DOI:
10.1039/C7RA11365J
(Paper)
RSC Adv., 2017,
7, 51986-51992
A new seco-pimarane diterpene and four new β-resorcylic acid lactones from a fungicolous Hypomyces subiculosus†
Received
15th October 2017
, Accepted 2nd November 2017
First published on 9th November 2017
Abstract
One new pimarane diterpene (1) and four new β-resorcylic acid lactones (2–5), together with eight known compounds (6–13) were isolated from the solid culture of the fungicolous Hypomyces subiculosus. The structures of the new compounds were elucidated using extensive spectroscopic methods, including 1D and 2D NMR, HRESIMS, CD experiments, and chemical means. Compound 1 is a ring cleavage pimarane diterpene possessing a bicyclic ring system. All compounds were evaluated for cytotoxic activities. Only compound 7 showed moderate cytotoxicity against the A549 cell line with an IC50 of 44.5 μM.
1. Introduction
Fungicolous fungi refer to a group of fungi growing on the mycelium or sporomes of other fungi as parasites, commensals or saprobes.1 They have been proven to be a rich source of new and biologically active natural products, with examples including lowdenic acid,2 thiersinines,3 solanapyrones,4 phomalevones,5 and botryolides.6 The fungi in the genus of Hypomyces are mycoparasitic and are distributed worldwide. Since the first report of sepedonin from Hypomyces in 1970,7 26 natural products possessing diverse biological activities, such as antitumor, antibacterial, antimalarial, antifungal and hyperglycemic activities, have been identified from Hypomyces sp.8–10
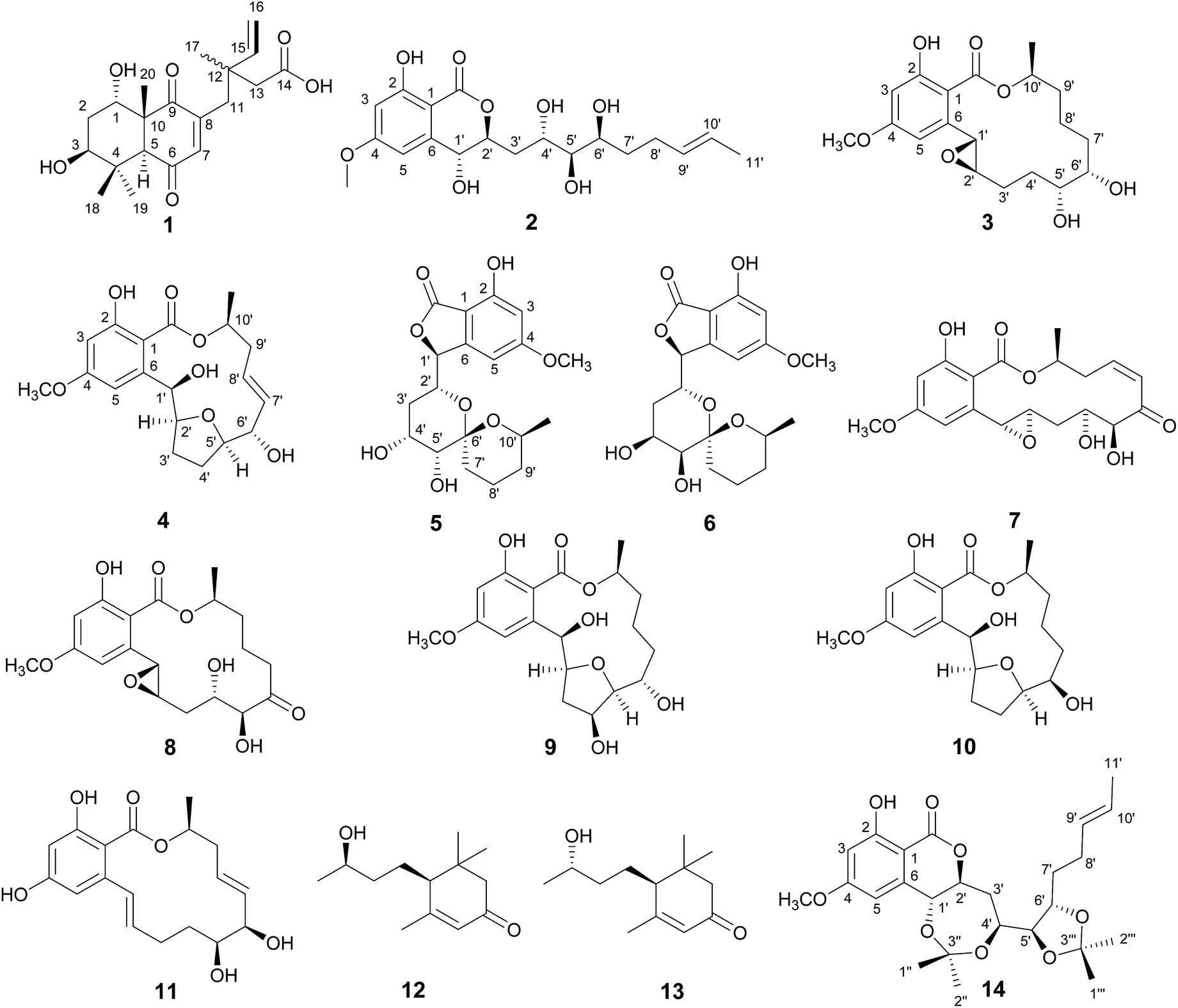
In our search for new bioactive natural products from fungi, a fungicolous fungus of Hypomyces subiculosus obtained from Polyporus versicolor was chemically investigated. This fungus was fermented on rice and extracted with ethyl acetate to afford the organic solvent extract. A detailed chemical investigation on EtOAc extract led to the isolations of five new compounds, subiculosin A (1), aigialomycin H (2), aigialomycin I (3), paecilomycin N (4), 4′R,5′R-dihydroaigialospirol (5), together with eight known compounds, 7′,8′-dihydroaigialospirol (6),11 hypothemycin (7),12 dihydrohypothemycin (8),13 paecilomycin J (9),14 paecilomycin L (10),14 aigialomycin D (11),13 blumenol C (12),15 9-epi-blumenol C (13).15 In this work, we reported the isolation, structural determination with the absolute stereochemistry, and bioactivity evaluation of these compounds (Fig. 1).
