (m, 1H, H2), 4.93 (m, 2H, Ha), 5.12 (s, 2H, H6), 5.79 (m, 1H, Hb), 6.56 (d, 3JH,H = 7.2 Hz, 1H, H13), 7.31–7.45 (m, 5H, H8 − H12).
N-Glu (1a): 13C-NMR (CDCl3, 27 °C, 100 MHz): δ [ppm] 25.4 (Ci), 26.5 (C3), 28.9–29.5 (Cd − Ch), 30.5 (C4), 33.7 (Cc), 36.3 (Cj), 52.3 (C2), 66.9 (C6), 114.1 (Ca), 128.2–128.7 (C8 − C12), 135.5 (C7), 139.1 (Cb), 171.5 (C1), 173.6 (C5), 174.1 (Ck).
N-Asp (1b): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 1.27 (m, 10H, Hd − Hh), 1.61 (m, 2H, Hi), 2.02 (m, 2H, Hc), 2.20 (m, 2H, Hj), 2.92 (m, 1H, H3), 3.09 (m, 1H, H3), 4.91 (m, 1H, H2), 4.96 (m, 2H, Ha), 5.14 (s, 2H, H5), 5.81 (m, 1H, Hb), 6.56 (d, 3JH,H = 7.2 Hz, 1H, H12), 7.31–7.45 (m, 5H, H7 − H11).
N-Asp (1b): 13C-NMR (CDCl3, 27 °C, 100 MHz): δ [ppm] 25.4 (Ci), 28.9–29.3 (Cd − Ch), 33.8 (Cc), 36.3 (Cj), 36.5 (C3), 48.5 (C2), 67.0 (C5), 114.1 (Ca), 128.2–128.6 (C7 − C11), 135.23 (C6), 139.1 (Cb), 171.1 (C1), 173.8 (C4), 174.1 (Ck).
General procedure for the amino acids without a protection group in the side chain (method B) (1c–1e). N-terminus functionalization was realized by Schotten–Baumann synthesis for the water soluble amino acids. The amino acid (1 eq.) and NaOH (2 eq.) was dissolved in water (20 mL) and the mixture was cooled in an ice-bath. 10-Undecenoyl chloride (1.1 eq.), dissolved in dichloromethane (20 mL), was added dropwise to the solution which was allowed to stir at room temperature for 24 h. The mixture was poured into cold 1 M HCl-solution (200 mL) and the organic phase were then separated, dried over Na2SO4, filtrated and concentrated in vacuum. The crude product was recrystallized three times in ethyl acetate
:n-hexane (1:40) mixture to yield a white solid 1c–1e (64–75%).N-Leu (1c): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 0.95 (m, 6H, H5 + H6), 1.28 (m, 10H, Hd − Hh), 1.60–1.71 (m, 5H, Hi + H3 + H4), 2.02 (m, 2H, Hc), 2.23 (m, 2H, Hj), 4.61 (m, 1H, H2), 4.94 (m, 2H, Ha), 5.79 (m, 1H, Hb), 5.96 (m, 1H, H7).
N-Leu (1c): 13C-NMR (CDCl3, 27 °C, 100 MHz): δ [ppm] 21.9–22.8 (C5 + C6), 24.9 (C4), 25.5 (Ci), 28.9–29.5 (Cd − Ch), 33.7 (Cc), 36.5 (Cj), 41.1 (C3), 50.8 (C2), 114.1 (Ca), 139.1 (Cb), 173.9 (C1), 176.3 (Ck).
N-Aib (1d): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 1.27–1.36 (m, 10H, Hd − Hh), 1.57–1.62 (m, 8H, H2 + H3 + Hi), 2.04 (m, 4H, Hc), 2.20 (m, 2H, Hj), 4.96 (m, 2H, Ha), 5.79 (m, 1H, Hb), 6.02 (m, 1H, H5).
N-Aib (1d): 13C-NMR (CDCl3, 27 °C, 100 MHz): δ [ppm] 24.9 (C2 + C3), 25.5 (Ci), 28.9–29.5 (Cd − Ch), 33.7 (Cc), 36.8 (Cj), 57.0 (C1), 114.1 (Ca), 139.1 (Cb), 174.4 (Ck), 177.2 (C4).
N-ACHC (1e): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 1.29–1.45 (m, 12H, Hd − Hi), 1.66 (m, 6H, H3 + H4 + H5), 1.89 (m, 2H, H2 + H6), 2.04 (m, 4H, Hc + H2 + H6), 2.26 (m, 2H, Hj), 4.94 (m, 2H, Ha), 5.69 (m, 1H, H7), 5.79 (m, 1H, Hb).
N-ACHC (1e): 13C-NMR (CDCl3, 27 °C, 100 MHz): δ [ppm] 21.3 (C3 + C5), 25.1–25.5 (Ci + C4), 28.9–29.5 (Cd − Ch), 32.0–33.7 (C2 + C6 + Cc), 36.6 (Cj), 59.7 (C1), 114.1 (Ca), 139.1 (Cb), 175.1 (Ck), 176.0 (C4).
General procedure for chiral AA (2a–2c). Reactions were performed under N2 atmosphere. N-terminus functionalized amino acid 1a–1c (1.0 eq.) was dissolved in dried dichloromethane (20 mL) and then A2 (1.0 eq.) was added to the mixture. The solution was cooled down to 0 °C and the coupling reagents DIPEA (3.6 eq.), HOBt (13% H2O; 1.6 eq.) and EDC·HCl (1.6 eq.) were added successively. The mixture was allowed to stir at room temperature for 4 hours before it was refluxed overnight. Then the solution was cooled and extracted with brine (20 mL), 1 M HCl (20 mL), brine (20 mL), saturated NaHCO3-solution (20 mL) and brine (20 mL). The organic phase was concentrated under vacuum and the crude product was dissolved in THF (5 mL), precipitated in cold NaHCO3 (150 mL) solution and isolated by filtration. Afterwards the obtained brown solid was dissolved in Et2O (20 mL), dried over Na2SO4, filtrated and concentrated in vacuum to yield products 2a–2c (63–86%).
N- + C-Glu (2a): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 1.26–1.34 (m, 20H, Hd − Hh), 1.44–1.60 (m, 4H, Hl + Hi), 2.03–2.20 (m, 6H, H4 + Hc + Hj), 2.40 (m, 1H, H3), 2.54 (m, 1H, H3), 3.18 (m, 2H, Hm) 4.42 (m, 1H, H2), 4.93 (m, 4H, Ha), 5.10 (s, 2H, H6), 5.77 (m, 2H, Hb), 6.43 (d, 3JH,H = 7.2 Hz, 1H, H13), 7.31–7.45 (m, 5H, H8 − H12).
N- + C-Glu (2a): 13C-NMR (CDCl3, 27 °C, 100 MHz): δ [ppm] 25.5 (Ci), 26.8 (Ci), 27.8 (C3), 28.9–29.5 (Cd − Ch + Cl), 30.6 (C4), 33.8 (Cc), 36.5 (Cj), 39.6 (Cm), 52.3 (C2), 66.6 (C6), 114.1 (Ca), 128.2–128.7 (C8 − C12), 135.8 (C7), 139.1 (Cb), 170.9 (C1), 173.4 (C5), 173.4 (Ck).
N- + C-Asp (2b): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 1.27–1.40 (m, 22H, Hd − Hh + Hl), 1.59 (m, 2H, Hi), 2.02–2.20 (m, 6H, H4 + Hc + Hj), 2.91 (m, 1H, H3), 3.08 (m, 1H, H3), 3.17 (m, 2H, Hm), 4.91 (m; 1H, H2), 4.94 (m, 2H, Ha), 5.14 (s, 2H, H5), 5.79 (m, 1H, Hb), 6.56 (d, 3JH,H = 7.2 Hz, 1H, H12), 7.31–7.45 (m, 5H, H7 − H11).
N- + C-Asp (2b): 13C-NMR (CDCl3, 27 °C, 100 MHz): δ [ppm] 25.5 (Ci), 26.8 (Ci), 28.9–29.3 (Cd − Ch + Cl), 33.7 (Cc), 35.7 (Cj), 36.5 (C3), 39.6 (Cm), 49.1 (C2), 66.9 (C5), 114.1 (Ca), 128.2–128.6 (C7 − C11), 135.4 (C6), 139.2 (Cb), 170.1 (C1), 172.1 (C4), 173.3 (Ck).
N- + C-Leu (2c): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 0.95 (m, 6H, H5 + H6), 1.28–1.43 (m, 22H, Hd − Hh + Hl), 1.60–1.71 (m, 5H, Hi + H3 + H4), 2.02 (m, 2H, Hc), 2.20 (m, 2H, Hj), 3.22 (m, 2H, Hm), 4.61 (m, 1H, H2), 4.94 (m, 2H, Ha), 5.79 (m, 1H, Hb), 5.96 (m, 1H, H7).
N- + C-Leu (2c): 13C-NMR (CDCl3, 27 °C, 100 MHz): δ [ppm] 21.9–22.6 (C5 + C6), 25.0 (C4), 25.6 (Ci), 26.8 (Ci), 28.9–29.5 (Cd − Ch), 33.7 (Cc), 36.6 (Cj), 39.5 (Cm), 41.1 (C3), 51.6 (C2), 114.1 (Ca), 139.1 (Cb), 172.0 (C1), 173.1 (Ck).
