
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of nitrogenated lignin-derived compounds and reactivity with laccases. Study of their application in mild chemoenzymatic oxidative processes†
Jesús Albarrán-Velo ,
María López-Iglesias,
Vicente Gotor,
Vicente Gotor-Fernández* and
Iván Lavandera*
,
María López-Iglesias,
Vicente Gotor,
Vicente Gotor-Fernández* and
Iván Lavandera*
Organic and Inorganic Chemistry Department, Biotechnology Institute of Asturias (IUBA), University of Oviedo, Avenida Julián Clavería 8, 33006 Oviedo, Spain. E-mail: vicgotfer@uniovi.es; lavanderaivan@uniovi.es
First published on 30th October 2017
Abstract
The chemical synthesis of a series of lignin-derived nitrogenated compounds was performed in high yields (73–99%) through simple conventional chemical transformations starting from natural monomers such as vanillin, syringaldehyde or 3,4-dihydroxybenzaldehyde. The study of the vanillin-derived compounds as substrates for commercially available laccases from Trametes versicolor and Myceliophthora thermophila in oxidative transformations, generally led to the isolation of several dimeric species in high to excellent conversions (>70%), while for hydrazone derivatives a more rapidly oxidative coupling was evidenced by the formation of oligomers and/or polymers. Remarkably, vanillin was obtained due to the hydrolysis of some of the nitrogenated functional groups, such as the hydrazone or the hydrazono tetrazole. The three families of lignin-derived compounds can provide a great source of new laccase-mediator systems (LMS), the possibility of employing them for lignin modification being particularly attractive. Preliminary experiments showed promising levels of activity towards the oxidation of a monomer (veratryl alcohol, up to 70% conversion) and a dimer (adlerol, up to 22% conversion) lignin models, higher than those achieved with the natural vanillin and syringaldehyde (up to 7% conversion with veratryl alcohol and almost negligible conversion with adlerol), these processes being also highly influenced by the pH of the reaction medium.
Introduction
Lignin is a highly abundant and irregular biopolymer which constitutes lignocellulosic biomass together with cellulose and hemicellulose. During the last decades, the scientific community has deeply studied the usability of these polymers as sources of energy and high-added value compounds. On the opposite, the complex structure of lignin has overshadowed its value as a renewable alternative to the fuel industry, since high amounts of energy could be involved to break down its cross-linked structure. Nevertheless, the use of lignin as feedstock has become an important field of research in recent years, finding multiple applications in the production of bio-based polymers such as adhesives, bioplastics or resins due to its polyphenolic composition.1 The lignin valorization is also moving to cleavage processes focused on degradation into smaller compounds,2 biocatalytic methodologies being currently booming.3Thus, it has been found that some bacterial and fungal microorganisms containing potent oxidative enzymes, have the capability to degrade the structure of lignin by promoting the cleavage of certain connections as the β-O-4-aryl ether bonds or the oxidation at Cα positions,3c these predominant linkages being illustrated in the dimeric species of Fig. 1 (left). Particularly, some peroxidases, peroxygenases and laccases have been applied in enzyme-catalyzed delignification processes.4
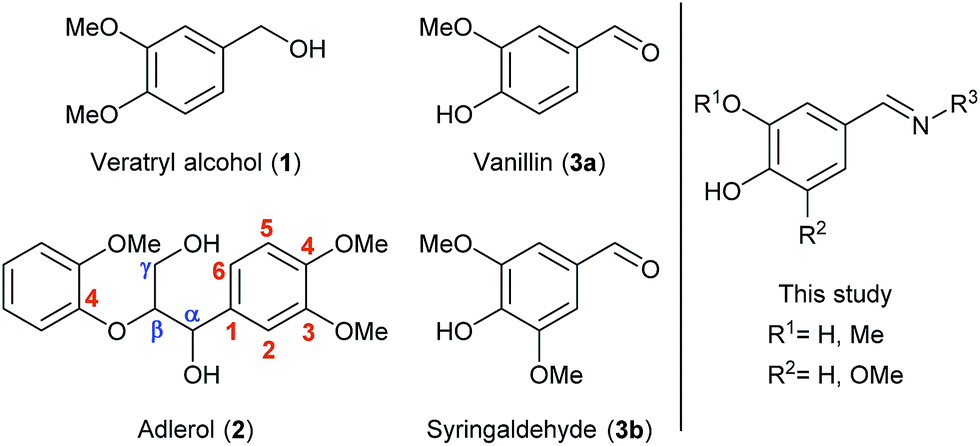 | ||
| Fig. 1 Lignin-derived model compounds, monomeric and dimeric species. | ||
Furthermore, lignin has attracted an increasing interest not only as source of energy but also because of the valuable low-molecular weight aromatic compounds contained in its structure, which can also serve as building blocks for synthetic processes.5 Within this area, many efforts have been intended to investigate the behavior of lignin-related compounds towards cleavage or oxidation processes as representative and handier models in which the most characteristic linkages are present. Among them, some dimeric species such as veratrylglycerol β-guaiacyl ether (adlerol, 2, Fig. 1) or similar β-1-type lignin model compounds have found wide application, for instance, for studying the mechanism of lignin C–C linkage cleavage by pyrolytic strategies6 or metal catalysis.2a Also Biocatalysis has been applied in lignin valorization by studying the cleavage or oxidation of β-O-4-linkages in dimeric species.7
Laccases (EC 1.10.3.2) are multi-copper oxidative enzymes that consume molecular oxygen as electron acceptor during the oxidation of a substrate producing water as the only by-product, which makes them an interesting and suitable tool working under mild reaction conditions.8 These enzymes have been employed in lignin degradation,4,9 and also in the opposite repolymerization processes or related lignin-surface modifications, in order to functionalize it obtaining valuable biomaterials, for example by single molecule grafting or copolymerization processes.10 Despite these enzymes present a suitable redox potential to abstract electrons from phenolic compounds, this is not high enough to transform other types of substrates such as aliphatic alcohols, so the use of laccase-mediator systems (LMS) is necessary to allow their oxidation.8c,11 Many synthetic compounds are commonly employed in these systems, such as TEMPO, HOBt or ABTS, but also some natural compounds obtained from lignin can be used. Among them, vanillin (3a, Fig. 1), syringaldehyde (3b, Fig. 1) and their derivatives are some of the most common lignin-derived monomers that have been extensively employed in different ways, for example as natural LMS.9a,12 In fact, laccases have proven to accept a large number of lignin-derived monomers due to the presence of a phenolic function within their structure,13 but still the product formation was not studied in many of those cases.
Herein, we aim to combine the use of compounds derived from renewable resources (e.g. lignocellulose) and the benefits that biocatalysis offers due to the capability of enzymes to selectively work under environmentally friendly conditions. Thus, we firstly report the chemical modification of vanillin and syringaldehyde towards the synthesis of nitrogenated derivatives whose broad structure is depicted in Fig. 1 (right). Later these compounds will be tested as suitable substrates in laccase-catalyzed processes. Overall, the synthetic strategies will provide novel systems that will be applied in the oxidation of lignin model compounds, which is the underlying purpose of the present research.
Results and discussion
Chemical modification of natural LMS
Vanillin (3a) and syringaldehyde (3b) were selected as starting materials for further modifications due to the presence of these compounds in high quantities when degrading lignin. As both substrates have a similar structure but a different number of methoxy groups in the aromatic ring, the choice of these two lignin-type monomers will allow us to compare their capacities in LMS. Moreover, anticipating the importance of both the methoxy and the hydroxyl groups in the substrate–laccase interaction, and in order to complete the study, we decided to include also 3,4-dihydroxybenzaldehyde (3c), which was synthesized starting from vanillin via demethylation in 85% yield as described in the literature (Scheme S1, see the ESI†).14In the interest of practical designs for the potential mediators, we focused on simple and easy synthetic procedures. Moreover, the rationale for the synthesis of the proposed compounds is based on the possibility of stabilizing the oxidized radical intermediates by aromatic systems via electron delocalization or by increasing the number of additional reactive points as in the case of an oxime derivative (Scheme 1), resembling in this particular case N-oxyl oxidants such as TEMPO.
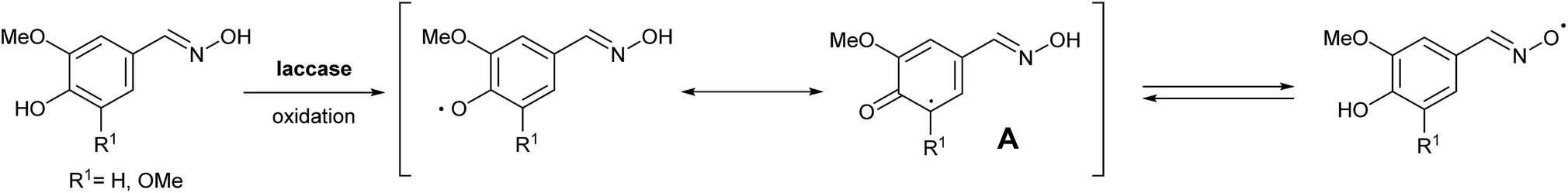 | ||
| Scheme 1 An oxime derivative presenting an increasing number of reacting points. | ||
In a first set of experiments, we evaluated the use of hydroxyl ammonium reagents to introduce both oxime and nitrile functions. Thus, oximes 5a–c were prepared in good to excellent yields following a described methodology15 in the presence of hydroxylammonium sulfate and sodium acetate in ethanol (Scheme 2). By varying the solvent to acetonitrile and using hydroxylamine hydrochloride, dehydration of the aldoxime intermediate took place yielding the nitrile derivative 4a from 3a after 4 h.