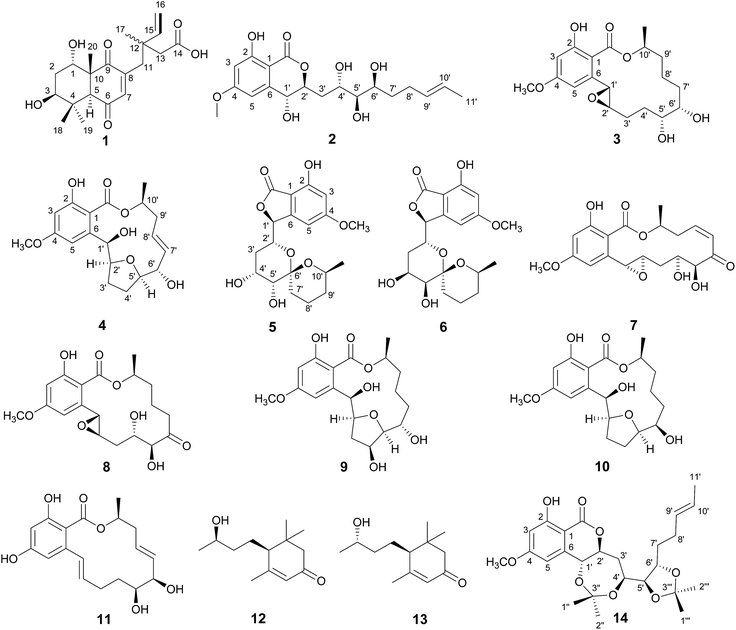 |
| | Fig. 1 Structures of compounds 1–14. | |
2. Results and discussion
Structural elucidation
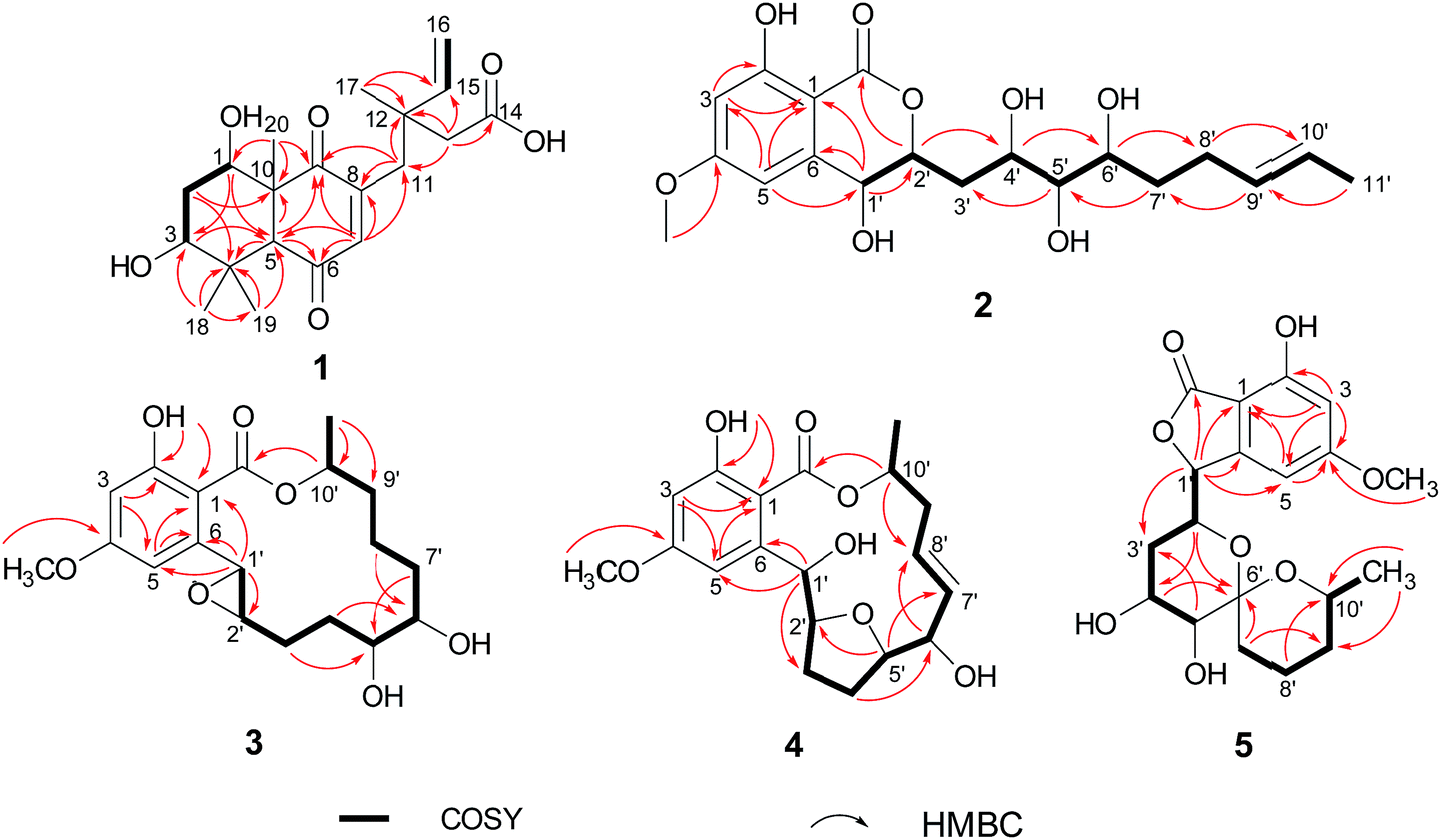
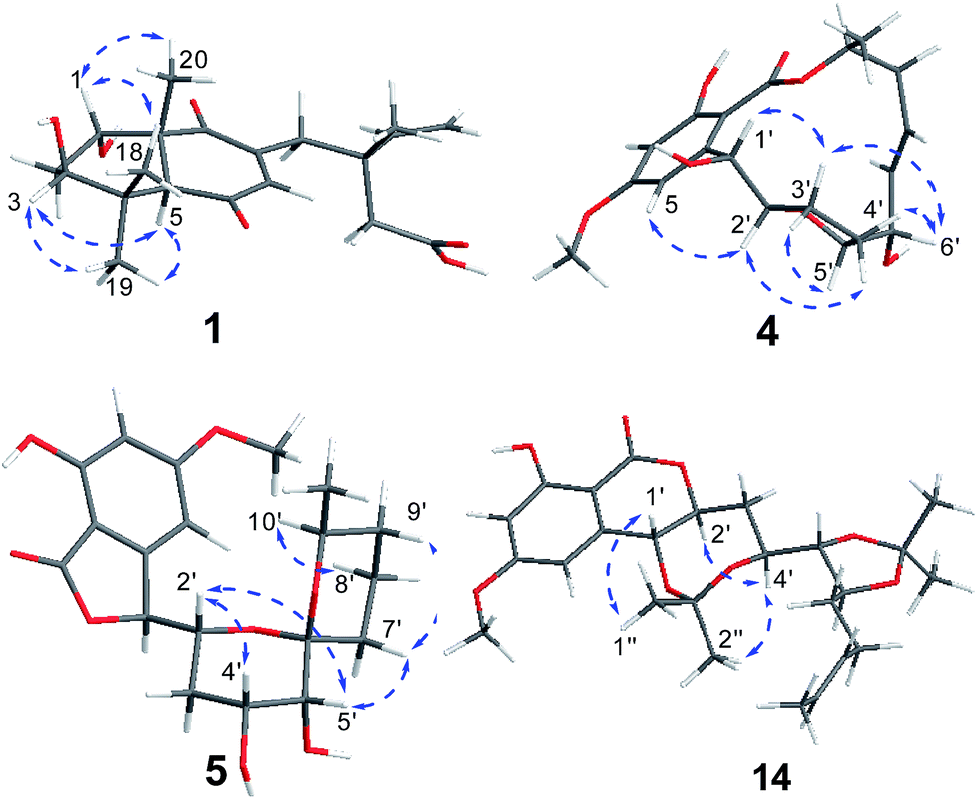
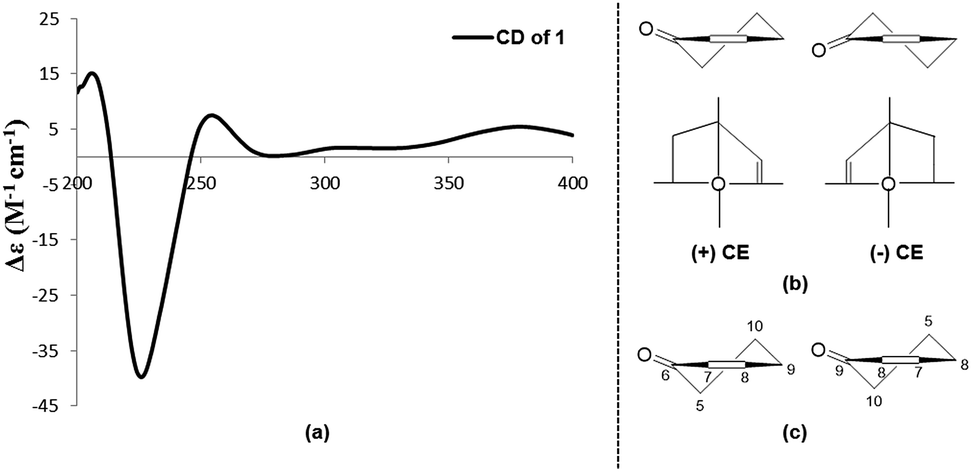
Subiculosin A (1) was obtained as pale yellow power. Its molecular formula was determined to be C20H28O6 with 7 degrees of unsaturation by HRESIMS spectrum at m/z [M + H]+ 365.1962. The 1H NMR (Table 1) of 1 showed signals due to four olefinic protons [δH 4.95 (m, 2H), 5.90 (dd, J = 17.5, 10.9 Hz), 6.60 s], three methylenes [δH 1.81 (dt, J = 10.1, 2.9 Hz); 2.37 (d, J = 3.1 Hz); 2.65 (d, J = 12.5 Hz), 2.54 (d, J = 12.5 Hz)], three methines [two oxygenated methines at xH 3.69 (dd, J = 10.1, 6.2 Hz), 4.23 (t, J = 3.1 Hz), respectively], and four methyl groups [δH 1.11 (s), 1.17 (s, 6H), 1.25 (s)]. The 13C NMR of 1 exhibited twenty carbon resonances corresponding to three carbonyl carbons (δC 172.8, 200.3, 204.0), four olefinic carbons (δC 112.9, 142.6, 146.0, 146.2), three sp3 quaternary carbons (δC 39.0, 40.8, 56.2), three methine carbons (δC 56.3, 71.5, 72.1) including two oxygenated methine carbons, three methylene carbons (δC 34.0, 37.5, 44.2), and four methyl carbons (δC 16.0, 21.8, 24.2, 28.3). Extensive analysis of the 1H–1H COSY correlations of H-1–H2-2–H-3, together with the key HMBC correlations (Fig. 2) from H-1 to C-3 and C-5, from H2-2 to C-4 and C-10, from H-3 to C-1 