General procedure for achiral AA (2d–2e). Reactions were performed under N2 atmosphere. To a solution of N-terminus functionalized amino acids 1d–1e (1.0 eq.) and A2 (1.0 eq.) in 0 °C cold dried dichloromethane (20 mL) DIPEA (4.5 eq.), DMAP (0.02 eq.) and EDC·HCl (2.6 eq.) were added successively. The solution was heated to reflux for 24 h followed by extraction with brine (20 mL), 1 M HCl (20 mL), brine (20 mL), saturated NaHCO3-solution (20 mL) and brine (20 mL). The organic layer was separated, dried over Na2SO4 and concentrated in vacuum to yield 2d–2e (78–82%).
N- + C-Aib (2d): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 1.27–1.36 (m, 20H, Hd − Hh), 1.50 (m, 2H, Hl), 1.55–1.62 (m, 8H, H2 + H3 + Hi), 2.04 (m, 4H, Hc), 2.16 (m, 2H, Hj), 3.23 (m, 2H, Hm), 4.96 (m, 2H, Ha), 5.81 (m, 1H, Hb), 6.08 (m, 1H, H5), 6.65 (m, 1H, H5).
N- + C-Aib (2d): 13C-NMR (CDCl3, 27 °C, 100 MHz): δ [ppm] 24.8 (C2 + C3), 25.5 (Ci), 26.8 (Ci), 28.9–29.5 (Cd − Ch), 33.8 (Cc), 37.4 (Cj), 39.8 (Cj), 57.4 (C1), 114.1 (Ca), 139.1 (Cb), 173.4 (Ck), 174.5 (C4).
N- + C-ACHC (2e): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 1.29–1.47 (m, 24H, Hd − Hi + Hl), 1.64 (m, 6H, H3 + H4 + H5), 1.88 (m, 2H, H2 + H6), 2.04 (m, 6H, Hc + H2 + H6), 2.21 (m, 2H, Hj), 3.20 (m, 2H, Hm), 4.97 (m, 4H, Ha), 5.30 (m, 1H, H7), 5.79 (m, 2H, Hb).
N- + C-ACHC (2e): 13C-NMR (CDCl3, 27 °C, 100 MHz): δ [ppm] 21.6 (C3 + C5), 25.1–25.5 (Ci + C4), 26.9 (Ci), 28.9–29.5 (Cd − Ch + Cl), 32.0–33.8 (C2 + C6 + Cc), 37.5 (Cj), 39.5 (Cm), 59.7 (C1), 114.1 (Ca), 139.1 (Cb), 175.1 (Ck), 176.0 (C4).
:1 monomer to catalyst ratio) was added to the bulk. Vacuum and repeated cycles flushing with nitrogen are applied during the reaction to remove evolving ethylene, further small amount of catalyst were additionally added to the bulk-reaction mixture after several hours. Due to increasing viscosity during polymerization the reaction temperature was increased stepwise starting from 65 °C up to 165 °C (for the last hour) and afterwards the reaction was quenched by adding THF and precipitating of the obtained polymer into cold MeOH to yield 3a–3e (61–87%).ADMET-Glu (3a): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 1.26–1.34 (m, Hrep.unit, Hd − Hh), 1.44–1.59 (m, Hrep.unit, Hi + Hl), 1.95–2.01 (m, Hrep.unit, H4 + Hc), 2.16 (m, Hrep.unit, Hj), 2.41 (m, Hrep.unit, H3), 2.55 (m, Hrep.unit, H3), 3.20 (m, Hrep.unit, Hm) 4.41 (m, Hrep.unit, H2), 4.96 (m, 4H, Ha), 5.10 (s, Hrep.unit, H6), 5.36 (m, Hrep.unit, Hx + Hy), 5.79 (m, 2H, Hb), 7.34 (m, Hrep.unit, H8 − H12).
ADMET-Asp (3b):1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 1.19–1.28 (m, Hrep.Einheit, Hd − Hh), 1.43 (m, Hrep.Einheit, Hl), 1.58 (m, Hrep.Einheit, Hi), 1.95 (m, Hrep.Einheit, Hc), 2.18 (m, Hrep.Einheit, Hj), 2.68 (m, Hrep.Einheit, H3), 2.90 (m, Hrep.Einheit, H3), 3.16 (m, Hrep.Einheit, Hm), 4.78 (m, Hrep.Einheit, H2), 4.96 (m, 4H, Ha), 5.13 (m, Hrep.Einheit, H5), 5.36 (m, Hrep.Einheit, Hx + Hy), 5.79 (m, 2H, Hb), 7.33 (m, Hrep.Einheit, H7 − H11).
ADMET-Leu (3c): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 0.87 (m, Hrep.Einheit, H5 + H6), 1.19–1.38 (m, 12H, Hd − Hh + Hl), 1.46–1.66 (m, Hrep.Einheit, H3 + H4 + Hi), 2.03 (m, Hrep.Einheit, Hc), 2.17 (m, Hrep.Einheit, Hj), 3.19 (m, Hrep.Einheit, Hl), 4.48 (m, Hrep.Einheit, H2), 4.90 (m, 4H, Ha), 5.35 (m, Hrep.Einheit, Hx + Hy), 5.78 (m, 2H, Hb).
ADMET-Aib (3d): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 1.27–1.36 (m, Hrep.unit, Hd − Hh), 1.48 (m, Hrep.unit, Hl), 1.55–1.59 (m, Hrep.unit, H2 + H3 + Hi), 1.94 (m, Hrep.unit, Hc), 2.16 (m, Hrep.unit, Hj), 3.23 (m, Hrep.unit, Hm), 4.96 (m, 2H, Ha), 5.36 (m, Hrep.unit, Hx + Hy), 5.76 (m, 1H, Hb), 6.08 (m, Hrep.unit, H5), 6.65 (m, Hrep.unit, H6).
ADMET-ACHC (3e): 1H-NMR (CDCl3, 27 °C, 400 MHz): δ [ppm] 1.23–1.47 (m, Hrep.unit, Hd − Hh + Hl + Hi), 1.62 (m, Hrep.unit, H3 + H4 + H5), 1.86 (m, Hrep.unit, H2 + H6), 1.94–2.12 (m, Hrep.unit, Hc + H2 + H6), 2.21 (m, Hrep.unit, Hj), 3.20 (m, Hrep.unit, Hm), 4.91 (m, 4H, Ha), 5.35 (m, Hrep.unit, Hx + Hy), 5.78 (m, 2H, Hb).
General procedure for hydrogenation and deprotection of the polymers bearing benzyl-protecting group in the side chain (4a–4b). To a solution of the polymer 3a–3b (1.0 eq.) in DMF TsNHNH2 (5.0 eq. per double bond) and DIPEA (5.0 eq.) were added. The solution was heated up to 150 °C for 6 hours. Then the solution was cooled to 85 °C, NaOH (2.0 eq. per protection group) was added and the resulting mixture was stirred overnight. The reaction mixture was cooled to room temperature and dialyzed against MeOH for 3 days to yield the hydrogenated and deprotected polymers 4a–4b (72–79%).
ADMET-Glu H (4a): 1H-NMR (CDCl3 + 15 vol% TFA, 27 °C, 400 MHz): δ [ppm] 0.87 (m, 6H, Hz), 1.25–1.34 (m, Hrep.unit, Hb − Hh + Hx + Hy), 1.54–1.60 (m, Hrep.unit, Hi + Hl), 2.00–2.16 (m, Hrep.unit, H3), 2.39 (m, Hrep.unit, H4), 2.54 (m, Hrep.unit, Hj), 3.29 (m, Hrep.unit, Hm), 4.73 (m, Hrep.unit, H2), 7.21 (m, Hrep.unit, H13), 7.81 (m, Hrep.unit, H14).
ADMET-Asp H (4b):1H-NMR (CDCl3 + 15 vol% TFA, 27 °C, 400 MHz): δ [ppm] 0.87 (m, 6H, Hz), 1.19–1.28 (m, Hrep.unit, Hb − Hh + Hx + Hy), 1.59 (m, Hrep.Einheit, Hi + Hl), 2.35 (m, Hrep.Einheit, Hj), 3.01 (m, Hrep.Einheit, H3), 3.24 (m, Hrep.Einheit, H3), 3.59 (m, Hrep.Einheit, Hm), 4.50 (m, Hrep.Einheit, H2), 7.05 (m, Hrep.unit, H12 + H13).
General procedure for hydrogenation of the polymers without a protecting group in the side chain (4c–4e). To a solution of the polymer 3c–3e (1.0 eq.) in DMF TsNHNH2 (5.0 eq. per double bond) and DIPEA (5.0 eq.) were added. The solution was heated up to 150 °C for 6 hours. The resulting mixture was cooled to room temperature and dialyzed against MeOH for 3 days to yield the hydrogenated polymers 4c–4e (67–83%).
ADMET-Leu H (4c): 1H-NMR (CDCl3 + 15 vol% TFA, 27 °C, 400 MHz): δ [ppm] 0.89 (m, Hrep.Einheit, H5 + H6 + Hz), 1.19–1.38 (m, 12H, Hb − Hh + Hx + Hy), 1.50–1.59 (m, Hrep.Einheit, H3 + H4 + Hi + Hl), 2.32 (m, Hrep.Einheit, Hj), 3.25 (m, Hrep.Einheit, Hm), 4.54 (m, Hrep.Einheit, H2), 7.19 (m, Hrep.Einheit, H7), 7.39 (m, Hrep.Einheit, H8).
ADMET-Aib H (4d): 1H-NMR (CDCl3 + 15 vol% TFA, 27 °C, 400 MHz): δ [ppm] 0.88 (m, 6H, Hz), 1.25 (m, Hrep.unit, Hb − Hh + Hl + Hx + Hy), 1.55–1.59 (m, Hrep.unit, H2 + H3 + Hi), 2.28 (m, Hrep.unit, Hj), 3.29 (m, Hrep.unit, Hm), 6.70 (m, Hrep.unit, H5), 6.99 (m, Hrep.unit, H6).