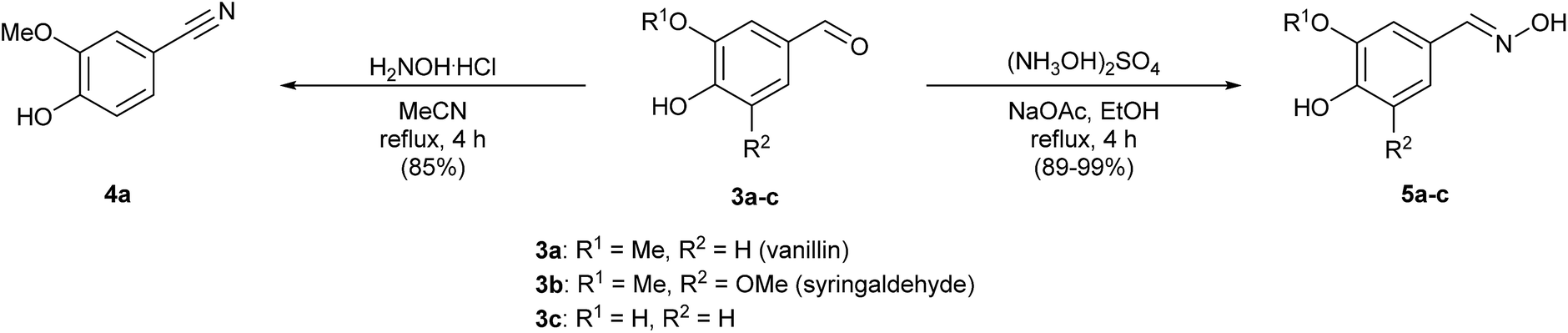 | ||
| Scheme 2 Synthesis of 4-hydroxy-3-methoxybenzonitrile (4a) and oximes 5a–c. | ||
On the other hand, a panel of nitrogenated substituents was introduced through the aldehyde function by tuning the strength of the acid catalyst and adjusting the reaction time (Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Reactant | Product | Acid | t (h) | Yielda (%) |
| a Yield obtained after isolation of the products by solvent evaporation (for 6a–c and 7a–c) or product filtration (for 8a–c and 9a–c). | ||||||
| 1 | 3a | N2H4·H2O (10) | 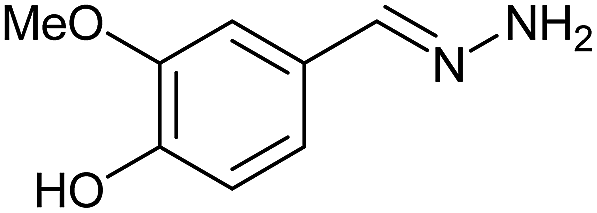 |
AcOH | 0.83 | 90 (6a) |
| 2 | 3b | N2H4·H2O (10) | 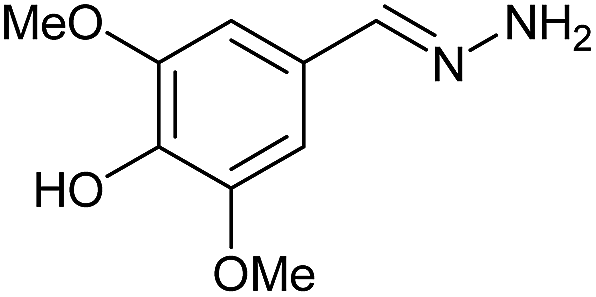 |
AcOH | 0.83 | 80 (6b) |
| 3 | 3c | N2H4·H2O (10) | 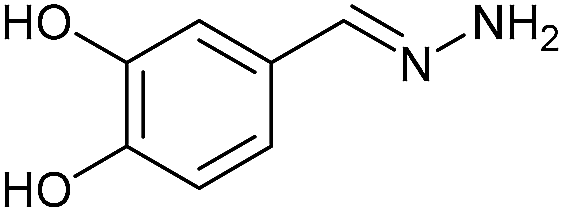 |
AcOH | 1 | 78 (6c) |
| 4 | 3a | 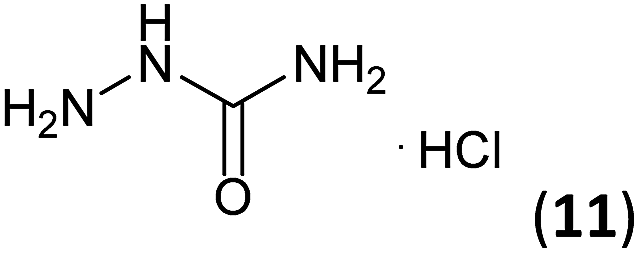 |
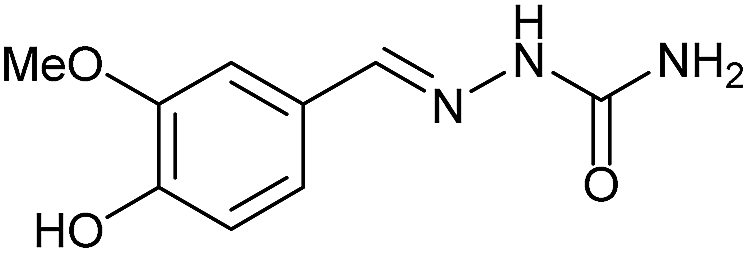 |
AcOH | 1.5 | 98 (7a) |
| 5 | 3b | 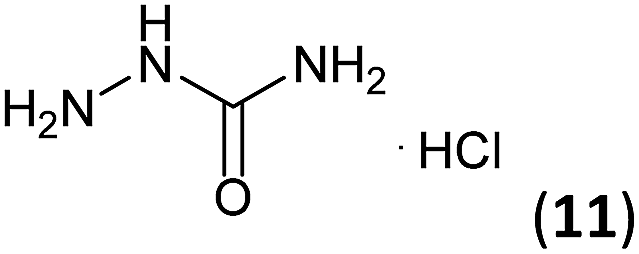 |
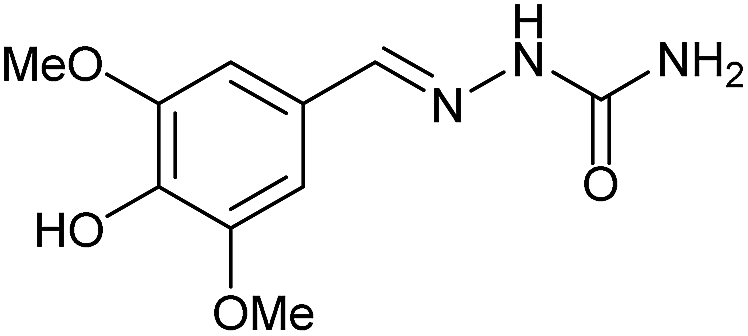 |
AcOH | 2 | 89 (7b) |
| 6 | 3c | 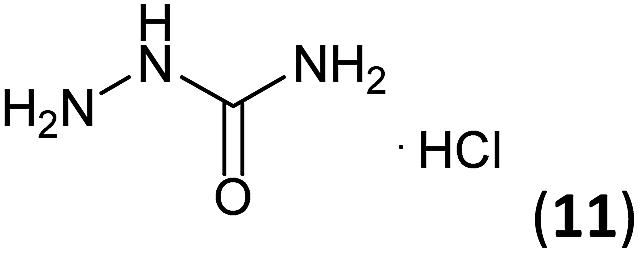 |
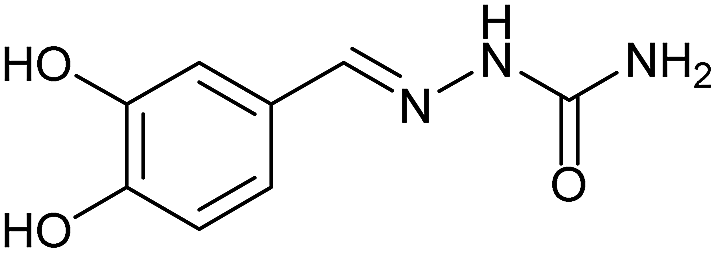 |
AcOH | 2.7 | 73 (7c) |
| 7 | 3a | 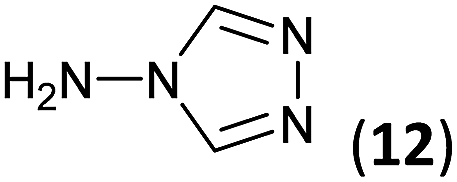 |
 |
H2SO4 conc. | 3 | 99 (8a) |
| 8 | 3b | 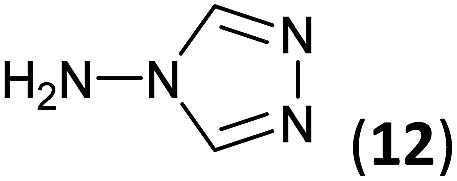 |
 |
H2SO4 conc. | 7.5 | 99 (8b) |
| 9 | 3c | 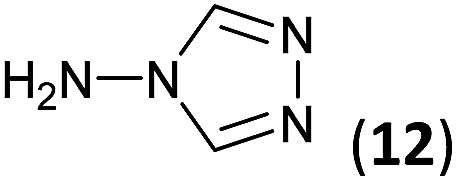 |
 |
H2SO4 conc. | 6.5 | 96 (8c) |
| 10 | 3a | 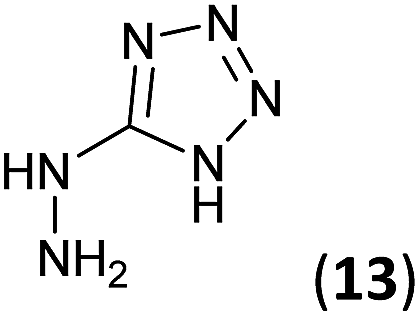 |
 |
HCl conc. | 4.5 | 94 (9a) |
| 11 | 3b | 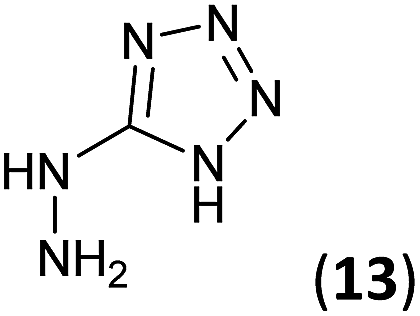 |
 |
HCl conc. | 5 | 97 (9b) |
| 12 | 3c | 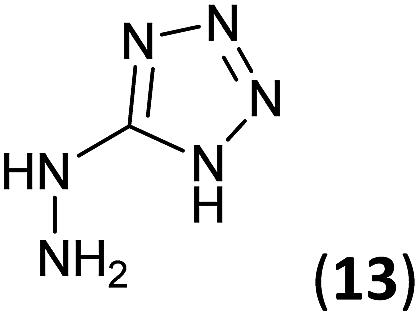 |
 |
HCl conc. | 1.75 | 80 (9c) |
Firstly, the reaction between hydrazine monohydrate (10) and 3a was performed under acidic conditions, observing full disappearance of the starting material by TLC analysis after 50 min. Hydrazone 6a was isolated in 90% yield after removing the solvent by distillation (entry 1). The procedure was extended to the dimethoxylated and dihydroxylated substrates 3b and 3c, leading to 6b and 6c also in good yields (entries 2 and 3). The reaction time was increased in the synthesis of semicarbazones 7a–c in the presence of semicarbazide hydrochloride (11), obtaining the desired products in good to excellent yields after solvent evaporation (entries 4–6).