and C-5, H-5 to C-1, C-3, C-4, C-6, C-7, C-9, and C-10, from H-7 to C-5, C-6, C-8, and C-9, from H3-18(19) to C-3, C-4, and C-5, and from H3-20 to C-1, C-5, C-9 and C-10 established 5,7-dihydroxy-4a,8,8-trimethyl-4a,5,6,7,8,8a-hexahydronaphthalene-1,4-dione skeleton (Fig. 3). The side chain of 3-ethyl-3-methylpent-4-enoic acid was deduced by the 1H–1H COSY correlation of H-15 with H2-16, as well as the HMBC correlations from H2-11 to C-12, C-13, C-15, C-16, and C-17, from H2-13 to C-11, C-12, C-14, C-15, and C-17, from H3-17 to C-11, C-12, C-13 and C-15. The connection of this side chain with C-8 was determined by HMBC correlations of H-7 with C-11, and H2-11 with C-7, C-8, and C-9. The NOE correlations (Fig. 3) of H-3 (δH 3.69) with H-5 and H3-19 placed them on the same side. The NOE correlations of H-1 (δH 4.23) with H3-18 and H3-20 indicated that H-1, H3-18 and H3-20 were on the other side of the rings. The absolute configuration of 1 was determined by applying the α, β-unsaturated cyclohexanone octant rule. In the α, β-unsaturated cyclohexanone octant rule, the conformation of cyclohexanone ring determines the hecility of enone moiety, which further establishes the sign of the Cotton effect. The positive Cotton effect (Fig. 4) deriving from n–π* transition at 380 nm in 1 allowed the unambiguous assignment of the absolute configuration of 1S,3S,5S,10R in 1.16,17 The absolute configuration at C-12 in 1 was left unsolved in this work.