ADMET-ACHC H (4e): 1H-NMR (CDCl3 + 15 vol% TFA, 27 °C, 400 MHz): δ [ppm] 0.88 (m, 6H, Hz), 1.27–1.46 (m, Hrep.unit, Hb − Hh + Hl + Hx + Hy + Hi), 1.74 (m, Hrep.unit, H3 + H4 + H5), 1.96–2.31 (m, Hrep.unit, Hj + H2 + H6), 3.37 (m, Hrep.unit, Hm), 7.12–7.26 (m, Hrep.unit, H7 + H8).
Results and discussion
The synthesis of the polymers was accomplished by ADMET polymerization of the chosen N- and C-terminus ω-alkene-chain functionalized amino acids. For the investigation of the influence on the chiral/achiral and polar/nonpolar nature of the amino acids on the crystallization behavior, chiral/polar amino acids glutamic (Glu) and aspartic acid (Asp), chiral/unpolar L-leucine (Leu) and achiral/unpolar 2-amino-2-methylpropanoic (Aib) acid and 1-aminocyclohexane-carboxylic acid (ACHC) were functionalized with terminal ω-alkenyl-group, polymerized by ADMET and hydrogenated (Fig. 1).Monomer synthesis
Conceptually, the amino acids were introduced in the polymer structure by the fixation of two ω-alkenyl-chains of equal length on either side. Therefore, N- and C-terminus functionalization was realized by reaction of the amino acid with 10-undecenoyl chloride and afterwards coupling with 10-undecen-1-amine (A2) (see Scheme 1).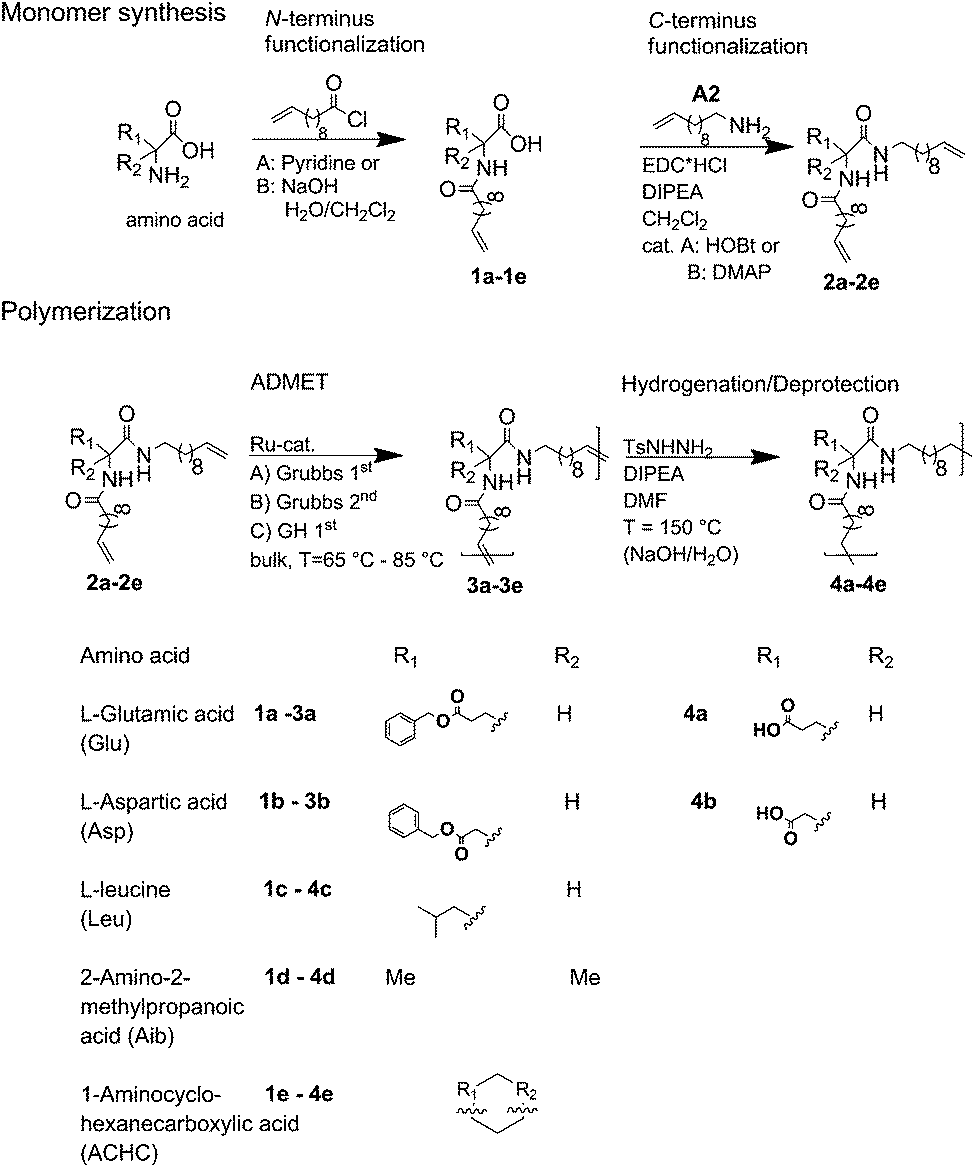 | ||
| Scheme 1 Synthesis for (a) N- + C-terminus functionalized monomers 2a–2e and (b) ADMET-polymers 3a–3e and hydrogenated/deprotected polymers 4a–4e. | ||
The chosen method and the yields of the functionalization reactions for the different amino acids are shown in Table 1.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Amino acid | R1 | R2 | N-terminus functionalization | C-terminus functionalization | ||||
| Method | Product | Yield (%) | Cat. | Product | Yield (%) | |||
| Glu |  |
H | A | 1a | 76 | A | 2a | 86 |
| Asp | 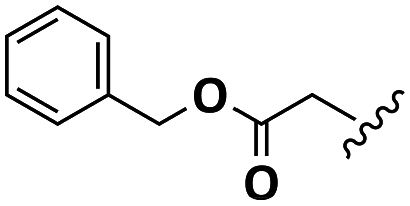 |
H | A | 1b | 72 | A | 2b | 79 |
| Leu | 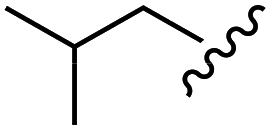 |
H | B | 1c | 75 | A | 2c | 63 |
| Aib | Me | Me | B | 1d | 67 | B | 2d | 78 |
| ACHC | 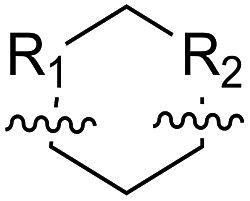 |
B | 1e | 64 | B | 2e | 82 | |
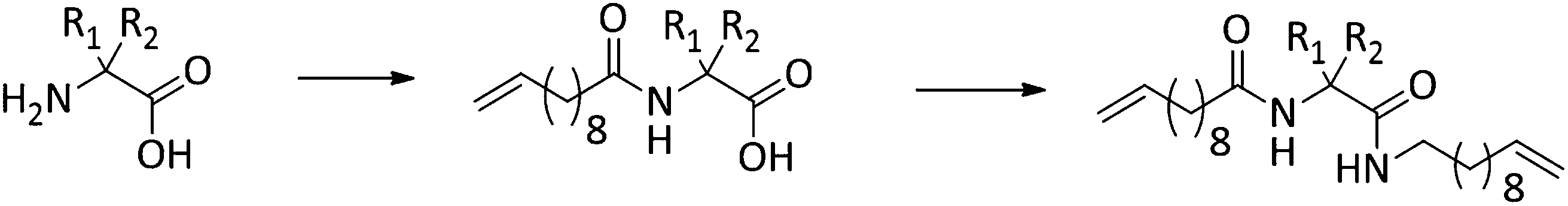
For N-terminus functionalization pyridine was used for the protected L-glutamic acid and L-aspartic acid 1a–1b or alternatively via a two phase system of aqueous NaOH/CH2Cl2 mixture (Schotten–Baumann-method) for the synthesis of 1c–1e. Free carboxylic group in products 1a–1e could then be reacted with 10-undecen-1-amine (A2), which was synthesized before in a two-step synthesis by reaction of 10-undecenoyl chloride with ammoniumhydroxid to the corresponding amide (A1), followed by reduction with LiAlH4 as described in the literature.45 All synthesized monomers 1a–1e and 2a–2b were analysed by 1H, 13C-NMR and ESI-ToF-MS spectroscopy (see Fig. S1–S28 ESI†), A1–A2 were analysed by 1H and 13C-NMR spectroscopy (see Fig. S29–S30 ESI†).
ADMET-polymerization and hydrogenation of the polymers
Synthesis of the precision PE-polymers was realized by ADMET-polymerization followed by hydrogenation with p-toulenesulfonhydrazide (TsNHNH2) as shown in Scheme 1.Polymerization of the monomers 2a–2e can be done as a bulk polymerization due to the low melting points. The solid monomers were heated up in an oil bath to 65 °C till they became completely liquid.16 The appropriate amount of catalyst (100:1 monomer to catalyst ratio) was added to the monomer-melt in which the same amount of catalyst was additionally added to the bulk after several hours. Due to the increasing viscosity during polymerization the reaction temperature was increased stepwise from 85 °C up to 165 °C. Finally the reaction was quenched by adding THF and precipitating the obtained polymer into cold MeOH.