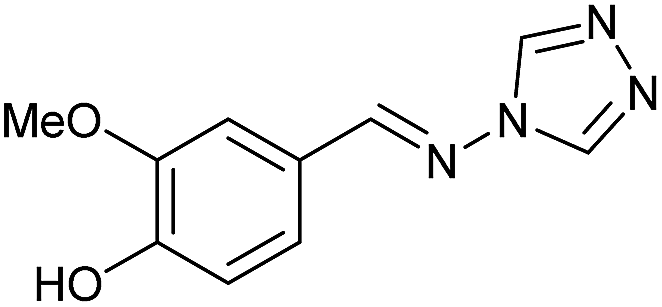
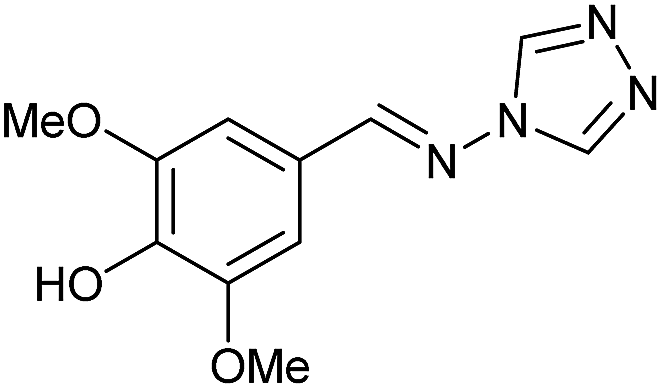
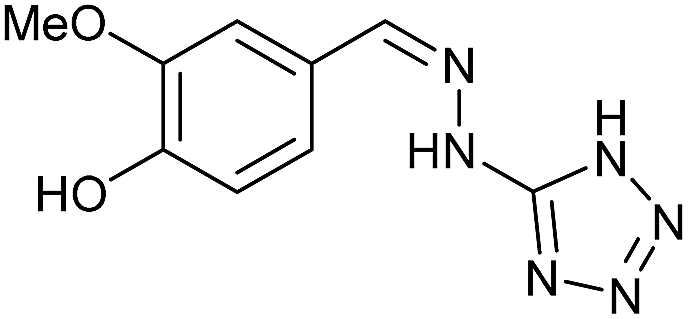
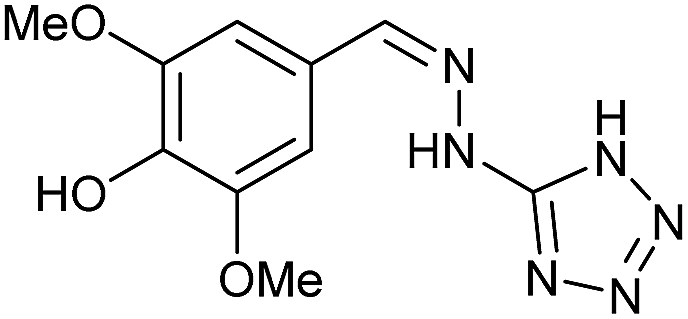
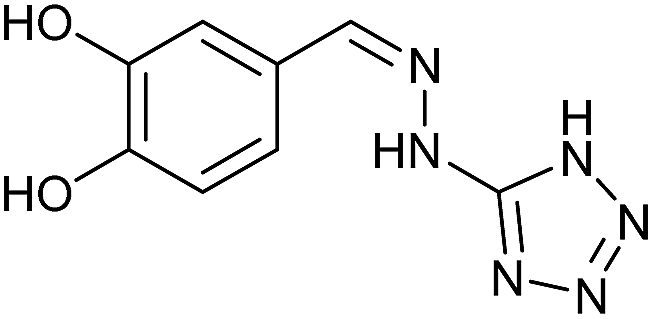
To prepare the triazole derivatives 8a–c, longer reaction times and harsher acidic conditions were needed, so concentrated H2SO4 was employed as catalyst.16 The reaction between 3b and 3c with 4H-1,2,4-triazol-4-amine (12) was completed after 7.5 and 6.5 h, respectively (entries 8 and 9), while starting from 3a only 3 h were necessary (entry 7), recovering 8a–c with excellent yields. Concentrated HCl was used in the reactions with tetrazole 13, obtaining 9a–c in 80–97% yield (entries 10–12). In these cases, the isolation was performed by filtration from the reaction crude, since both families of poly-nitrogenated compounds 8a–c and 9a–c were insoluble in the reaction medium. In the latter reactions, it was necessary to synthesize the reagent 13 through a two-step process (see Experimental section), as it was not commercially available.
Laccase-catalyzed oxidation reactions of the synthesized derivatives
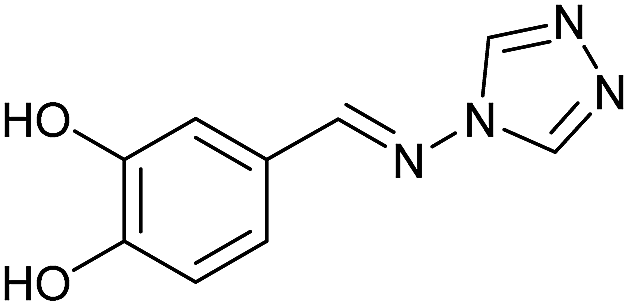
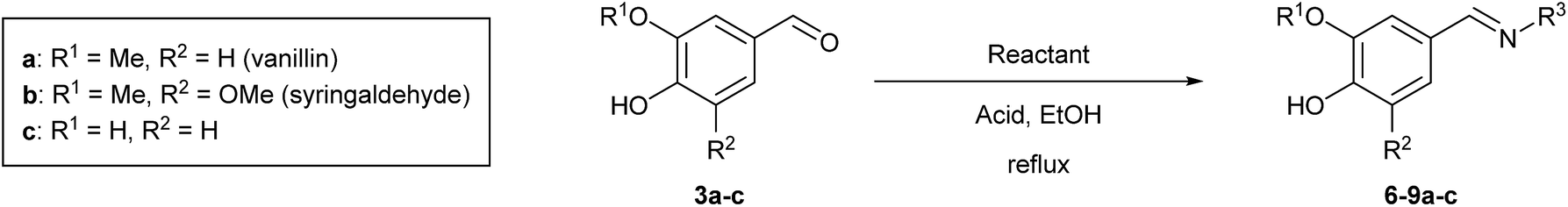
To develop an efficient LMS with these compounds, first we investigated if they could react with laccases. For this, we chose commercially available laccases from Trametes versicolor (LTv)17 and Myceliophthora thermophila (Novozym 51003),18 and the enzymatic experiments were conducted with the family of vanillin-modified derivatives, in order to illustrate the oxidative processes that could take place. This study would verify the validity of the new synthesized compounds as potential laccase mediators. Following previous described protocols with these biocatalysts, enzymatic reactions were carried out in acetate buffer at pH 4.7, varying the substrate concentration from 10 to 50 mM. Due to solubility issues, 10% (v/v) of EtOH or DMSO19 were added as water-miscible organic solvents in some cases. The control reactions in the absence of enzyme led to no conversion in all cases, proving the direct role of the laccase as the only catalyst.When the laccases and the vanillin derivatives were mixed, three processes were mainly observed. In most cases the C–C dimerization reaction of species-type A (Scheme 1) was favored, but also the formation of vanillin (via hydrolysis) and the polymerization/oligomerization of these substrates were observed (Table 2). From now on, no distinction between polymerization and oligomerization will be made, since this was not the main goal of this research, so the polymeric structures were not analyzed such depth to make that differentiation. Firstly, the enzymatic experiments were conducted with the vanillin itself (3a), observing it was susceptible to dimerization in a very high extent (entries 1 and 2). Similarly, dimeric species 15–18 were isolated and properly characterized from the reaction crudes when starting from nitrile 4a, oxime 5a, semicarbazone 7a, and triazole 8a (entries 3–6, 9, 11 and 12). Particularly, 4a reacted at 10 mM concentration, achieving a quantitative conversion with LTv and Novozym 51003 (entries 3 and 4). Oxime 5a also led to full conversions at 50 mM (entries 5 and 6), observing with both biocatalysts the formation of a minimum amount of 3a (3%) which was no detected in the absence of the enzyme. Semicarbazone 7a was converted at 20 mM concentration in the presence of LTv conducting to dimer 17 (72% conversion, entry 9). However, the