Table 1 1H (500 MHz) and 13C NMR (125 MHz) data of 1 in acetone-d6
| No. |
δC |
δH (mult. J in Hz) |
| 1 |
71.5 |
4.23, t (3.1) |
| 2 |
34.0 |
1.81, dt (10.1, 2.9) |
| 3 |
72.1 |
3.69, dd (10.1, 6.2) |
| 4 |
39.0 |
|
| 5 |
56.3 |
3.27, s |
| 6 |
200.3 |
|
| 7 |
142.6 |
6.60, s |
| 8 |
146.2 |
|
| 9 |
204.0 |
|
| 10 |
56.2 |
|
| 11 |
37.5 |
2.65, d (12.5) |
| |
|
2.54, d (12.5) |
| 12 |
40.8 |
|
| 13 |
44.2 |
2.37, d (3.1) |
| 14 |
172.8 |
|
| 15 |
146.0 |
5.90, dd (17.5, 10.9) |
| 16 |
112.9 |
4.95, m |
| 17 |
24.2 |
1.11, s |
| 18 |
16.0 |
1.17, s |
| 19 |
28.3 |
1.25, s |
| 20 |
21.8 |
1.17, s |
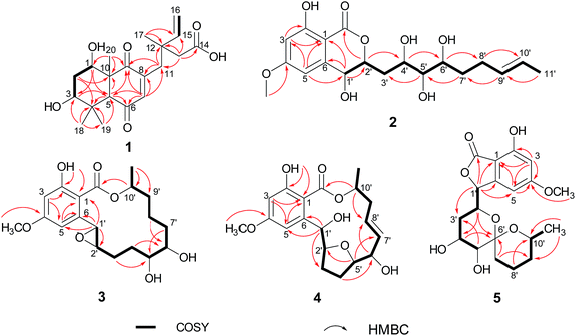 |
| | Fig. 2 Key 1H–1H COSY and HMBC correlations of compounds 1–5. | |
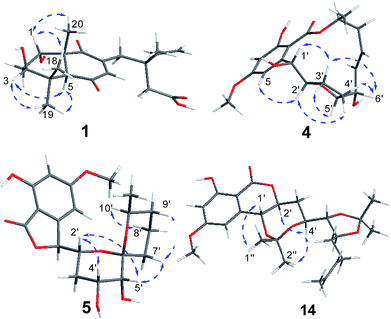 |
| | Fig. 3 Key ROESY correlations of compounds 1, 4–5 and 14. | |
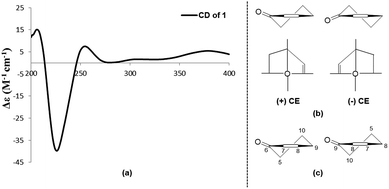 |
| | Fig. 4 (a) CD spectrum of 1. (b) Standard conformation of α, β-unsaturated cyclohexanone and its application of the octant rule. (c) The application of α, β-unsaturated cyclohexanone octant rule in 1. | |
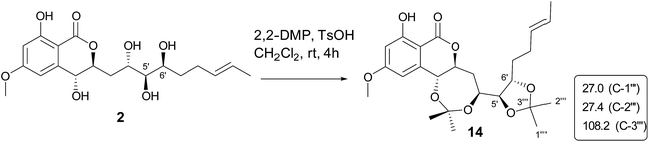
Aigialomycin H (2) possessed the molecular formula of C19H26O8 with 7 degrees of unsaturation based on HRESIMS and NMR data. Further analysis of its 1D NMR data (Table 2) and HSQC revealed an ester carbonyl group (δC 168.1), a 1, 2, 3, 5- tetrasubstituted benzene ring, a double bond (δH 5.40, δC 124.2; δH 5.42, δC 131.5), five oxygenated methines (δH 3.71, δC 65.9; δH 4.52, δC 66.7; δH 3.64, δC 68.7; δH 2.96, δC 76.1; δH 4.60, δC 80.3), three methylenes (δH 2.05, 1.96, δC 28.7; δH 1.90, 1.69, δC 36.1; δH 1.47, 1.40, δC 33.5), a doublet methyl [δH 1.60 (d, J = 4.8 Hz), δC 17.8], a methoxy group (δH 3.83, δC 55.8) and five exchangeable hydrogens [δH 4.05 (d, J = 7.0), 4.29 (d, J = 7.4), 4.76 (d, J = 6.4), 6.04 (d, J = 6.2), 11.12 (s)]. The HMBC correlations from H-3 to C-1, C-2, C-4, and C-5, from H-5 to C-1, C-3, C-4, and C-6, and from CH3O-4 to C-4 revealed the substructure moiety of 3-methoxyphenol (Fig. 2). The side chain structure from C-1′ to C-11′ was established primarily by COSY correlations of H-1′–H-2′–H2-3′–H-4′–H-5′–H-6′–H2-7′–H2-8′–H-9′–H-10′–H3-11′, OH-1′–H-1′, OH-4′–H-4′, OH-5′–H-5′, and OH-6′–H-6′. The connection of C-1′ and C-6 was elucidated by HMBC correlations from H-1′ to C-1, C-5, and C-6. Considering a remaining unsaturation and the chemical shift of C-2′ (δC 80.3), as well as the HMBC correlations from H-2′ (δH 4.60) to the ester carbonyl group (δC 168.1), C-2′ was determined to connect with carbonyl carbon via an oxygen atom to form a δ-lactone. The trans-double bond between C-9′ and C-10′ was ascertained by the NOE correlation of H3-11′ with H-9′. The stereochemistry of 2 was deduced by analysis of coupling constant values and conversion to acetonide derivatives as previous reported for aigialomycin F.11 The 1H–1H J1′,2′ value of 6.6 Hz indicated a trans relation of these protons. Treatment of 2 with 2, 2-DMP in the presence of TsOH (rt, 4 h) gave a bis-acetonide derivative 14 as the sole product (Scheme 1). Compound 14 was determined to possess the molecular formula of C25H34O8 by HRESIMS at m/z [M + H]+ 463.2335, thus indicating two more C3H6 units than that of 2. The HMBC correlations from H-1′ (δH 5.05) and H-4′ (δH 3.90) to C-3′′ (δC 101.7) revealed the seven-membered acetonide moiety in 14. Although the HMBC corresponding signals between H-5′ (6′) and C-3′′′ were not observed, another C3H6 unit was most likely to be linked with C-5′ and C-6′ via oxygen by considering the chemical shifts of C-5′ (δC 81.6) and C-6′ (δC 67.9). The NOE correlation (Fig. 3) of H-4′ (δH 3.90) with H-2′ (δH 4.45) placed them on the same side of the seven membered-ring. The vicinal coupling constants (3J4′,5′ = 7.1 Hz) between H-4′ and H-5′ implied a trans relation of them. The Dana's method could be applied to assign the relative configuration of the 1,2-diols via their 1,2-dioxolanes. In the 1,2-diol acetonides, the chemical shifts of methyl carbons for 1,2-anti configuration were at about 25 ppm (Meβ) and 28 ppm (Meα); for 1,2-syn configuration, both of the chemical shifts were at about 27 ppm.18–20 Hence, 1,2-diols at C5/C6 position in 2 was determined to adopt the 1,2-syn based on the 13C NMR signals of C-1′′′ (δC 27.4) and C-2′′′ (δC 27.0) in the 1,2-diol acetonide (Scheme 1). Therefore, the relative configuration of 2 was determined. Furthermore, the similar NMR data between 2 and aigialomycin F at stereogenic centers also supported the same relative stereochemistry of 2 as that of aigialomycin F. The absolute configuration at C-1′ of 2 was confirmed as R by considering the same biosynthetic pathway for the known β-resorcylic acid lactones.11 Thus, the absolute configuration of 2 was deduced to be 1′R,2′S,4′S,5′R,6′S.