Due to different activity and isomerization rates46–49 Grubbs Catalyst first generation (Grubbs 1st), second generation (Grubbs 2nd) and Hoveyda–Grubbs Catalyst first generation (GH 1st) were tested for the chosen reaction conditions. In Table 2 the results of the molecular weights (obtained by NMR and GPC as well as the PDI's), the yields and the obtained trans:cis ratio of the internal double bonds for the different catalyst are displayed. A low isomerization rate48 and good yields for monomers 3a–3c by using Grubbs 1st catalyst were obtained. GH 1st catalyst under complete oxygen-free conditions also achieved good results with respect to yield and molecular weights of the products, whereas Grubbs 2nd catalyst was found to be the catalyst resulting in the lowest molecular weights. Probing the optimized conditions described by Wagener et al.16 molecular weights as high as 22 kDa were obtained – higher molecular weights were not obtained. This might be due to the high temperature at which most of the monomers are melting (often above 165 °C), which is limiting the activity of the catalysts. Even though such high reaction temperatures could lead to various side reactions, only a low isomerization rate was observed in MALDI-Tof-MS analysis.
:cis ratio of the internal double bonds for ADMET-polymerization of the monomers 2a–2e to polymers 3a–3e by using different types of catalyst
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Polymer | R1 | R2 | Cat. | Mn GPC (Da) | Mn NMR (Da) | DP (n) | PDI | trans:cis ratio |
Yield (%) |
| 3a | 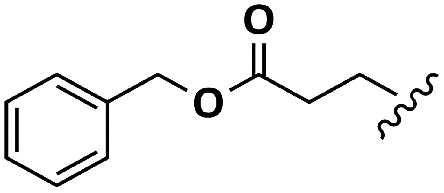 |
H | Grubbs 1st | 7600 | 9200 | 17 | 1.7 | 68:32 |
77 |
| Grubbs 2nd | 700 | 800 | 2 | 1.5 | 89:11 |
67 | |||
| GH 1st | 2300 | 2400 | 5 | 2.2 | 78:22 |
70 | |||
| 3b | 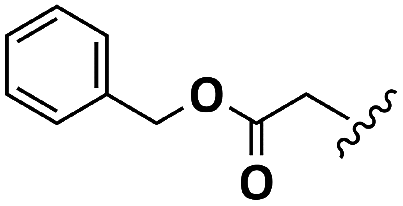 |
H | Grubbs 1st | 10500 |
22000 |
41 | 1.9 | 68:32 |
87 |
| Grubbs 2nd | 800 | 700 | 2 | 1.4 | 82:18 |
61 | |||
| GH 1st | 4000 | 5400 | 10 | 1.5 | 72:28 |
72 | |||
| 3c | 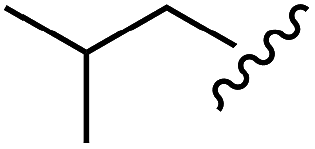 |
H | Grubbs 1st | 3500 | 4300 | 10 | 1.8 | 75:25 |
73 |
| Grubbs 2nd | 1000 | 700 | 2 | 1.4 | 78:22 |
69 | |||
| GH 1st | 4100 | 5500 | 12 | 1.6 | 76:24 |
71 | |||
| 3d | Me | Me | Grubbs 1st | 2500 | 3000 | 7 | 1.4 | 79:21 |
73 |
| 3e | 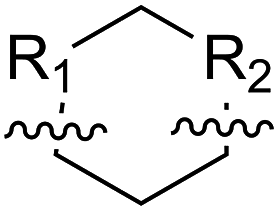 |
Grubbs 1st | 9400 | 14600 |
32 | 1.9 | 76:24 |
70 | |
| GH 1st | 11500 |
18200 |
40 | 1.9 | 79:21 |
62 | |||
Hydrogenation of the terminal and internal double bonds in the backbone to yield the polymer 4a–4e was realized by reaction of the polymers with p-toluenesulfonhydrazide (TsNHNH2) and DIPEA in DMF at 150 °C according to literature50–53 (see Scheme 1). For the hydrogenation the ADMET polymers 3a–3e synthesized with Grubbs 1st generation catalyst were used and molecular weights from 3100 g mol−1 to 18500 g mol−1 were obtained (Table 3). Reactions were stopped after 6 h, complete deprotection of the asparagine and glutamic acid unit in the polymer backbone could be realized by subsequent ester hydrolysis with aqueous NaOH-solution. The resulting mixture was dialysed against MeOH for 3 days to remove the obtained p-toluenesulfonic acid and benzyl alcohol in case of 4a and 4b. During dialysis, the obtained hydrogenated polymers precipitated due to their complete insolubility in the organic solvents used. The monomer and polymer synthesis as well as hydrogenation/deprotection can be followed by 1H-NMR, MALDI-TOF-MS and IR as shown in Fig. 2 for the 1H-NMR data of monomer 2c (top), the ADMET-polymer 3c (middle) and the hydrogenated polymer 4c (bottom). Successful polymerization of the monomer can be proven by detection of the internal double bonds, which occur during ADMET-polymerization and can be found at 5.3 ppm. All other signals become broader and show higher intensity in relation to the terminal double bonds, which are still present after polymerization at 5.8 ppm and 4.9. Thus, this resonance can be used for calculation of the molecular weight by comparison of the ratio of the integrals for the terminal and internal double bonds, which can be determined as 10:1 which leads to a molecular weight of 4200 g mol−1 in 1H-NMR for 3c and is agreeable with the molecular weight of 3500 g mol−1 determined by GPC.
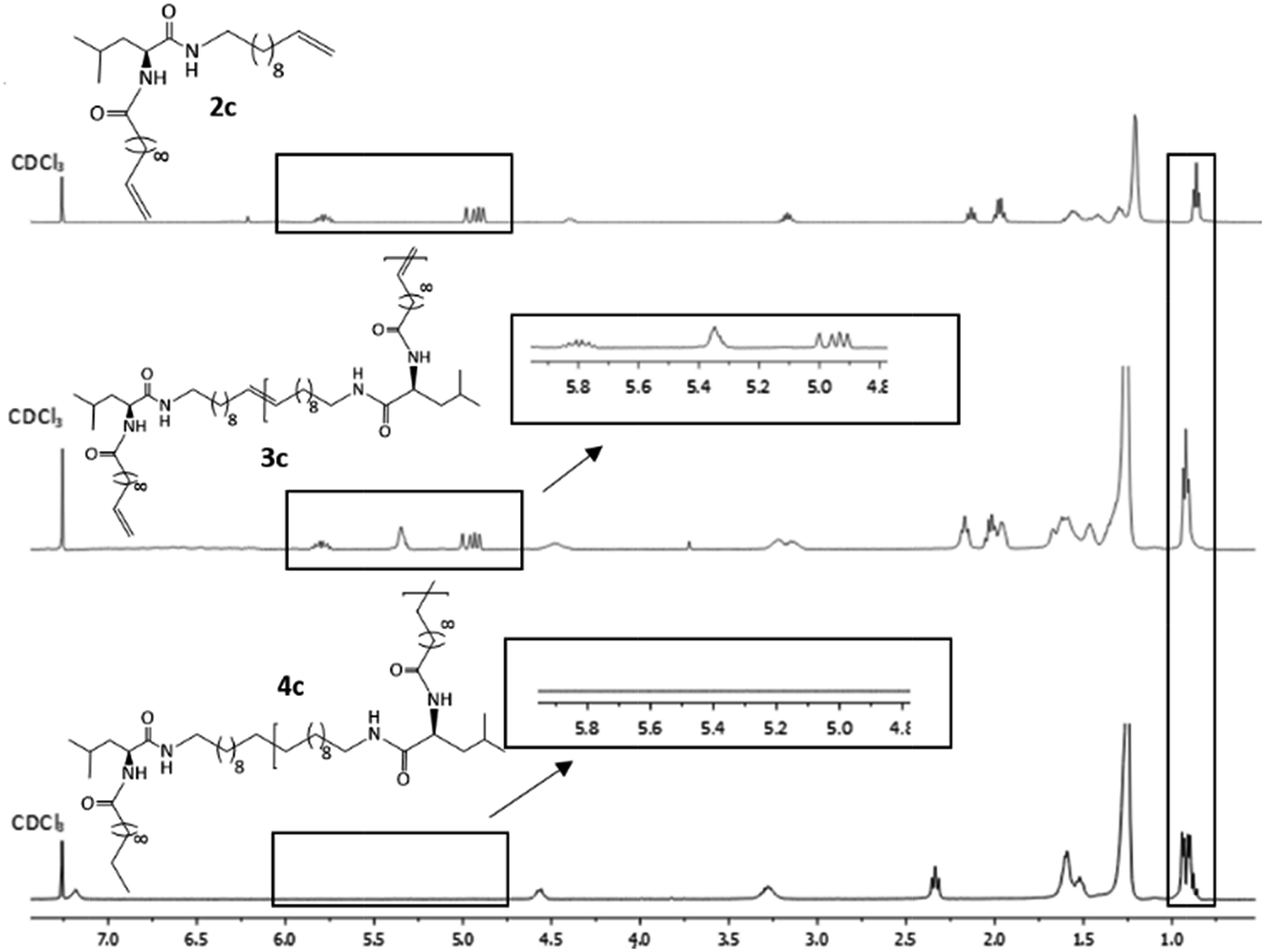 | ||
| Fig. 2 1H-NMR spectra of monomer 2c (top), ADMET-polymer 3c (middle) and hydrogenated polymer 4c (bottom) in CDCl3. For 4c 15 vol%. TFA was added. | ||
Due to complete insolubility of the final product in common organic solvents complete hydrogenation and deprotection can be demonstrated by 1H-NMR in CDCl3 only after adding 15 vol% TFA to the mixture. The characteristic signals for the internal double bonds at 5.3 ppm as well as for the terminal double bond at 5.8 ppm and 4.9 ppm are no longer present in the 1H-NMR spectra of 4c. Furthermore, protons next to the terminal double bonds at 2.0 ppm are disappearing and signals for the methyl groups at 0.9 ppm become broader due to protons of the resulting methyl end group. The successful deprotection of the benzyl-group in the case of 4a and 4b can be proven by the disappearance of the signals of the CH2-group next to the aromatic system at 5.1 ppm as well as the characteristic aromatic system signals at 7.3 ppm which is shown in Fig. 3. New methyl end-group can be detected at 0.9 ppm and was used for calculation of the molecular weight, indicating successful hydrogenation of the double bonds, whereas all other signals are still present showing slightly changed chemical shifts due to interaction with TFA molecules. Degree of polymerization (DP) calculated for the hydrogenated polymers 4a, 4b, 4d and 4e are always higher as for unsaturated polymers.