product formation differed in the case of Novozym 51003 (entry 10), in which the presence of a polymeric species, even at short reaction time, was detected due to the broad signals observed in the 1H-NMR experiments. The generation of polymeric products in laccase-catalyzed reactions has already been reported in the literature for this type of lignin monomer.13b,20 Finally, dimer 18 was obtained as a precipitate from the reaction crude when starting from triazole 8a (20 mM, entries 11 and 12) in almost full conversion with both enzymes. These results can be related with the stability of the radical species-type A (Scheme 1). If this intermediate is stable enough, the dimerization process will be favored. Nitrogenated electron-withdrawing groups enhanced the stability of this radical as the C–C dimerization reaction was prominent.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate, concentration | Cosolvent | Enzyme | t (h) | T (°C) | Product | cb (%) |
| a Reaction conditions: substrate 3–9a, cosolvent (10% v/v), acetate buffer pH 4.7 (total volume: 3 mL) saturated with O2, laccase from Trametes versicolor (LTv, 25 U) or Novozym 51003 (31.4 U).b Conversion values were measured by 1H-NMR analysis of the reaction crudes.c Vanillin and a mixture of dimeric species were obtained.d Vanillin and a mixture of unidentified products were attained. | |||||||
| 1 | 3a, 20 mM | EtOH | LTv | 2 | 30 | 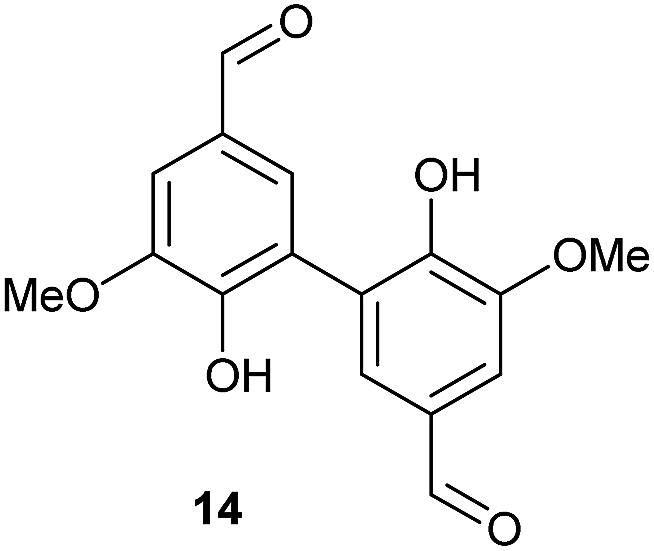 |
94 |
| 2 | 3a, 20 mM | — | Novozym 51003 | 2 | 50 | 93 | |
| 3 | 4a, 10 mM | — | LTv | 2 | 30 | 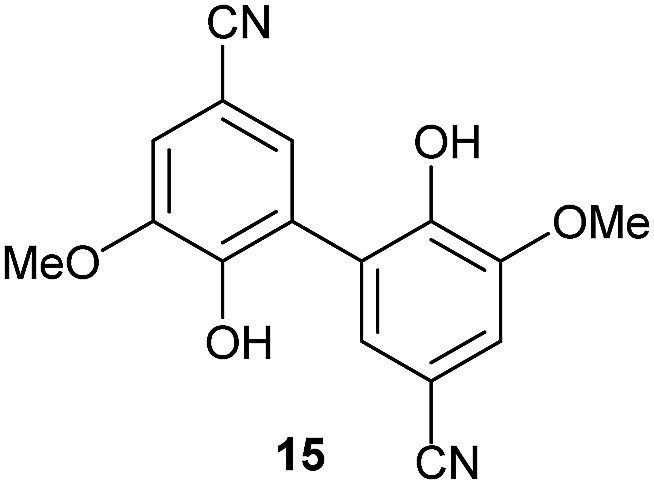 |
>99 |
| 4 | 4a, 10 mM | — | Novozym 51003 | 2 | 50 | >99 | |
| 5 | 5a, 50 mM | EtOH | LTv | 16 | 30 | 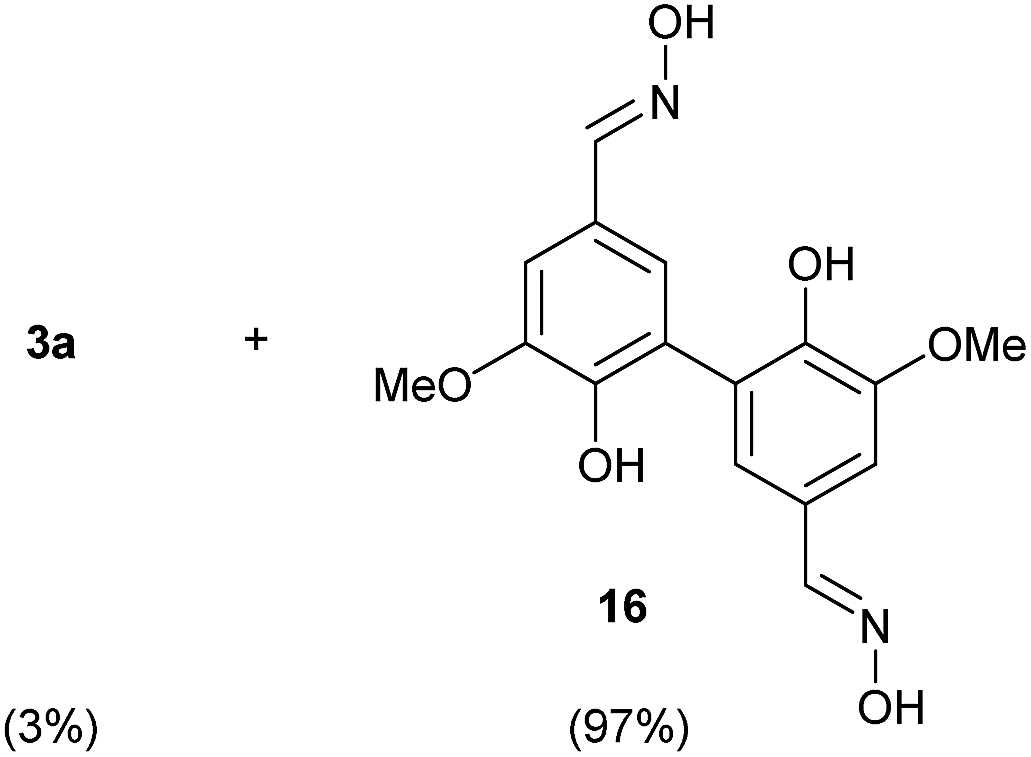 |
>99 |
| 6 | 5a, 50 mM | EtOH | Novozym 51003 | 16 | 50 | >99 | |
| 7 | 6a, 50 mM | DMSO | LTv | 16 | 30 | 3ac | 51 |
| 8 | 6a, 50 mM | DMSO | Novozym 51003 | 16 | 50 | 3ac | 69 |
| 9 | 7a, 20 mM | — | LTv | 16 | 30 | 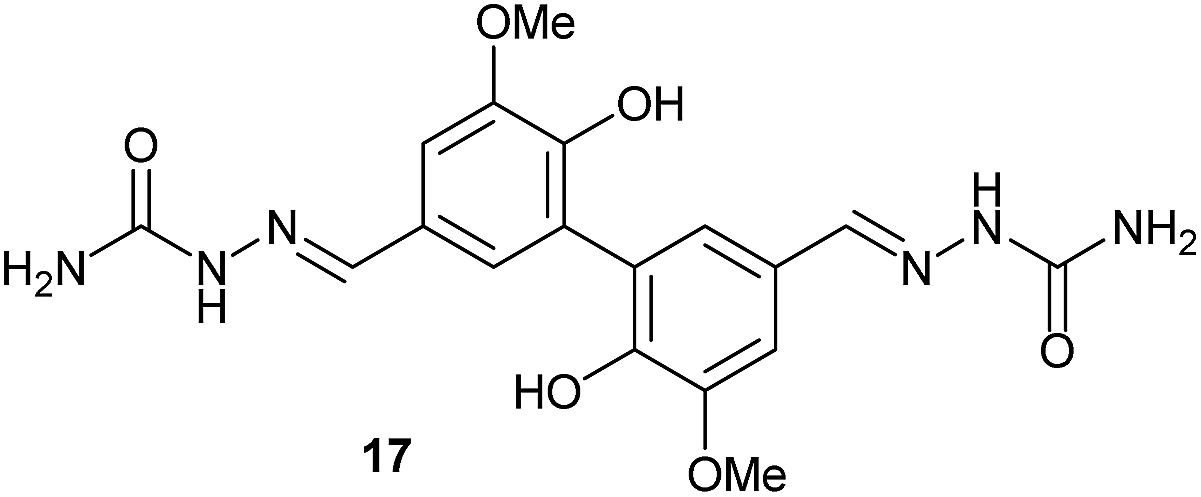 |
72 |
| 10 | 7a, 20 mM | — | Novozym 51003 | 2 | 50 | 17 + polymerization | 25 |
| 11 | 8a, 20 mM | EtOH | LTv | 2 | 30 | 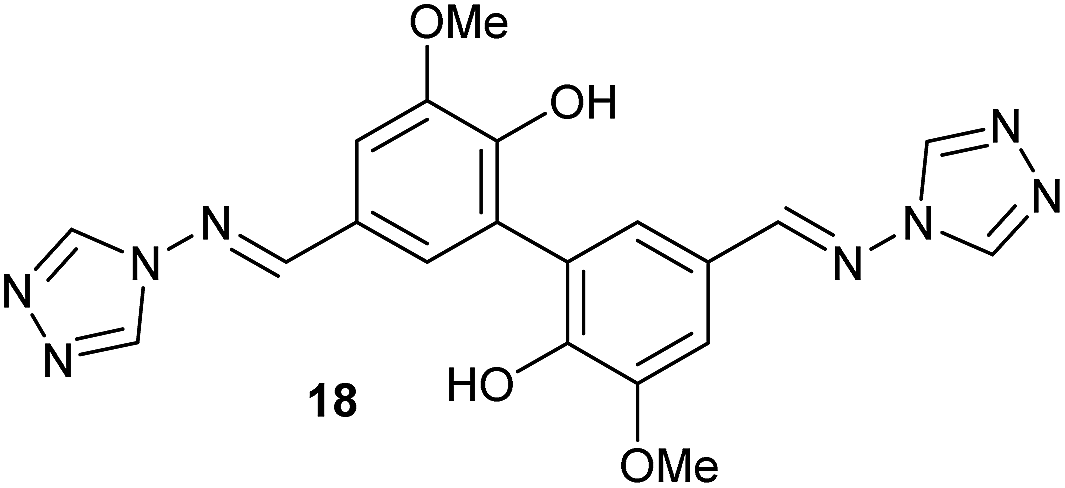 |
93 |
| 12 | 8a, 20 mM | EtOH | Novozym 51003 | 2 | 50 | 95 | |
| 13 | 9a, 10 mM | EtOH | LTv | 2 | 30 | 3ad | 69 |
| 14 | 9a, 10 mM | EtOH | Novozym 51003 | 2 | 50 | 3ad | 97 |
The hydrazone bond of 6a was hydrolyzed during the enzymatic process, so vanillin was detected in significant amounts in the 1H-NMR of the reaction crude (entries 7 and 8), a 4% of the hydrolysis product 3a being also formed in the absence of enzyme. Moreover, after aldehyde formation also dimerization took place, so a mixture of starting material, 3a and several dimeric species were detected in the reaction crude in a proportion varying depending on the biocatalyst used. Despite of the presence of unreacted hydrazone in the crude, it was noticed a fast reaction as the mixture acquired a dark purple color just after the addition of the enzyme. When the N-tetrazole substituted hydrazone derivative 9a (10 mM) was submitted to oxidation, the hydrolysis of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond was also observed. Thus, a mixture of products was extracted from the reaction crude when LTv or Novozym 51003 were used, in which the starting material, vanillin and other unidentified products were already present at 2 h. Nevertheless, polymerization might also be occurring, as a significant loss of weight in the crude recovered by extraction was observed.