Table 2 1H (500 MHz) and 13C NMR (125 MHz) data of 2 in DMSO-d6
| No. |
δC |
δH (mult. J in Hz) |
| 1 |
100.1 |
|
| 2 |
163.1 |
|
| 3 |
100.0 |
6.48, d (2.4) |
| 4 |
165.7 |
|
| 5 |
104.8 |
6.60, d (2.3) |
| 6 |
145.1 |
|
| 1′ |
66.7 |
4.52, t (6.6) |
| 2′ |
80.3 |
4.60, m |
| 3′ |
36.1 |
1.90, m |
| 1.69, t (13.0) |
| 4′ |
65.9 |
3.71, m |
| 5′ |
76.1 |
2.96, td (7.7, 2.1) |
| 6′ |
68.7 |
3.64, m |
| 7′ |
33.5 |
1.47, m |
| 1.40, m |
| 8′ |
28.7 |
2.05, m |
| 1.96, m |
| 9′ |
131.5 |
5.42, m |
| 10′ |
124.2 |
5.40, m |
| 11′ |
17.8 |
1.60, d (4.8) |
| –COO– |
168.1 |
|
| 2-OH |
|
11.12, s |
| 4-OCH3 |
55.8 |
3.83, s |
| 1′-OH |
|
6.04, d (6.2) |
| 4′-OH |
|
4.76, d (6.4) |
| 5′-OH |
|
4.29, d (7.4) |
| 6′-OH |
|
4.05, d (7.0) |
 |
| | Scheme 1 Preparation of acetonide derivatives of 14 and 13C NMR signals of acetonide carbon atoms; 2,2-DMP = 2,2-dimethoxypropane, TsOH = para-toluenesulfonic acid. | |
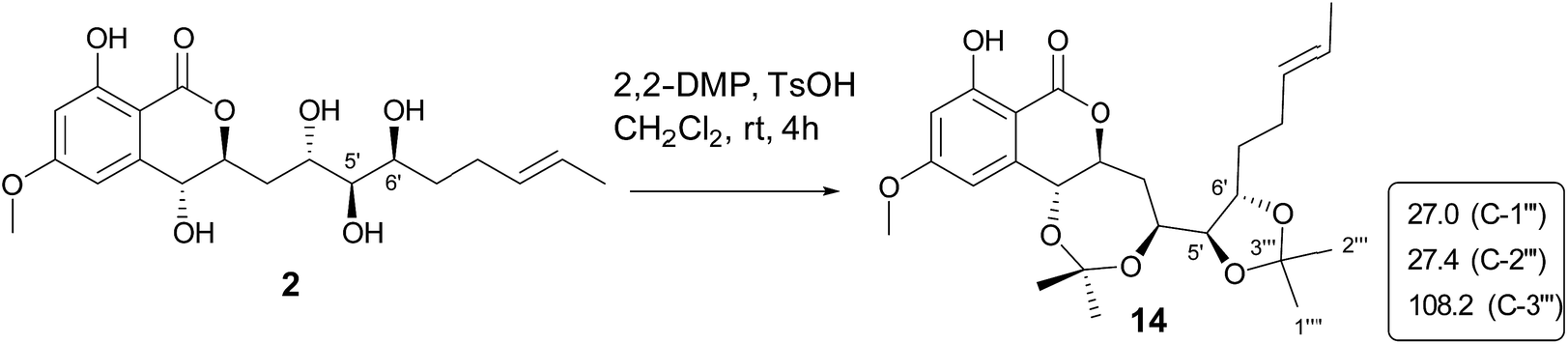
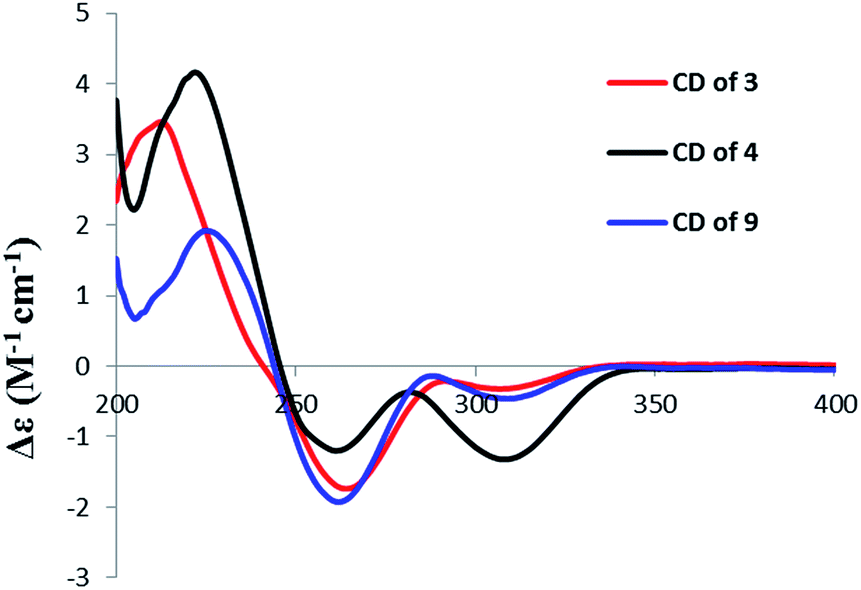
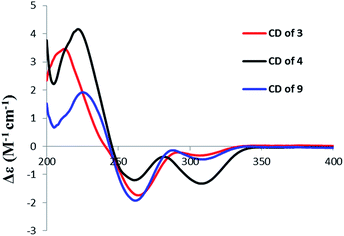
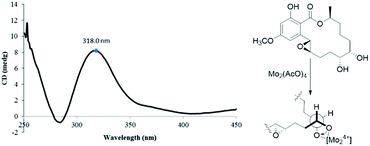
The molecular formula of aigialomycin I (3) was established as C19H26O7 with 7 degrees of unsaturation, determined by HRESIMS at m/z [M + H]+ 367.1757. The 1H and 13C NMR data of 3 showed similar structure with those of dihydrohypothemycin (8),13 except for the absence of one ketone carbon, and the presence of a methylene (δH 1.76, 2.07, δC 27.9) that was assigned at C-4′ by the 1H–1H COSY correlations of H-1′–H-2′–H2-3′–H2-4′–H-5′–H-6′–H2-7′–H2-8′ and the HMBC correlations of H2-4′with C-2′, C-3′, C-5′ and C-6′. Considering the chemical shifts of C-5′ (δC 71.2) and C-6′ (δC 69.0), the two hydroxyl moieties were attached at C-5′ and C-6′, respectively. The stereochemistry of C-5′ and C-6′ were assigned as 5′R, 6′S by applying the Snatzke's method21 base on an observed positive Cotton effect at 318 nm in the Mo4(OAc)4-induced CD spectrum (Fig. 6). The absolute configurations of C-1′, C-2′ and C-10′ in 3 were deduced to be 1′R, 2′R, 10′S by the similar negative Cotton effects around 264 nm and 307 nm with those of compound 9 (Fig. 5). Therefore, the absolute configuration of 3 was determined to be 1′R,2′R,5′R,6′S,10′S.
 |
| | Fig. 5 CD spectrum of 3, 4, and 9. | |
 |
| | Fig. 6 CD spectrum of Mo-complex of 3 with Mo2(AcO)4. | |
Paecilomycin N (4) was obtained as a white power. Its molecular formula was determined as C19H24O7 (8 degrees of unsaturation) on the basis of HRESIMS ion peak at m/z [M + H]+ 407.2138. The structure of 4 was confirmed by analysis of 1D and 2D NMR spectra (Table 3). A trans-configuration between H-7′ and H-8′ was determined on the basis of the larger coupling constant (J7′,8′ = 15.7 Hz) in 4. In the ROESY spectrum, the correlations of H-1′ (δH 5.60) with H-3′β (δH 2.02) as well as H-6′ (δH 3.98) with H-3′β and H-4′β (δH 1.76) indicated that H-1′, H-3′β, H-4′β, and H-6′ were on the same side. The correlations of H-2′ (δH 4.10) with H-4′α (δH 2.07), and H-3′α (δH 2.25) with H-5′ (δH 4.15) placed H-2′, H-3′α, H-4′α and H-5′ on the opposite side (Fig. 3). The absolute configuration of 4 was assigned as 1′R,2′S,5′R,6′S,10′S by comparing the CD spectrum between 4 and 9 (Fig. 5).