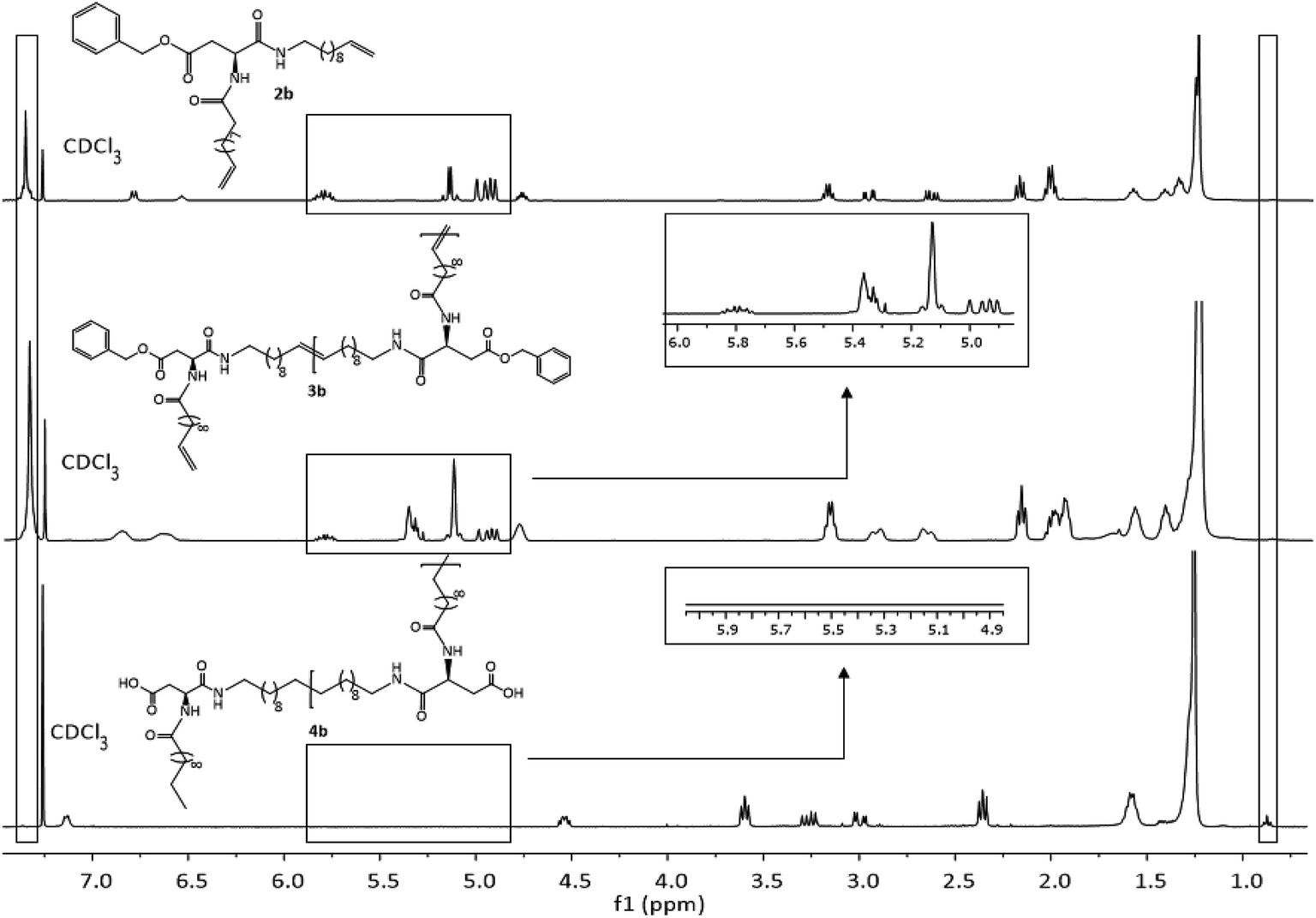 | ||
| Fig. 3 1H-NMR spectra of monomer 2b (top), ADMET-polymer 3b (middle) and hydrogenated polymer 4b (bottom) in CDCl3. For 4b 15 vol%. TFA was added. | ||
All other NMR-spectra of the ADMET- and hydrogenated polymers are shown in the ESI (Fig. S31–S36†). The successful hydrogenation can also be proven in IR-spectroscopy by disappearance of the deformation vibration band of the double bonds at 990 cm−1 and 915 cm−1 in the IR spectrum (Fig. S42–S51†).
MALDI-ToF-MS analysis
In Fig. 4 and 5 MALDi-ToF-MS spectra of the ADMET-polymer 3b and the hydrogenated polymer 4e are shown. All other MALDI-Tof-MS spectra are shown in Fig. S42–S51† with the exception of 3d, 4a and 4d, which cannot be detected under the chosen conditions. The MS spectra of the ADMET-polymer 3b (Fig. 4) shows a mass distribution from 1091 g mol−1 to 4165 g mol−1 (Fig. 4a) with a maximum peak at 1604.041 g mol−1. Two series can be assigned and the distance between signals of the same series amounts to 512 g mol−1. The distance between signals of different series is 14 g mol−1, in which the signal with the lower molecular weight has always a lower intensity (Fig. 4b). This signal indicates the loss of a CH2-group during ADMET-polymerization due to olefin isomerization, leading to polymers having a slightly different chain length of the alkyl chain,46,54 with a small amount of isomerization in e.g. 3b. The low isomerization rate can be proven by the appearance of only one series displaying isomerization products for 3b and three series for 4e (Fig. 5), whereas high isomerization rates have been observed at cysteine functionalized polymers, resulting in 6 series of isomerized side products.54 The signal at 2117 g mol−1 can be assigned to the polymer [E-M2-EK]+ with potassium as counter ion, which is confirmed by the agreement of the experimental and simulated data (see Fig. 4c). Thus, the obtained main series definitely indicates the expected polymer structure by ADMET polymerization including internal and external double bonds and the loss of ethylene in the repeating unit.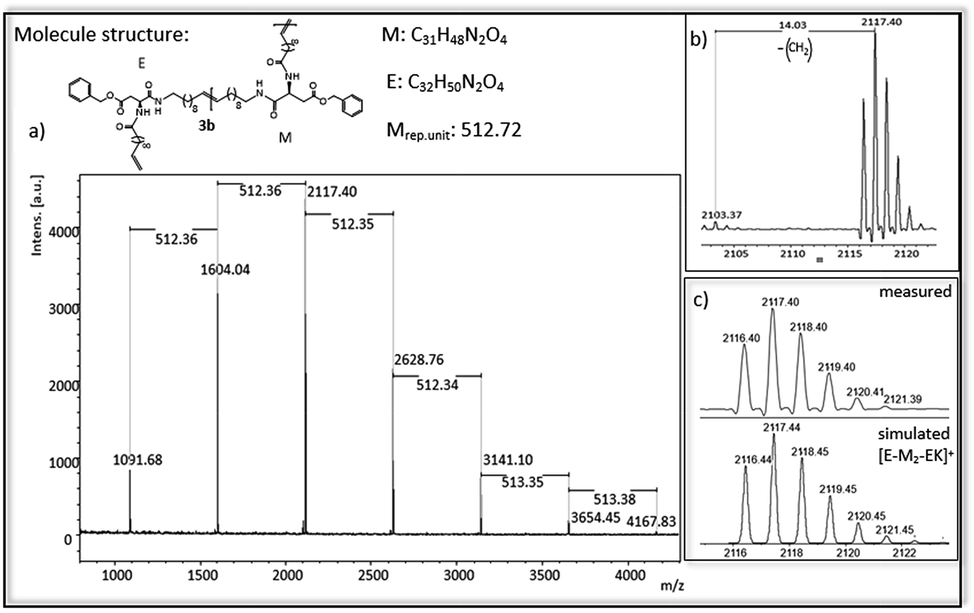 | ||
| Fig. 4 (a) MALDI-ToF-MS of ADMET polymer 3b (b) zoom in to [E-M2-EK]+ to show isomerization of the olefin (c) measured and simulated pattern for [E-M2-EK]+. | ||
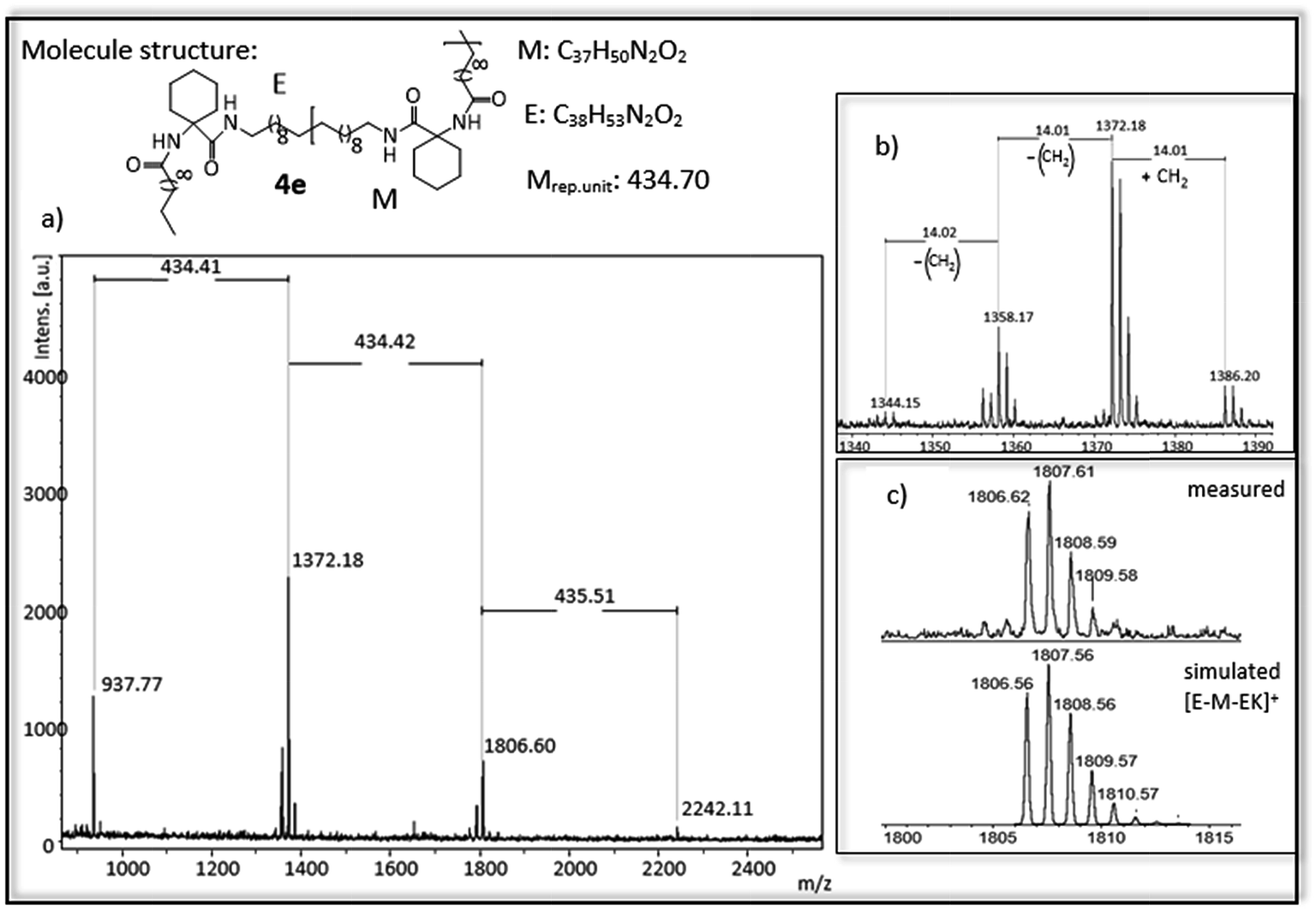 | ||
| Fig. 5 (a) MALDI-ToF-MS of ADMET polymer 4e (b) zoom in to [E-M-EK]+ to show isomerization of the olefin (c) measured and simulated pattern for [E-M-EK]+. | ||
For polymer 4e (Fig. 5) a mass distribution from 937 g mol−1 to 2242 g mol−1 is obtained, in which the highest absorption peak at 1372 g mol−1 indicates the polymer structure [E-M-EK]+. Distance between the main series is 434 g mol−1, which is in agreement with the molecular weight of the repetitive unit. In addition to the obtained main series up to three different series with a distance of 14 g mol−1 can be observed, again indicating the isomerization of the olefin. In comparison to 3b, the isomerization is higher and occurs at more than one site. The main series however proofs the complete hydrogenation of the polymer.
DSC analysis
The thermal properties of the hydrogenated polymer in comparison to the monomer and the unsaturated polymers were investigated by DSC-measurements. Fig. 6 shows the cooling and heating curve for the monomers 2a and 2b (dotted lines) and the polymers 4a and 4b (straight lines) after ADMET and hydrogenation.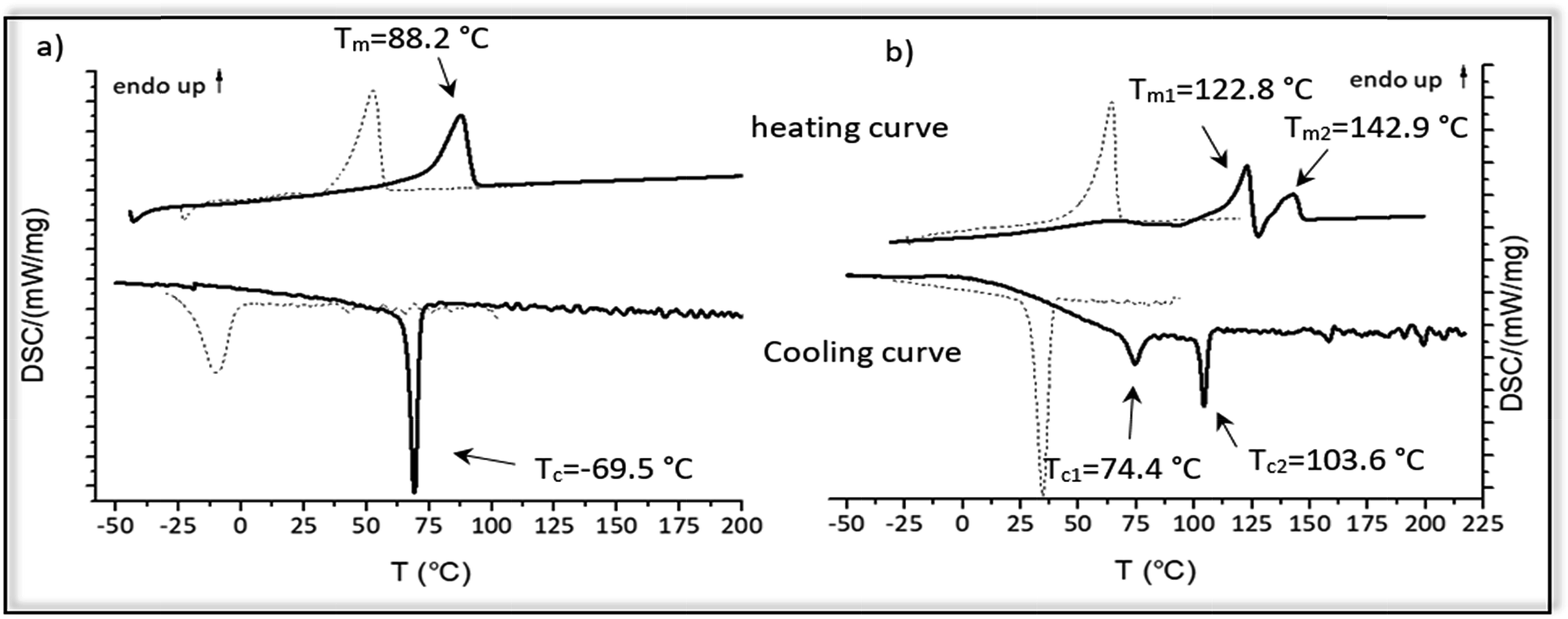 | ||