N bond was also observed. Thus, a mixture of products was extracted from the reaction crude when LTv or Novozym 51003 were used, in which the starting material, vanillin and other unidentified products were already present at 2 h. Nevertheless, polymerization might also be occurring, as a significant loss of weight in the crude recovered by extraction was observed.
Since laccases are not expected to hydrolyze hydrazones, we evaluated if unspecific catalysis of this protein or other impurities that could be present in the commercial laccase preparation might be responsible of this transformation. So the LTv was denatured by a heat treatment (for details, see the ESI†). The conversion towards 3a formation was just 4%, demonstrating the role of the biocatalytic system in the hydrolysis of 6a. While it has been described the hydrolysis of hydrazones under oxidative conditions,21 there is only one precedent in the literature showing a similar behavior of LMS, the authors observing bio-deoximation processes in the presence of TEMPO as mediator.11a In our experiments, we assume that the laccase is able to catalyze the synthesis of unstable radical intermediates which can undergo hydrolysis releasing vanillin.
New LMS applied to the oxidation of lignin-model compounds
After checking that these compounds were suitable substrates for the studied laccases, they were tested as potential mediators to oxidize two different lignin model compounds. The syringyl and 3,4-dihydroxybenzaldehyde families were also included in this study. Thus, veratryl alcohol (1) and adlerol (2) were employed as representative monomeric and dimeric structures of lignin units,22 respectively, the alcohol moiety being in both cases at benzylic position (Fig. 1). Control experiments in the absence of laccase and/or nitrogenated compound revealed that both must be present for an efficient catalysis.Beginning with the veratryl alcohol oxidation, we selected 3 mM as starting substrate concentration according to some reports found in the literature,22d evaluating both commercial laccases. A deep optimization was performed in order to determine the best reaction conditions, including the reaction medium, temperature, amount of catalyst and concentration of the nitrogenated derivative, and the use of different cosolvents (see Tables S1–S18 in the ESI†). Among them, the pH was identified as key parameter, and depending on the phenolic compound employed, the optimum value varied. Similarly, changes in the amount of the co-oxidant implied important variations in the conversion degree, detecting the higher values at 8 mM concentration. The first experiments were stopped at 48 h but, after observing that the laccase was still active, the reactions were followed until 72 h. The best results were obtained in the acidic pH range of 4.7–7, as described for these laccases,17,18 and are summarized in Table 3. LTv works better under acidic conditions while Novozym 51003 accepts higher pHs.
|
|||||
|---|---|---|---|---|---|
| Entry | Compound | Laccase | T (°C) | pH | Conversionb (%) |
| a Reaction conditions: natural or nitrogenated derivative (16 μmol), buffer (2 mL), veratryl alcohol (0.9 μL, 6 μmol), laccase from Trametes versicolor (LTv, 25 U, 30 °C) or Novozym 51003 (31.4 U, 50 °C), 72 h. Buffers used: acetate buffer 100 mM pH 4.7; citrate buffer 50 mM pH 5; citrate buffer 50 mM pH 5.5; citrate buffer 50 mM pH 6; phosphate buffer 100 mM pH 7.b Conversion values were measured by GC analysis of the reaction crudes. | |||||
| 1 | 3a | LTv | 30 | 5 | 4 |
| 2 | 3b | LTv | 30 | 6 | 6 |
| 3 | 3a | Novozym 51003 | 50 | 6 | 6 |
| 4 | 3b | Novozym 51003 | 50 | 6 | 7 |
| 5 | 4a | LTv | 30 | 4.7 | 3 |
| 6 | 4a | Novozym 51003 | 50 | 6 | 6 |
| 7 | 5a | LTv | 30 | 6 | 37 |
| 8 | 5b | LTv | 30 | 4.7 | 11 |
| 9 | 5c | LTv | 30 | 5 | 70 |
| 10 | 5a | Novozym 51003 | 50 | 6 | 34 |
| 11 | 5b | Novozym 51003 | 50 | 7 | 35 |
| 12 | 5c | Novozym 51003 | 50 | 6 | 21 |
| 13 | 6a | LTv | 30 | 5 | 39 |
| 14 | 6b | LTv | 30 | 5 | <1 |
| 15 | 6c | LTv | 30 | 5.5 | 8 |
| 16 | 6a | Novozym 51003 | 50 | 6 | 54 |
| 17 | 6b | Novozym 51003 | 50 | 6 | 3 |
| 18 | 6c | Novozym 51003 | 50 | 5 | 10 |
| 19 | 7a | LTv | 30 | 6 | 14 |
| 20 | 7b | LTv | 30 | 4.7 | 23 |
| 21 | 7c | LTv | 30 | 4.7 | 8 |
| 22 | 7a | Novozym 51003 | 50 | 6 | 16 |
| 23 | 7b | Novozym 51003 | 50 | 7 | 23 |
| 24 | 7c | Novozym 51003 | 50 | 5 | 5 |
| 25 | 8a | LTv | 30 | 4.7 | 8 |
| 26 | 8b | LTv | 30 | 4.7 | 5 |
| 27 | 8c | LTv | 30 | 4.7 | 4 |
| 28 | 8a | Novozym 51003 | 50 | 6 | 9 |
| 29 | 8b | Novozym 51003 | 50 | 6 | 16 |
| 30 | 8c | Novozym 51003 | 50 | 6 | 2 |
| 31 | 9a | LTv | 30 | 6 | 39 |
| 32 | 9b | LTv | 30 | 5 | 36 |
| 33 | 9c | LTv | 30 | 5 | 11 |
| 34 | 9a | Novozym 51003 | 50 | 6 | 24 |
| 35 | 9b | Novozym 51003 | 50 | 6 | 32 |
| 36 | 9c | Novozym 51003 | 50 | 6 | 3 |
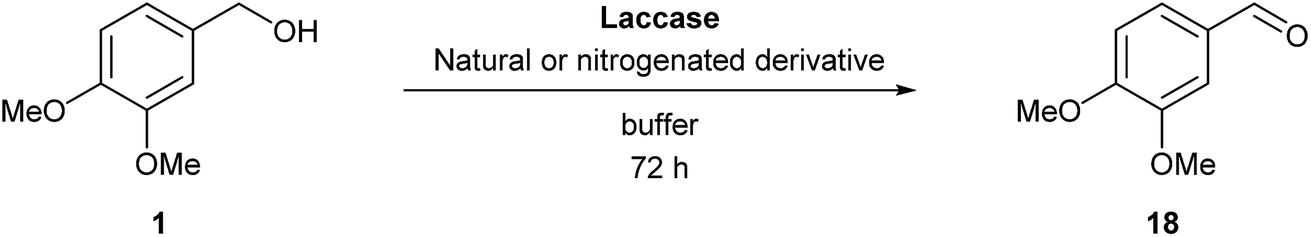
The process was also carried out in the presence of vanillin and syringaldehyde (entries 1–4), to compare the behavior of these natural derivatives with the chemically modified ones, finding an improvement in the conversion values for several of the newly nitrogenated synthesized compounds. Still some of them displayed very low activity even at these long reaction times, as was the case of the nitrile derivative 4a, which led to less than 10% conversion with both enzymes LTv and Novozym 51003 (entries 5 and 6). On the contrary, oximes 5a–c exhibited remarkable activities, 5c providing with LTv the highest value of all derivatives tested with 70% conversion (entry 9). The reaction exceeded 30% conversion when employing vanillin and syringyl oximes 5a and 5b, the process being promoted to 37% conversion at pH 6 with 5a and LTv, and 34% when Novozym 51003 was employed under the same pH conditions at 50 °C (entries 7 and 10). In an attempt to increase those values, the use of different cosolvents was tested but resulted ineffective (see Table S4 in the ESI†), and longer reaction times led to the disappearance of the product signal in the GC chromatogram. We hypothesized that the product was degraded, partially to form the veratryl acid, which was identified in the reaction analysis.
The presence of an additional methoxy group in oxime 5b did not affect significantly the performance of Novozym 51003, obtaining a conversion of 35% at pH 7 (entry 11), while the reaction was disfavored with LTv (entry 8), especially at higher pHs (see Table S9 in the ESI†).
When the vanillin hydrazone 6a was studied, good values of conversion were observed (entries 13 and 16), obtaining 54% in combination with Novozym 51003 at pH 6 (entry 16). On the opposite, 6b led to low or null conversions with both enzymes (entries 14 and 17). The results were slightly better but still poor with the dihydroxylated derivative 6c (entries 15 and 18), not improving in the presence of an organic cosolvent (see Table S15 in the ESI†). Conversely to the hydrazones, the semicarbazone derived from syringaldehyde 7b (entries 20 and 23) showed higher activities than the corresponding analogue from vanillin 7a (entries 19 and 22). As for the hydrazone family, the dihydroxylated semicarbazone 7c displayed the lowest conversions (entries 21 and 24).