Table 3 1H (500 MHz) and 13C NMR (125 MHz) data of 3–5 in CDCl3
| No. |
3 |
4 |
5 |
| δC |
δH (mult. J in Hz) |
δC |
δH (mult. J in Hz) |
δC |
δH (mult. J in Hz) |
| 1 |
104.4 |
|
105.5 |
|
104.6 |
|
| 2 |
166.0 |
|
165.0 |
|
158.0 |
|
| 3 |
100.9 |
6.41, d (2.6) |
101.1 |
6.42, d (2.6) |
101.5 |
6.48, s |
| 4 |
164.9 |
|
164.4 |
|
167.4 |
|
| 5 |
104.1 |
6.43, d (2.6) |
107.4 |
6.72, d (2.6) |
101.9 |
6.65, s |
| 6 |
142.4 |
|
148.0 |
|
149.2 |
|
| 1′ |
57.1 |
4.61, s |
71.9 |
5.60, d (9.0) |
82.9 |
5.36, d (7.5) |
| 2′ |
64.8 |
2.67, d (9.1) |
84.8 |
4.10, dt (9.0, 6.3) |
66.3 |
4.01, dt (7.5, 2.1) |
| 3′ |
26.7 |
2.27, m |
30.1 |
2.25, m |
28.9 |
2.01, m |
| 1.38, m |
2.02, m |
1.84, dd (14.0, 3.5) |
| 4′ |
29.4 |
2.07, m |
27.9 |
2.07, m |
69.0 |
3.98, d (3.5) |
| 1.64, m |
1.76, m |
| 5′ |
71.2 |
3.61, m |
84.3 |
4.15, m |
70.0 |
3.49, d (3.5) |
| 6′ |
69.0 |
3.72, m |
75.0 |
3.98, m |
100.4 |
|
| 7′ |
32.4 |
1.66, m |
133.6 |
5.44, ddd (15.7, 4.7, 1.5) |
31.0 |
1.97, m |
| 1.43, m |
| 8′ |
21.5 |
1.58, m |
125.9 |
5.66, ddd (15.7, 8.7, 5.7, 1.5) |
18.5 |
1.84, m |
| 1.70, m |
| 9′ |
34.2 |
1.76, m |
37.9 |
2.33, dt (14.3, 8.1) |
32.1 |
1.61, m |
| 1.66, m |
2.74, m |
1.28, m |
| 10′ |
73.6 |
5.31, m |
71.9 |
5.50, m |
67.5 |
3.63, m |
| 10′-CH3 |
20.9 |
1.38, d (6.3) |
19.8 |
1.40, d (6.5) |
21.8 |
1.12, d (6.2) |
| 2-OH |
|
12.10, s |
|
11.77, s |
|
|
| 4-OCH3 |
55.5 |
3.80, s |
55.5 |
3.80, s |
56.2 |
3.86, s |
| –COO– |
171.2 |
|
171.3 |
|
171.5 |
|
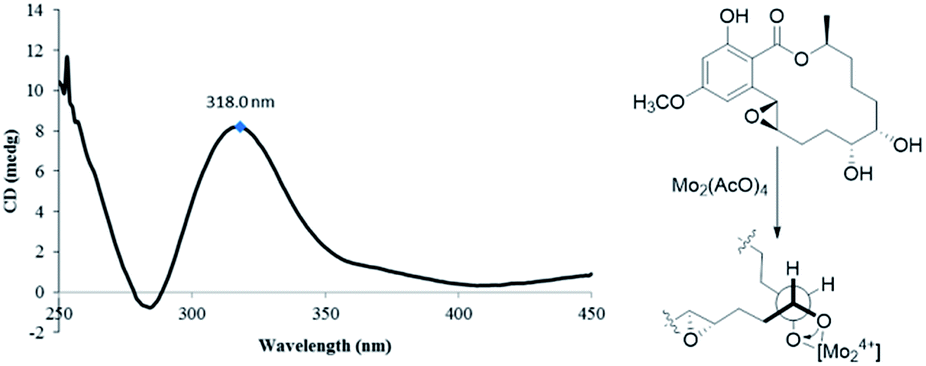
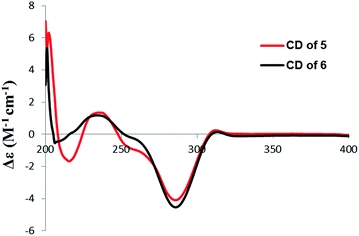
4′R,5′R-Dihydroaigialospirol (5) was isolated as a colorless solid with the molecular formula C19H24O8 (7 degrees of unsaturation). Careful analysis of 1D and 2D NMR data (Table 3), particularly 1H–1H COSY and HMBC revealed the same planar structure with the known compound 7′,8′-dihydroaigialospirol (6) (Fig. 2). The smaller coupling constant (J4′,5′ = 3.5 Hz) between H-4′ and H-5′ in 5 implied a syn configuration. The NOE correlations of H-2′ (δH 4.01) with H-4′ (δH 3.98) and H-5′ (δH 3.49) placed these protons on the same face of the rings. 1D NOE correlations of H-10′ to H-2′ and H-5 revealed their proximity in space, and the relative configuration of the spiroketal carbon C-6′ also was determined meanwhile. In the previous report, the absolute configuration of compound 6 has been determined by chemical derivation and X-ray analysis. The identical negative Cotton effect at 216 and 284 nm, and positive Cotton effect at 240 nm between 5 and the co-isolated known compound 6 (Fig. 7), combined with the almost same chemical shifts and coupling constant of H-1′ and H-2′ between 5 and 6, implied the 1′S,2′R configuration at C-1′ and C-2′. Thus, the absolute configuration of 5 was determined as 1′S,2′R,4′R,5′R,6′R,10′S.