| Fig. 6 (a) DSC thermograms of monomer 2a (dotted lines) and the hydrogenated polymer 4a (straight lines) (b) DSC thermograms of monomer 2b (dotted lines) and the hydrogenated polymer 4b (straight lines). | ||
For monomer 2a melting at 53 °C and crystallization at −10 °C could be observed, whereas monomer 2b has a significant higher melting and crystallization point (Tm = 64.4 °C; Tc = 34.7 °C). After ADMET-polymerization the investigated compounds 3a–3e become completely amorphous, presumably due to sterically hindrance caused by incorporation of the internal double bonds into the polymer. Crystallization of such polymers strongly depends on the packing of the alkyl chains, which in this case is limited due to the presence of the thus different olefin isomers and the incorporated amino acid in the polymer backbone.
After hydrogenation and deprotection the polymers 4a and 4b display a crystalline structure as visible by their melting points. In comparison to the corresponding monomer, melting and crystallization of 4a and 4b shifts to significantly higher temperatures, in which 4b has a higher melting and crystallization temperature than 4a. However, in comparison to the unsaturated monomers and polymer 4a, DSC-curves of 4b are more complex. Melting at 122.8 °C and 142.9 °C indicate a melting-recrystallization, which can be due to formation of metastable crystals during the cooling process.55 Such crystals are melting first and reorganizing again into more stable areas resulting in an exothermal signal in the DSC curve. These areas are melting later, similar to the behavior found for precision polyolefines with different functional groups acting as defects.56 Furthermore, two crystallization temperatures can be observed at 103.6 °C and 74.4 °C, which may be due to microphase separation. In Table 4 thermal behavior as well as characteristic melting and crystallization data of these samples are summarized.
| Amino acid | R1 | Tm (°C) | Tc (°C) | ΔH (J g−1) | α (%) | |
|---|---|---|---|---|---|---|
| Glu | 2a | 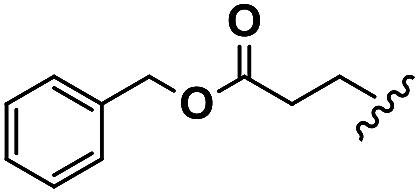 |
52.8 | −10.1 | 60.9 | 25 |
| 3a | — | — | — | — | ||
| 4a | 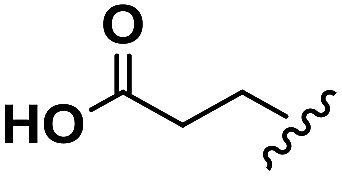 |
88.2 | 69.5 | 26.1 | 11 | |
| Asp | 2b | 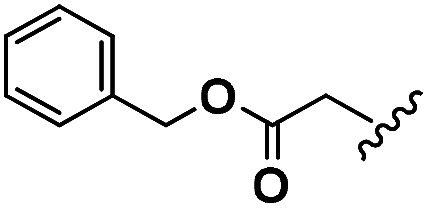 |
64.4 | 34.7 | 51.4 | 21 |
| 3b | — | — | — | — | ||
| 4b | 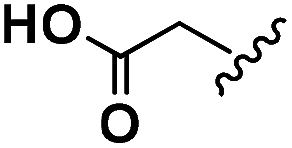 |
142.8 | 103.6 | 60.9 | 25 |
These observations indicate the big influence of the molecular size and hydrogen-bonding of the amino acids acting as defects, in which the polymer 4b bearing aspartic acid after every 19th carbon shows a higher melting and crystallization temperatures than 4a. Such effects of the defect size on the melting and crystallization were reported early e.g. for alkyl branches27 and butyl branched polyethylene's and polyphosphoester's.28
For all other monomers 2c–2e and polymers 4c–4e no crystalline behavior could be observed as those amino acids with an aliphatic backbone disturbed crystallinity, whereas amino acids with a functional group able to form defined crystal structures. Thus, an additional intermolecular interaction of the carboxylic-acid-defects is proposed for crystallization of the samples, e.g. by dimerization of the carboxylic-acid moieties.