In general, the triazole family was poorly active, affording the dimethoxylated derivative 8b the highest conversion value in the reaction with Novozym 51003 at pH 6 (entry 29). Interestingly, tetrazole derivatives 9a and 9b showed enhanced conversions (entries 31–32 and 34–35). As for the case of semicarbazones 7a–b and triazoles 8a–b, Novozym 51003 exhibited higher acceptance to the syringyl-derived substrate than to the corresponding vanillin-derived one (entries 34 and 35), behaving positively the additional methoxy group. On the contrary, when the reaction was carried out in the presence of the dihydroxylated derivative 9c, the conversions decayed significantly, especially for Novozym 51003 acting as catalyst (entry 36).
As the next step, we decided to increase the veratryl alcohol concentration for those cases where higher conversions were found. This would allow us to study the effect of this parameter in the reaction outcome and also to obtain a higher productivity of the aldehyde 18 in the process. Remarkably, at 10 mM substrate concentration, conversions suffered just a slight decrease in some cases, maintaining the previous values in other cases, resulting in an enhancement of the relative activity for all derivatives tested (Fig. 2).
 | ||
| Fig. 2 Relative activity representing micromoles of alcohol 1 converted into aldehyde 18 at different substrate concentrations, using the new synthesized compounds as co-oxidants, and Novozym 51003 (up) or LTv (down) as biocatalyst. 100% relative activity corresponds to 0.42 μmol 18 per μL mg−1 of enzyme (entry 9, Table 3). Reactions with relative activities higher than 200% were performed by duplicate, showing similar results. | ||
Noteworthy, it was possible to rise the substrate concentration until 15 mM and 20 mM, maintaining good levels of catalytic activity and, therefore, improving the productivities. For instance, relative activity values of 186–295% were found with oxime 5b, hydrazone 6a and tetrazole 9b within the Novozym 51003-catalyzed reactions (Fig. 2, top). In general, LTv showed less alcohol concentration tolerance, but even so oxime 5c displayed an excellent activity in the presence of this enzyme (286%, Fig. 2, bottom).
Finally, the methodology was extended to the oxidation of the benzylic position of the dimeric lignin model compound adlerol 2 using the derivatives that displayed the higher activities with the laccases for the oxidation of 1 (Fig. 3). The reaction conditions were taken in analogy to the previously shown for veratryl alcohol.
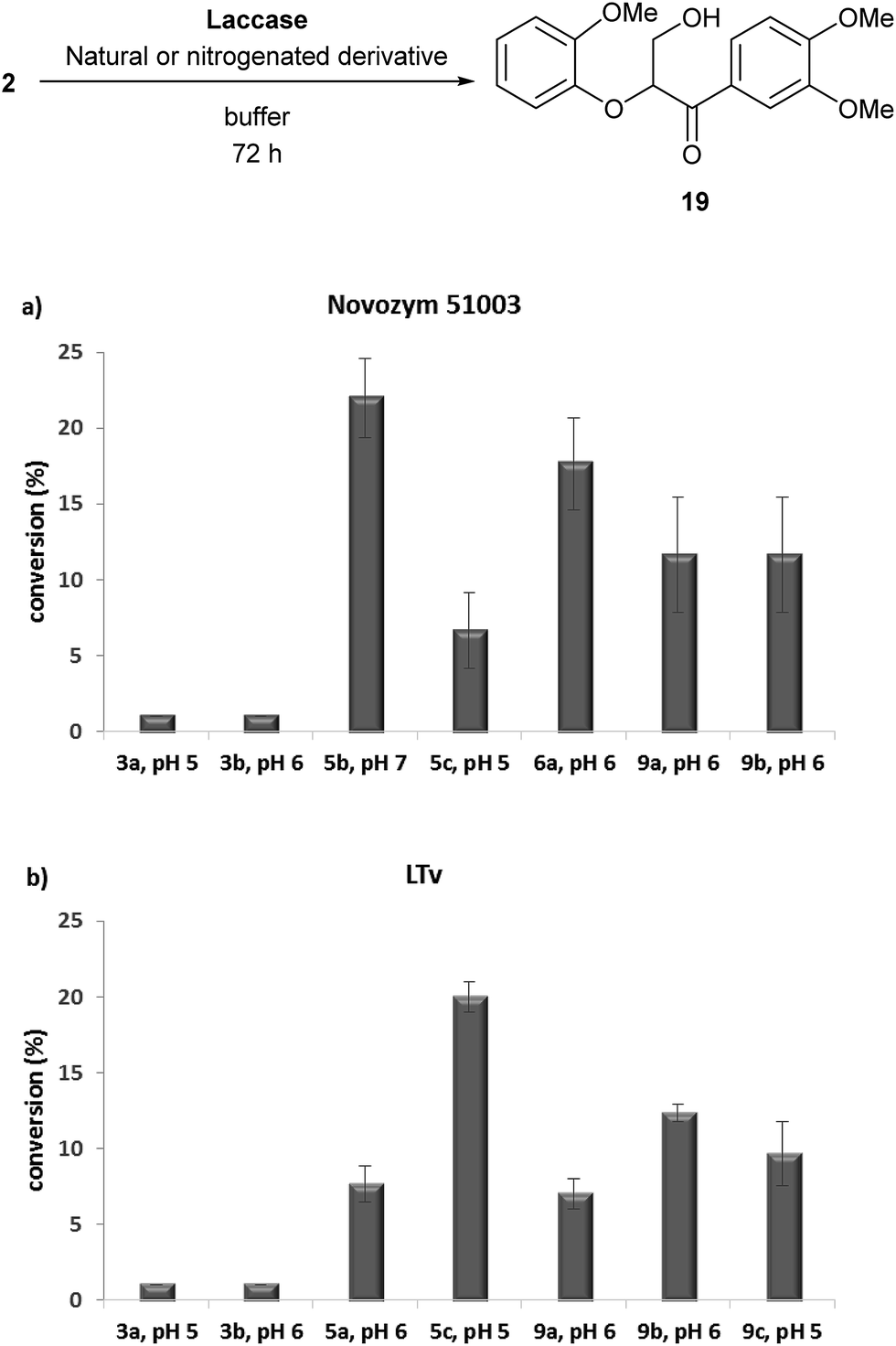 | ||
| Fig. 3 Oxidation of adlerol (2) using natural or nitrogenated lignin-derived compounds with: (a) Novozym 51003; or (b) LTv. All the experiments were performed by triplicate. | ||
Despite the conversions were lower than for the case of 1, significant improvements were accomplished with some derivatives, obtaining the best results (22% conversion) for the combination of oxime 5b with Novozym 51003, which is a remarkable value when considering the examples reported in the literature using vanillin or syringyl-type natural mediators in this kind of process.22 In order to verify the improvement over the natural compounds, 3a and 3b were tested, observing no formation of hydroxy ketone 19.
Oxime 5c showed significant conversions in the LTv-catalyzed process, as well as tetrazoles 9b and 9c (Fig. 3b). On the contrary, the use of Novozym 51003 as catalyst resulted in high conversion values with derivatives 6a, 9a and 9b (Fig. 3a). We figure out that the lower conversion values may be caused by a fast reaction of the phenolic compound with the laccase, leading to the formation of inactive species and disabling the oxidative capacity in the adlerol transformation. As a final remark, it must be mentioned that coupling between the synthesized nitrogenated derivatives and the substrates (veratryl alcohol and adlerol) was never observed.
Conclusions
The chemical modification of some lignin-type monomers such as vanillin and syringaldehyde has been carried out through simple methodologies toward the synthesis of several families of nitrogenated compounds. These derivatives were envisaged as suitable candidates for LMS due to the stabilization of the phenoxy radicals formed. Their combination with two commercially available laccases, the one from Trametes versicolor and the one from Myceliophthora thermophila, allowed their dimerization or oligomerization, showing that they could act as substrates for laccases.The application of these compounds in oxidative processes of lignin model units in laccase-mediator systems has been investigated employing these commercial laccases, showing much better conversions than the natural compounds vanillin and syringaldehyde. Remarkably, both the substitution pattern of the aromatic ring and the pH have shown a dramatic influence in the reactivity behavior, conducting to higher conversions according to the presence of additional methoxy and/or hydroxyl groups. On the one hand, veratryl alcohol was transformed into veratraldehyde in high conversions at 3 mM, and the increase in the substrate concentration up to 20 mM led to higher relative activity values (up to 295%). On the other hand, lower conversions were obtained in the oxidation of the lignin model dimer adlerol, but these results are a good starting point for future studies in the enzymatic modification of lignin surfaces.
Experimental section
General methods
Chemical reagents employed in the synthesis of starting and reference materials were purchased from Sigma-Aldrich. Solvents for chemical reactions were used without further treatment.Laccase from Trametes versicolor (2.5 U mg−1) was purchased from Sigma, while the laccase from Myceliophthora thermophila (Novozym 51003, 3.14 U μL−1) was acquired from Novozymes. Enzymatic activities were measured using the ABTS assay.23
Thin-layer chromatography (TLC) was conducted using Merck Silica Gel 60 F254 precoated plates and visualized with UV and potassium permanganate stain. Column chromatographies were performed using silica gel 60 (230–400 mesh). 1H-NMR, 13C-NMR, and DEPT experiments were registered using different Bruker spectrometers (1H, 300.13 MHz and 13C, 75.5 MHz). The chemical shifts (δ) are given in ppm and the coupling constants (J) in Hertz (Hz). High resolution mass spectra (HRMS) experiments were carried out by ESI+ or ESI− using an Impact II ESI-Q-Tof spectrometer.