 |
| | Fig. 7 CD spectrum of 5 and 6. | |
Compounds 1–13 were tested for their cytotoxic activities against A549, K562 and ASPC cell lines using MTT method.22 Only compound 7 showed moderate cytotoxicity against A549 cell line with IC50 values of 44.5 μM ± 3.1 (positive control: IC50 of cisplatin against A549 was 25.60 μM ± 2.8). None of them exhibited cytotoxicities against K562 and ASPC at the concentration of 100 μM.
In conclusion, the investigation of the fungicolous fungus H. subiculosus obtained from Polyporus versicolor resulted in the isolation and identification of five new compounds and eight known compounds. Compound 1 is a ring cleavage pimarane diterpene with new structural type. Compounds 2–5 are new β-resorcylic acid lactone derivatives. The cytotoxic activities for these compounds against a small panel of tumor cell lines were evaluated. The discovery of compounds 1–5 expands the resources for natural products.
3. Experimental section
General experimental procedures
The NMR spectroscopic data were measured on a Bruker Avance-500 spectrometer. UV and IR spectroscopic data were recorded on a Thermo Genesys-10S UV/Vis and Nicolet IS5FT-IR spectrophotometer, respectively. HRTOFMS data were obtained with an Agilent Accurate-Mass-Q-TOF LC/MS 6520 spectrometer. CD spectra were measured with a Chirascan circular dichroism spectrometer (Applied Photophysics, Surrey, UK). TLC were performed on plates precoated with Silica gel HSGF254 and the spots were detected under UV lights or by heating after spraying with 10% H2SO4. Silica gel (200–300 mesh, Qingdao Haiyang Chemical Co., Ltd., People's Republic of China), and Sephadex LH-20 (Amersham Biosciences) were used for column chromatography. Semi-preparative HPLC was performed with an Waters 2489 HPLC system using an octadecylsilyl (ODS) column (YMC-Pack, ODS-A, 10 × 250 mm, 5 μm).
Fungal material
The fungal H. subiculosus was isolated from Polyporus versicolor. The strain was identified based on combined data of morphology, anatomy, culture characteristics, type of substrate, and the DNA sequences of ITS1-5.8S-ITS2, RPB2 and TEF regions.
Extraction and isolation
H. subiculosus was cultured on PDB-agar plates at 25 °C for 7 days. Seed culture Agar plugs were inoculated into a 250 mL Erlenmeyer flask containing 100 mL PDB medium cultured at 25 °C for 10 days on a rotary shaker at 180 rpm. Mass scale fermentation was carried out in 20 × 500 mL Fernbach culture flasks each containing 80 g of rice and 100 mL of distilled water. Each flask was inoculated with 5 mL of seed culture medium and incubated at 25 °C for 40 days in dark. After 40 days, the fermentated rice sample was harvested and extracted using EtOAc (3 × 10 L), and 14 g crude extract was obtained by evaporating solvent under vaccum.