XRD-analysis
Crystalline structure of polymers 4a and 4b was pre-investigated by WAXS measurements (Fig. 7), observing four reflections for both polymers (Table 5). The first reflection in the small angle area at a 2θ of 3.09° for 4a and 3.60° and 4b is the (001) reflection, indicative of the distance between two amino acid groups along the chain of 2.86 nm for 4a and 2.45 nm for 4b, which is in good agreement with the length of the precision polymers. At almost exactly the doubled value of 2θ higher order reflection (002) can be detected which was also found for precision sulfone polyethylenes,31 demonstrating that the reflection intensity for 4a is not as intensive as for 4b. These (002) reflections are describing the evolution of a lamellar morphology and both (001) and (002) reflections could have also been observed for other precision PE-polymers of different methylene sequences lengths bearing DAP-units at 16th, 18th or 20th carbon atom, shifting to higher 2θ values with decreasing length of the alkyl chain.57 Formation of lamellae in 4a could be more hindered than in 4b, caused by the glutamic acid as a major defect, so that the intensity of this reflex for 4a is significantly smaller than for 4b. As previously reported for ADMET-PE and high density polyethylene, two characteristic reflections at scattering angles of 21.7° and 24.0° can be detected, containing information about crystal morphology of the alkyl chains and corresponding to (110) and (200) reflections of a orthorhombic crystal system.58 For both polymers 4a and 4b, broad reflections with maxima at 2θ of 19.58° and 21.33° and 18.98° and 20.68° can be observed and thus the crystalline structure is definitely dissimilar to ADMET-PE.19,27 The width of the signals can be explained due to the low crystallinity as the defects are large, in accordance with DSC data and literature.27 Similar to other precision PE-polymers of different methylene spacer length bearing alkyl moieties19,27 chlorines25 and meta-substituted aryl ether branches56 in the main backbone it is likely that orthorhombic, triclinic and other metastable crystal systems are coexistent as supported by the strong asymmetric distribution of the reflections.19,27 Corresponding to the observations for ADMET-polyethylene containing m-substituted arylene the amino acid defect presumably can either be excluded from or included into the PE crystals.56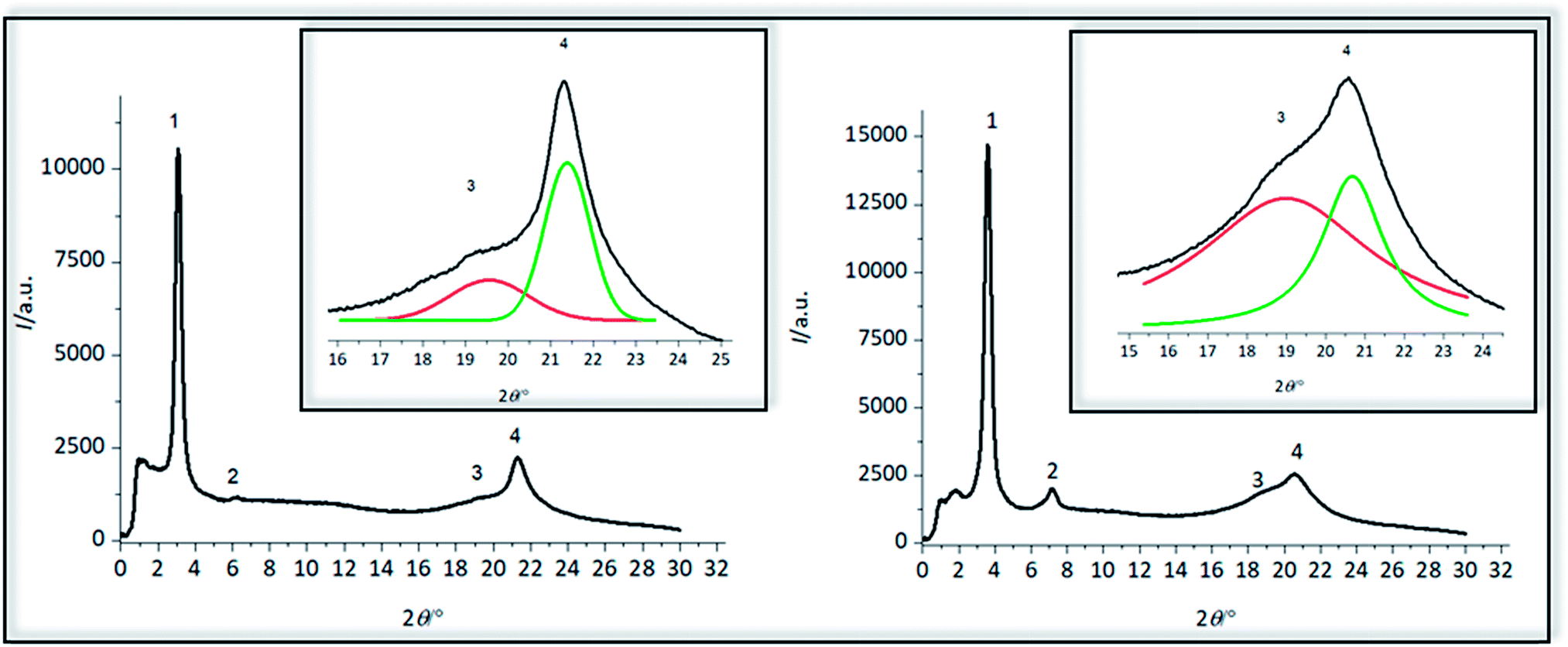 | ||
| Fig. 7 Wide-angle-X-ray diffraction patterns for precision oligomers 4a and 4b. | ||
| Sample | Reflection | 2θ/° | q/nm−1 | d/nm |
|---|---|---|---|---|
| 4a | 1 | 3.09 | 2.20 | 2.86 |
| 2 | 6.21 | 4.42 | 1.42 | |
| 3 | 19.58 | 13.87 | 0.45 | |
| 4 | 21.33 | 15.10 | 0.42 | |
| 4b | 1 | 3.60 | 2.56 | 2.45 |
| 2 | 7.14 | 5.08 | 1.24 | |
| 3 | 18.98 | 13.46 | 0.47 | |
| 4 | 20.68 | 14.64 | 0.43 |
Conclusion
We here report a strategy for the synthesis and characterization of newly developed precision polyalkyl-polymers with amino acid defects at every 19th CH2 carbon atom. Chiral/achiral as well as polar/nonpolar amino acids were functionalized on the N- and C-terminus with alkyl branches, bearing terminal double bonds at each end. The synthesis of precision polymers with molecular weights up to 22 kDa was realized by ADMET polymerization under melt-polymerization conditions, followed by complete hydrogenation of the internal double bonds with p-toluenesulfonhydrazide (TsNHNH2). For ADMET polymerization Grubbs 1st, Grubbs 2nd and Grubbs Hoveyda 1st catalyst were tested, the former was found to be the best catalyst in handling and in synthesizing low isomerized, high molecular weight polymers in good yields, proving the structures of the obtained products by 1H-NMR, GPC, MALDI-ToF-Ms and IR. Thermal investigation of the monomers and polymers demonstrated that all compounds having unpolar amino acids (L-leucine, 2-amino-2-methylpropanoic acid, 1-aminocyclohexane-carboxylic acid) in the chain are amorphous, whereas the monomers and the hydrogenated polymers of polar amino acids (glutamic and aspartic acid) show crystalline morphologies. Melting points for glutamic acid modified monomers and polymers are lower in comparison to aspartic acid, demonstrating that the bigger the size of the defect the lower the ability to pack into an ordered structure. The hydrogenated polymer bearing aspartic acid moieties (4b) shows a complex thermal behavior, which is represented by melting-recrystallization and two crystallization points. WAXD-measurements show four reflections for both 4a and 4b, indicating a distance between the two amino acid groups of 2.86 nm for 4a and 2.45 nm for 4b along the PE-chain, together with the formation of a lamellar structure. We also assume an orthorhombic crystal in which amino acids can either be excluded from or included into the PE crystals – a final statement however cannot be accomplished on basis of the current data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author grateful acknowledge the grant within the Sonderforschungsbereich SFB TRR 102 (Polymers under multiple constraints) (SR, WHB, Project A03). We also thank Sophie Reimann for MALDI-ToF-MS and Silvio Poppe and Matthias Fischer for providing WAXS measurement.References
- R. M. Michell, A. Mugica, M. Zubitur and A. J. Müller, in Polymer Crystallization I: From Chain Microstructure to Processing, ed. F. Auriemma, G. C. Alfonso and C. de Rosa, Springer International Publishing, Cham, 2017, pp. 215–256, DOI:10.1007/12_2015_327
.