Synthesis of 4-hydroxy-3-methoxybenzonitrile (4a)24
Hydroxylamine hydrochloride (82 mg, 1.18 mmol) was added to a solution of vanillin (3a, 150 mg, 0.986 mmol) in acetonitrile (1 mL) and the mixture was stirred under reflux for 4 h. After cooling the mixture at room temperature, H2O was added and the reaction was extracted with Et2O (3 × 10 mL). The organic layers were combined, washed with an aqueous saturated NaHCO3 solution (15 mL) and dried over Na2SO4. The solvent was removed by distillation under reduced pressure and the reaction crude was purified by column chromatography on silica gel (eluent: 30% EtOAc/hexane) yielding the nitrile derivative 4a in 85% isolated yield. White solid. Rf (50% EtOAc/hexane): 0.80. Mp: 86–88 °C. 1H NMR (300.13 MHz, MeOD-d4): δ 3.87 (s, 3H), 4.91 (br s, 1H), 6.87 (d, 3JHH = 8.1 Hz, 1H), 7.18 (dd, 3JHH = 8.1, 4JHH = 1.9 Hz, 1H), 7.21 (d, 3JHH = 1.7 Hz, 1H). 13C NMR (75.5 MHz, MeOD-d4): δ 56.6 (CH3), 103.1 (C), 115.7 (CH), 117.0 (CH), 120.5 (C), 127.7 (CH), 149.3 (C), 152.7 (C). HRMS (ESI+, m/z): calcd for (C8H8NO2)+ (M + H)+ 150.0550, found: 150.0548.General procedure for the synthesis of oximes 5a–c
Hydroxylamine sulfate (650 mg, 3.95 mmol) and sodium acetate (447 mg, 3.29 mmol) were successively added to a solution of 3a–c (3.29 mmol) in ethanol (3.3 mL). The mixture was stirred under reflux for 4 h, cooled at room temperature, resuspended in CH2Cl2 (10 mL), filtered and washed with CH2Cl2 (2 × 2 mL). The solvent of the filtrate was evaporated under reduced pressure obtaining pure oxime 5a. Oxime 5b was submitted to column chromatography (eluent: 30% EtOAc/hexane) for further purification.For the isolation of oxime 5c, the solvents were evaporated and the residue was redissolved in H2O. The mixture was extracted with EtOAc (3 × 5 mL), the organic layers were combined, dried over Na2SO4 and concentrated in vacuo, affording pure 5c.
General procedure for the synthesis of hydrazones 6a–c
Hydrazine monohydrate (10, 207 μL, 6.57 mmol) and glacial acetic acid (10 drops) were successively added to a solution of 3a–c (6.57 mmol) in ethanol (10 mL). The mixture was stirred under reflux for 50 min or 1 h (see Table 1), and the solvent was removed by distillation under reduced pressure isolating 6a–c in 78–90% yield.General procedure for the synthesis of semicarbazones 7a–c
Semicarbazide hydrochloride (11, 733 mg, 6.57 mmol) and glacial acetic acid (10 drops) were successively added to a solution of 3a–c (6.57 mmol) in ethanol (10 mL). The mixture was stirred under reflux for 1.5–2.7 h (see Table 1) and the solvent was removed by distillation under reduced pressure, isolating semicarbazones 7a–c in good to excellent yields (73–98%).General procedure for the synthesis of triazoles 8a–c
4-Aminotriazole (12, 55 mg, 0.657 mmol) and an aqueous concentrated sulfuric acid solution (5 drops) were successively added to a solution of 3a–c (0.657 mmol) in ethanol (2 mL). The mixture was stirred under reflux for 3–7.5 h (see Table 1) and the resulting precipitate was filtrated from the reaction media, yielding 8a–c in 96–99% yield.General procedure for the synthesis of hydrazono tetrazoles 9a–c
5-Hydrazinyl-1H-tetrazole (13, 227 mg, 2.27 mmol) and an aqueous concentrated HCl solution (5 drops) were successively added to a solution of 3a–c (2.27 mmol) in ethanol (4 mL). The mixture was stirred under reflux for 1.75–5 h (see Table 1) and the resulting precipitate was filtrated from the reaction media, yielding the corresponding tetrazoles 9a–c in 80–97% yield.Synthesis of 5-hydrazinyl-1H-tetrazole 13
5-Aminotetrazole monohydrate (5 g, 48.51 mmol) was dissolved in a 15% aqueous NaOH solution (50 mL). The mixture was heated at 100 °C and KMnO4 (7 g) was slowly added by portions until the colour of the solution no longer changed. After stirring 30 min, ethanol was added to eliminate the excess of KMnO4. The warm solution was filtered and concentrated under reduced pressure to 1/3 of the initial volume. Sodium 5,5′-azotetrazolate was crystallized as a yellow solid (6.2 g, 61% yield).To a suspension of sodium 5,5′-azotetrazolate (6.2 g, 29.51 mmol) in water (62 mL), an aqueous HCl 6 N solution (13 mL) was added. The mixture was warmed until the gas evolution finished, then evaporated to dryness from water three times to ensure the removal of HCl. The residue was dissolved in the minimum volume of hot water and a hot solution of NaOAc (6.2 g, 75.58 mmol) in water (6.2 mL) was added. The apparatus was flushed out with carbon dioxide and the solution was allowed to cool to give 5-hydrazinyl-1H-tetrazole (13) as a white solid (987 mg, 20% yield). Mp: 192–194 °C. 1H NMR (300.13 MHz, DMSO-d6): δ 8.17 (s, 4H). 13C NMR (75.5 MHz, DMSO-d6): δ 161.4 (C).
General procedure for the laccase-catalyzed oxidation of the new synthesized compounds
In a test tube, the corresponding derivative was dissolved in an organic cosolvent (EtOH or DMSO, 300 μL). Then, the corresponding buffer saturated with O2 prior to the experiment (2.7 mL) and the commercial laccases from Trametes versicolor (25 U) or the laccase from Myceliophthora thermophila (Novozym 51003, 31.4 U), were successively added. A balloon that was filled with oxygen was attached to the test tube and the mixture was stirred at 30 °C or 50 °C for the time shown in Table 2. Then, the reaction was finished by extraction with EtOAc (3 × 2 mL), and the organic layers were combined and dried with Na2SO4. After solvent evaporation under reduced pressure, the reaction crude was analyzed by 1H-NMR. In those cases in which an abundant precipitate was detected in the reaction medium, this was separated by decantation and analyzed by 1H-NMR.General procedure for the laccase-catalyzed oxidation of lignin model compounds in the presence of natural or nitrogenated compounds
The corresponding derivative (16 μmol) was placed in a test tube. The buffer (2 mL) saturated with O2 prior to the experiment (pH 4.7: acetate buffer 100 mM; pH 5–6: citric buffer 50 mM; pH 7: KPi buffer 100 mM) and the veratryl alcohol (1, 6 μmol) or adlerol (2, 6 μmol) were added. After stirring, the commercial laccases from Trametes versicolor (25 U) or the laccase from Myceliophthora thermophila (Novozym 51003, 31.4 U), were added and the reaction stirred at 30 or 50 °C, respectively, during 72 h. Then, the reaction was acidified with an aqueous HCl 2 N solution (4 drops) and extracted with EtOAc (2 × 1 mL). The organic layers were combined and dried with Na2SO4. For the oxidations of veratryl alcohol, an aliquot was taken and analyzed by GC. In the case of the oxidation of adlerol, the solvent of the organic layers was evaporated and the reaction crudes were analyzed by 1H-NMR. Thus, the conversions were calculated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Ministry of Economy and Competitiveness and the European Union (Project OXYPOL, PCIN-2014-134 from an ERA-NET ERA-IB 2 Call).Notes and references
- (a) O. Y. Abdelaziz, D. P. Brink, J. Prothmann, K. Ravi, M. Sun, J. García-Hidalgo, M. Sandahl, C. P. Hulteberg, C. Turner, G. Lidén and M. F. Gorwa-Grauslund, Biotechnol. Adv., 2016, 34, 1318–1346 CrossRef CAS PubMed; (b) S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 4862–4887 RSC; (c) K. Wang, H. Yang, S. Guo, X. Yao and R.-C. Sun, J. Appl. Polym. Sci., 2014, 131, 39673 CrossRef.
- (a) X. Huang, O. M. M. Gonzalez, J. Zhu, T. I. Korányi, M. D. Boot and E. J. M. Hensen, Green Chem., 2017, 19, 175–187 RSC; (b) H. Yang, J. Li, J. Xu and L. Mo, BioResources, 2017, 12, 344–361 CAS; (c) H. Zhu, J. P. Max, C. L. Marcum, H. Luo, M. M. Abu-Omar and H. I. Kenttämaa, J. Am. Soc. Mass Spectrom., 2016, 27, 1813–1823 CrossRef CAS PubMed; (d) M. D. Kärkäs, B. S. Matsuura, T. M. Monos, G. Magallanes and C. R. J. Stephenson, Org. Biomol. Chem., 2016, 14, 1853–1914 RSC; (e) S. Huang, N. Mahmood, M. Tymchyshyn, Z. Yuan and C. Xu, Bioresour. Technol., 2014, 171, 95–102 CrossRef CAS PubMed.
- (a) G. de Gonzalo, D. I. Colpa, M. H. M. Habib and M. W. Fraaije, J. Biotechnol., 2016, 236, 110–119 CrossRef CAS PubMed; (b) F. Asina, I. Brzonova, K. Voeller, E. Kozliak, A. Kubátová, B. Yao and Y. Ji, Bioresour. Technol., 2016, 220, 414–424 CrossRef CAS PubMed; (c) L. Zhang, T. You, T. Zhou, L. Zhang and F. Xu, BioResources, 2016, 11, 3972–3986 CAS; (d) D. Salvachúa, R. Katahira, E. N. S. Cleveland, P. Khanna, M. G. Resch, B. A. Black, S. O. Purvine, M. Zink, A. Prieto, M. J. Martínez, A. T. Martínez, B. A. Simmons, J. M. Gladden and G. T. Beckham, Green Chem., 2016, 18, 6046–6062 RSC; (e) T. D. H. Bugg, M. Ahmad, E. M. Hardiman and R. Rahmanpour, Nat. Prod. Rep., 2011, 28, 1883–1896 RSC.
- (a) Z. Ahmad, M. Asgher, F. Hussain and M. A. Randhawa, BioResources, 2016, 11, 10511–10527 CrossRef CAS; (b) C. Zhao, S. Xie, Y. Pu, R. Zhang, F. Huang, A. J. Ragauskas and J. S. Yuan, Green Chem., 2016, 18, 1306–1312 RSC; (c) P. Picart, L. Wiermans, M. Pérez-Sánchez, P. M. Grande, A. Schallmey and P. Domínguez de María, ACS Sustainable Chem. Eng., 2016, 4, 651–655 CrossRef CAS.