- F. S. Bates, Macromolecules, 1984, 17, 2607–2613 CrossRef CAS
- G. Strobl, in The Physics of Polymers Concepts for Understanding Their Structures and Behavior, Springer International Publishing, Berlin, Heidelberg, New york, 2007, pp. 215–256, DOI:10.1007/12_2015_327
- E. Haber and C. B. Anfinsen, J. Biol. Chem., 1961, 236, 1361 Search PubMed
- V. Percec, G. Ungar and M. Peterca, Science, 2006, 313, 55–56 CrossRef PubMed
- P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS PubMed
- B. Heck, T. Hugel, M. Iijima and G. Strobl, Polymer, 2000, 41, 8839–8848 CrossRef CAS
- J. Adamcik and R. Mezzenga, Macromolecules, 2012, 45, 1137–1150 CrossRef CAS
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed
- F. J. Baltá-Calleja and R. Hosemann, J. Polym. Sci., Polym. Phys. Ed., 1980, 18, 1159–1165 CrossRef
- J. Martinez De Salazar and F. J. BaltáCalleja, J. Cryst. Growth, 1980, 48, 283–294 CrossRef CAS
- F. Gutzler and G. Wegner, Colloid Polym. Sci., 1980, 258, 776–786 CAS
- J. M. Salazar and F. J. B. Calleja, Polym. Bull., 1980, 2, 163–167 CrossRef
- E. Martuscelli and M. Pracella, Polymer, 1974, 15, 306–314 CrossRef CAS
- C. L. F. De Ten Hove, J. Penelle, D. A. Ivanov and A. M. Jonas, Nat. Mater., 2004, 3, 33–37 CrossRef PubMed
- H. Li, L. Caire da Silva, M. D. Schulz, G. Rojas and K. B. Wagener, Polym. Int., 2017, 66, 7–12 CrossRef CAS
- P. Atallah, K. B. Wagener and M. D. Schulz, Macromolecules, 2013, 46, 4735–4741 CrossRef CAS
- H. Mutlu, L. M. de Espinosa and M. A. R. Meier, Chem. Soc. Rev., 2011, 40, 1404–1445 RSC
- B. Inci, I. Lieberwirth, W. Steffen, M. Mezger, R. Graf, K. Landfester and K. B. Wagener, Macromolecules, 2012, 45, 3367–3376 CrossRef CAS
- G. Lieser, G. Wegner, J. A. Smith and K. B. Wagener, Colloid Polym. Sci., 2004, 282, 773–781 CAS
- W. Qiu, J. Sworen, M. Pyda, E. Nowak-Pyda, A. Habenschuss, K. B. Wagener and B. Wunderlich, Macromolecules, 2006, 39, 204–217 CrossRef CAS
- M. Tasaki, H. Yamamoto, M. Hanesaka, K. Tashiro, E. Boz, K. B. Wagener, C. Ruiz-Orta and R. G. Alamo, Macromolecules, 2014, 47, 4738–4749 CrossRef CAS
- P. Kaner, C. Ruiz-Orta, E. Boz, K. B. Wagener, M. Tasaki, K. Tashiro and R. G. Alamo, Macromolecules, 2014, 47, 236–245 CrossRef CAS
- E. Boz, A. J. Nemeth, K. B. Wagener, K. Jeon, R. Smith, F. Nazirov, M. R. Bockstaller and R. G. Alamo, Macromolecules, 2008, 41, 1647–1653 CrossRef CAS
- R. G. Alamo, K. Jeon, R. L. Smith, E. Boz, K. B. Wagener and M. R. Bockstaller, Macromolecules, 2008, 41, 7141–7151 CrossRef CAS
- E. Boz, K. B. Wagener, A. Ghosal, R. Fu and R. G. Alamo, Macromolecules, 2006, 39, 4437–4447 CrossRef CAS
- G. Rojas, B. Inci, Y. Wei and K. B. Wagener, J. Am. Chem. Soc., 2009, 131, 17376–17386 CrossRef CAS PubMed
- Y.-R. Zheng, H. T. Tee, Y. Wei, X.-L. Wu, M. Mezger, S. Yan, K. Landfester, K. Wagener, F. R. Wurm and I. Lieberwirth, Macromolecules, 2016, 49, 1321–1330 CrossRef CAS
- K. N. Bauer, H. T. Tee, I. Lieberwirth and F. R. Wurm, Macromolecules, 2016, 49, 3761–3768 CrossRef CAS
- M. Pulst, M. H. Samiullah, U. Baumeister, M. Prehm, J. Balko, T. Thurn-Albrecht, K. Busse, Y. Golitsyn, D. Reichert and J. Kressler, Macromolecules, 2016, 49, 6609–6620 CrossRef CAS
- T. W. Gaines, E. B. Trigg, K. I. Winey and K. B. Wagener, Macromol. Chem. Phys., 2016, 217, 2351–2359 CrossRef CAS
- T. W. Baughman and K. B. Wagener, in Metathesis Polymerization, ed. M. R. Buchmeiser, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 1–42, DOI:10.1007/b101318
- F. Sanda and T. Endo, Macromol. Chem. Phys., 1999, 200, 2651–2661 CrossRef CAS
- H. Kudo, F. Sanda and T. Endo, Macromolecules, 1999, 32, 8370–8375 CrossRef CAS
- F. Sanda, T. Abe and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 2619–2629 CrossRef CAS
- J. Morcellet-Sauvage, M. Morcellet and C. Loucheux, Makromol. Chem., 1981, 182, 949–963 CrossRef CAS
- J. Morcellet-Sauvage, M. Morcellet and C. Loucheux, Makromol. Chem., 1982, 183, 821–829 CrossRef CAS
- H. D. Maynard, S. Y. Okada and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 1275–1279 CrossRef CAS PubMed
- H. D. Maynard, S. Y. Okada and R. H. Grubbs, Macromolecules, 2000, 33, 6239–6248 CrossRef CAS
- J. K. Kammeyer, A. P. Blum, L. Adamiak, M. E. Hahn and N. C. Gianneschi, Polym. Chem., 2013, 41, 3929–3933 RSC
- R. B. Breitenkamp, Z. Ou, K. Breitenkamp, M. Muthukumar and T. Emrick, Macromolecules, 2007, 40, 7617–7624 CrossRef CAS
- T. E. Hopkins, J. H. Pawlow, D. L. Koren, K. S. Deters, S. M. Solivan, J. A. Davis, F. J. Gómez and K. B. Wagener, Macromolecules, 2001, 34, 7920–7922 CrossRef CAS
- F. N. Behrendt and H. Schlaad, Polym. Chem., 2017, 8, 366–369 RSC
- J. K. Leonard, T. E. Hopkins, K. Chaffin and K. B. Wagener, Macromol. Chem. Phys., 2008, 209, 1485–1494 CrossRef CAS
- K. M. Lambert, J. M. Bobbitt, S. A. Eldirany, L. E. Kissane, R.
K. Sheridan, Z. D. Stempel, F. H. Sternberg and W. F. Bailey, Chem.–Eur. J., 2016, 22, 5156–5159 CrossRef CAS PubMed
- V. I. Petkovska, T. E. Hopkins, D. H. Powell and K. B. Wagener, Anal. Chem., 2006, 78, 3624–3631 CrossRef CAS PubMed
- F. C. Courchay, J. C. Sworen, I. Ghiviriga, K. A. Abboud and K. B. Wagener, Organometallics, 2006, 25, 6074–6086 CrossRef CAS
- S. E. Lehman and K. B. Wagener, Macromolecules, 2002, 35, 48–53 CrossRef CAS
- Y. Schrodi, Aldrichimica Acta, 2007, 40, 45 CAS
- S. Kobayashi, L. M. Pitet and M. A. Hillmyer, J. Am. Chem. Soc., 2011, 133, 5794–5797 CrossRef CAS PubMed
- Z.-L. Li, L. Li, X.-X. Deng, L.-J. Zhang, B.-T. Dong, F.-S. Du and Z.-C. Li, Macromolecules, 2012, 45, 4590–4598 CrossRef CAS
- E. B. Berda and K. B. Wagener, Macromolecules, 2008, 41, 5116–5122 CrossRef CAS
- S. Reimann, U. Baumeister and W. H. Binder, Macromol. Chem. Phys., 2014, 215, 1929 CrossRef
- V. I. Petkovska, T. E. Hopkins, D. H. Powell and K. B. Wagener, Macromolecules, 2005, 38, 5878–5885 CrossRef CAS
- S. Song, W. Miao, Z. Wang, D. Gong and Z.-R. Chen, Polymer, 2015, 64, 76–83 CrossRef CAS
- S.-F. Song, Y.-T. Guo, R.-Y. Wang, Z.-S. Fu, J.-T. Xu and Z.-Q. Fan, Macromolecules, 2016, 49, 6001–6011 CrossRef CAS
- S. Reimann, V. Danke, M. Beiner and W. H. Binder, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3736–3748 CrossRef CAS
- M. F. Butler, A. M. Donald, W. Bras, G. R. Mant, G. E. Derbyshire and A. J. Ryan, Macromolecules, 1995, 28, 6383–6393 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR data of monomers as well as 1H- and 13C-NMR-, IR-data and MALDI-ToF-MS-data of polymers. See DOI: 10.1039/c7ra10485e |
| This journal is © The Royal Society of Chemistry 2017 |