- (a) Y. Ohta, R. Hasegawa, K. Kurosawa, A. H. Maeda, T. Koizumi, H. Nishimura, H. Okada, C. Qu, K. Saito, T. Watanabe and Y. Hatada, ChemSusChem, 2017, 10, 425–433 CrossRef CAS PubMed; (b) L. Yang, K. Seshan and Y. Li, Catal. Today, 2017, 298, 276–297 CrossRef CAS; (c) T. D. Bugg and R. Rahmanpour, Curr. Opin. Chem. Biol., 2015, 29, 10–17 CrossRef CAS PubMed.
- (a) X.-Y. Jiang, Q. Lu, X.-N. Ye, B. Hu and C.-Q. Dong, BioResources, 2016, 11, 6232–6243 CAS; (b) M. Akazawa, Y. Kato and Y. Kojima, J. Anal. Appl. Pyrolysis, 2016, 122, 355–364 CrossRef CAS.
- (a) L. T. M. Pham, S. J. Kim and Y. H. Kim, Biotechnol. Biofuels, 2016, 9, 247 CrossRef PubMed; (b) E. Rosini, C. Allegretti, R. Melis, L. Cerioli, G. Conti, L. Pollegioni and P. D'Arrigo, Catal. Sci. Technol., 2016, 6, 2195–2205 RSC.
- (a) M. D. Cannatelli and A. J. Ragauskas, Chem. Rec., 2017, 17, 122–140 CrossRef CAS PubMed; (b) T. Kudanga, B. Nemadziva and M. L. Roes-Hill, Appl. Microbiol. Biotechnol., 2017, 101, 13–33 CrossRef CAS PubMed; (c) M. Mogharabi and M. A. Faramarzi, Adv. Synth. Catal., 2014, 356, 897–927 CrossRef CAS; (d) S. Witayakran and A. J. Ragauskas, Adv. Synth. Catal., 2009, 351, 1187–1209 CrossRef CAS; (e) S. Riva, Trends Biotechnol., 2005, 24, 219–226 CrossRef PubMed.
- (a) B. Yao, P. Kolla, R. Koodali, Y. Ding, S. Balaranjan, S. Shrestha and A. Smirnova, New J. Chem., 2017, 41, 958–964 RSC; (b) S. Xie, Q. Sun, Y. Pu, F. Lin, S. Sun, X. Wang, A. J. Ragauskas and J. S. Yuan, ACS Sustainable Chem. Eng., 2017, 5, 2215–2223 CrossRef CAS; (c) Q. Wang, S. Liu, G. Yang and J. Chen, Bioresour. Technol., 2015, 175, 638–641 CrossRef CAS PubMed.
- (a) L. Munk, A. M. Punt, M. A. Kabel and A. S. Meyer, RSC Adv., 2017, 7, 3358–3368 RSC; (b) M. D. Cannatelli and A. J. Ragauskas, Appl. Microbiol. Biotechnol., 2016, 100, 8685–8691 CrossRef CAS PubMed; (c) D. Huber, A. Pellis, A. Daxbacher, G. S. Nyanhongo and G. M. Guebitz, Polymers, 2016, 8, 280/1–280/10 CrossRef CAS; (d) H. Wu, J. Noro, Q. Wang, X. Fan, C. Silva and A. Cavaco-Paulo, RSC Adv., 2016, 6, 90427–90434 RSC; (e) I. F. Fiţigău, C. G. Boeriu and F. Peter, Macromol. Symp., 2015, 352, 78–86 CrossRef.
- (a) J. González-Sabín, N. Ríos-Lombardía, I. García, N. M. Vior, A. F. Braña, C. Méndez, J. A. Salas and F. Morís, Green Chem., 2016, 18, 989–994 RSC; (b) A. Díaz-Rodríguez, L. Martínez-Montero, I. Lavandera, V. Gotor and V. Gotor-Fernández, Adv. Synth. Catal., 2014, 356, 2321–2329 CrossRef; (c) K. Kędziora, A. Díaz-Rodríguez, I. Lavandera, V. Gotor-Fernández and V. Gotor, Green Chem., 2014, 16, 2448–2453 RSC.
- (a) A. Kirsch, K. Ostendorf, A. Kharazipour and M. Euring, BioResources, 2016, 11, 7091–7101 CrossRef CAS; (b) A. Rico, J. Rencoret, J. C. del Río, A. T. Martínez and A. Gutiérrez, Biotechnol. Biofuels, 2014, 7, 6 CrossRef PubMed; (c) P. Könst, S. Kara, S. Kochius, D. Holtmann, I. W. C. E. Arends, R. Ludwig and F. Hollmann, ChemCatChem, 2013, 5, 3027–3032 CrossRef; (d) T. Rosado, P. Bernardo, K. Koci, A. V. Coelho, M. P. Robalo and L. O. Martins, Bioresour. Technol., 2012, 124, 371–378 CrossRef CAS PubMed; (e) A. I. Cañas and S. Camarero, Biotechnol. Adv., 2010, 28, 694–705 CrossRef PubMed.
- (a) T. Saravanakumar, H.-S. Park, A.-Y. Mo, M.-S. Choi, D.-H. Kim and S.-M. Park, J. Mol. Catal. B: Enzym., 2016, 134, 196–205 CrossRef CAS; (b) S. Slagman, J. Escorihuela, H. Zuilhof and M. C. R. Franssen, RSC Adv., 2016, 6, 99367–99375 RSC; (c) R. Reiss, J. Ihssen, M. Richter, E. Eichhorn, B. Schilling and L. Thöny-Meyer, PLoS One, 2013, 8, e65633 CAS.
- J. Ravindran, G. V. Subbaraju, M. V. Ramani, B. Sung and B. B. Aggarwal, Biochem. Pharmacol., 2010, 79, 1658–1666 CrossRef CAS PubMed.
- H. E. Master, S. I. Khan and K. A. Poojari, Bioorg. Med. Chem., 2005, 13, 4891–4899 CrossRef CAS PubMed.
- H. S. Patel and K. B. Patel, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 2443–2452 CrossRef CAS.
- (a) M.-J. Han, H.-T. Choi and H.-G. Song, J. Microbiol., 2006, 43, 555–560 Search PubMed; (b) P. Cassland and L. J. Jönsson, Appl. Microbiol. Biotechnol., 1999, 52, 393–400 CrossRef CAS PubMed.
- R. M. Berka, P. Schneider, E. J. Golightly, S. H. Brown, M. Madden, K. M. Brown, T. Halkier, K. Mondorf and F. Xu, Appl. Environ. Microbiol., 1997, 63, 3151–3157 CAS.
- Dimethylsulfoxide is known as radical scavenger, but it has been described as a useful organic co-solvent in laccase-catalyzed reactions. See, for instance ref. 11c.
- (a) B. Ramalingam, B. Sana, J. Seayad, F. J. Ghadessy and M. B. Sullivan, RSC Adv., 2017, 7, 11951–11958 RSC; (b) N. H. Pham, F. Hollmann, D. Kracher, M. Preims, D. Haltrich and R. Ludwig, J. Mol. Catal. B: Enzym., 2015, 120, 38–46 CrossRef CAS; (c) K. Rittstieg, A. Suurnakki, T. Suortti, K. Kruus, G. Guebitz and J. Buchert, Enzyme Microb. Technol., 2002, 31, 403–410 CrossRef CAS.
- See, for instance: (a) J. Safaei-Ghomi and A. R. Hajipour, Org. Prep. Proced. Int., 2011, 43, 372–376 CrossRef CAS; (b) D. Enders, L. Wortmann and R. Peters, Acc. Chem. Res., 2000, 33, 157–169 CrossRef CAS PubMed.
- (a) P. Nousiainen, J. Kontro, H. Manner, A. Hatakka and J. Sipilä, Fungal Genet. Biol., 2014, 72, 137–149 CrossRef CAS PubMed; (b) T. M. Larson, A. M. Anderson and J. O. Rich, Biotechnol. Lett., 2013, 35, 225–231 CrossRef CAS PubMed; (c) M. Díaz-González, T. Vidal and T. Tzanov, Appl. Microbiol. Biotechnol., 2011, 89, 1693–1700 CrossRef PubMed; (d) R. Bourbonnais and M. G. Paice, FEBS Lett., 1990, 267, 99–102 CrossRef CAS PubMed.
- M. L. Niku-Paavola, L. Raaska and M. Itävaara, Mycol. Res., 1990, 94, 27–31 CrossRef.
- V. R. Kanetkar, D. D. Zope and Y. V. V. Subrahmanyam, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2005, 44, 1317–1318 Search PubMed.
- R. B. de Oliveira, E. M. de Souza-Fagundes, R. P. P. Soares, A. A. Andrade, A. U. Krettli and C. L. Zani, Eur. J. Med. Chem., 2008, 43, 1983–1988 CrossRef CAS PubMed.
- M. Delomenède, F. Bedos-Belval, H. Duran, C. Vindis, M. Baldas and A. Nègre-Salvayre, J. Med. Chem., 2008, 51, 3171–3181 CrossRef PubMed.
- S. Grelier, H. Cramail, A. Llevot, S. Carlotti and E. Grau, Patent WO 2016/050988 A1, 2016.
- D. R. Kelly, S. C. Baker, D. S. King, D. S. de Silva, G. Lord and J. P. Taylor, Org. Biomol. Chem., 2008, 6, 787–796 CAS.
- A. M. Costero, S. Gil, M. Parra, P. M. E. Mancini, M. N. Kneeteman and M. I. Quindt, Tetrahedron Lett., 2015, 56, 3988–3991 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: This file includes extensive enzymatic optimization studies, analytical data for the oxidation of adlerol and copy of 1H, 13C and DEPT NMR spectra for novel synthesized compounds. See DOI: 10.1039/c7ra10497a |
| This journal is © The Royal Society of Chemistry 2017 |