The crude extract was subjected to a silica gel column eluted with petroleum ether–EtOAc in a gradient eluent (v/v, 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 50:1, 20:1, 10:1, 5:1), followed by CH2Cl2–MeOH (v/v, 100:0, 100:1, 80:1, 50:1, 20:1, 10:1, 0:100) to obtain 16 fractions (fr.1–fr.16) on the basis of TLC. The fr.9 (2.5 g) was separated by ODS CC using a gradient of MeOH–H2O (25–100%) to afford 19 subfractions (fr.9.1–fr.9.19). The fr.9.7 was further purified by RP-HPLC (43% MeOH in H2O for 30 min) to obtain 4 (3.5 mg, tR = 27.5 min). The fr.9.9 was further purified by Sephadex LH-20 CC eluted with 100% MeOH to afford 6 (91.5 mg). The fr.9.8 was further separated by Sephadex LH-20 CC eluted with 100% MeOH, followed by RP-HPLC (40% MeOH for 55 min) to afford 12 (15.7, tR = 51.5 min), 13 (16.2, tR = 52.8 min). The fr.9.19 was further purified by semipreparative reversed-phase (RP) HPLC (YMC-ODS C18 column; 5 μm; 9.4 × 250 mm; 60% MeOH in H2O for 35 min) to afford 2 (3.8 mg, tR = 32.2 min), 7 (37.8 mg, tR = 15.9 min), 8 (43.7 mg, tR = 28.8 min). The fr.11 (2.8 g) was subjected to ODS CC eluted with a gradient of MeOH–H2O (10–100%) further separated by Sephadex LH-20 CC eluted with 100% MeOH, followed by RP-HPLC (58% MeOH for 55 min) to afford 1 (2.3 mg, tR = 19.0 min), 5 (2.8 mg, tR = 24.3 min), 9 (4.5 mg, tR = 28.0 min), 6 (42.0 mg, tR = 50.8 min). The fr.12 (1.1 g) was subjected to ODS CC eluted with MeOH–H2O in a gradient eluent (10–100%), followed by RP-HPLC (50% MeOH for 40 min) to obtain 10 (5.2 mg, tR = 24.7 min), 3 (2.3 mg, tR = 33.5 min), 11 (20.0 mg, tR = 36.0 min).
:0, 50:1, 20:1, 10:1, 5:1), followed by CH2Cl2–MeOH (v/v, 100:0, 100:1, 80:1, 50:1, 20:1, 10:1, 0:100) to obtain 16 fractions (fr.1–fr.16) on the basis of TLC. The fr.9 (2.5 g) was separated by ODS CC using a gradient of MeOH–H2O (25–100%) to afford 19 subfractions (fr.9.1–fr.9.19). The fr.9.7 was further purified by RP-HPLC (43% MeOH in H2O for 30 min) to obtain 4 (3.5 mg, tR = 27.5 min). The fr.9.9 was further purified by Sephadex LH-20 CC eluted with 100% MeOH to afford 6 (91.5 mg). The fr.9.8 was further separated by Sephadex LH-20 CC eluted with 100% MeOH, followed by RP-HPLC (40% MeOH for 55 min) to afford 12 (15.7, tR = 51.5 min), 13 (16.2, tR = 52.8 min). The fr.9.19 was further purified by semipreparative reversed-phase (RP) HPLC (YMC-ODS C18 column; 5 μm; 9.4 × 250 mm; 60% MeOH in H2O for 35 min) to afford 2 (3.8 mg, tR = 32.2 min), 7 (37.8 mg, tR = 15.9 min), 8 (43.7 mg, tR = 28.8 min). The fr.11 (2.8 g) was subjected to ODS CC eluted with a gradient of MeOH–H2O (10–100%) further separated by Sephadex LH-20 CC eluted with 100% MeOH, followed by RP-HPLC (58% MeOH for 55 min) to afford 1 (2.3 mg, tR = 19.0 min), 5 (2.8 mg, tR = 24.3 min), 9 (4.5 mg, tR = 28.0 min), 6 (42.0 mg, tR = 50.8 min). The fr.12 (1.1 g) was subjected to ODS CC eluted with MeOH–H2O in a gradient eluent (10–100%), followed by RP-HPLC (50% MeOH for 40 min) to obtain 10 (5.2 mg, tR = 24.7 min), 3 (2.3 mg, tR = 33.5 min), 11 (20.0 mg, tR = 36.0 min).
Subiculosin A (1). C20H28O6, [α]20.0D +55.60 (c 0.13, CH2Cl2); UV (MeOH) λmax (logε) 245 (4.12) nm; CD (c 9.5 × 10−4 M, MeOH) λmax (Δε) 226 (−39.7), 254 (+7.5), 380 (+5.4); IR (neat) νmax 3419, 2930, 1677, 1378, 1203, 1054, 997 cm−1; 1H NMR (500 MHz, acetone-d6) and 13C NMR (125 MHz, acetone-d6) data, see Table 1; positive HRESIMS m/z 365.1962 [M + H]+ (calcd for C20H29O6, 365.1964).
Aigialomycin H (2). Colorless solid; [α]20.0D −13.33 (c 0.03, CH2Cl2); UV (MeOH) λmax (logε) 212 (4.40), 268 (4.17), 303 (3.89) nm; IR (neat) νmax 2926, 1681, 1380, 1210, 1058, 1034, 706 cm−1; 1H NMR (500 MHz, DMSO-d6) and 13C NMR (125 MHz, DMSO-d6) data, see Table 2; positive HRESIMS m/z 383.1704 [M + H]+ (calcd for C19H27O8, 383.1706).
Aigialomycin I (3). Colorless solid; [α]20.0D –1.54 (c 0.12, MeOH/CH2Cl2 = 1:1); UV (MeOH) λmax (logε) 220 (4.59), 264 (4.28), 305 (4.01) nm; CD (c 5.5 × 10−4 M, MeOH) λmax (Δε) 212 (+3.4), 264 (−1.7), 307 (−0.3); IR (neat) νmax 3418, 1647, 1613, 1357, 1318, 1257, 1205, 1160, 1111 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) data, see Table 3; positive HRESIMS m/z 367.1757 [M + H]+ (calcd for C19H27O7, 367.1757).
Paecilomycin N (4). White powder; [α]20.0D –15.29 (c 0.34, CH2Cl2); UV (MeOH) λmax (logε) 220 (4.63), 263 (4.34), 303 (3.99) nm; CD (c 5.5 × 10−4 M, MeOH) λmax (Δε) 222 (+4.2), 260 (−1.2), 308 (−1.3); IR (neat) νmax 3393, 1645, 1613, 1254, 1160, 1037 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) data, see Table 3; positive HRESIMS m/z 365.1601 [M + H]+ (calcd for C19H25O7, 365.1600).
4′R,5′R-Dihydroaigialospirol (5). Colorless solid; [α]20.0D +14.19 (c 0.08, CH2Cl2); UV (MeOH) λmax (logε) 212 (4.47), 256 (4.21), 291 (3.80) nm; CD (c 8.0 × 10−4 M, MeOH) λmax (Δε) 216 (−1.7), 240 (+1.4), 282 (−4.1); IR (neat) νmax 3420, 1742, 1613, 1217, 1157, 1069 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) data, see Table 3; HRESIMS m/z 381.1547 [M + H]+ (calcd for C19H25O8, 381.1549).
Compound 14. Colorless solid; 1H NMR (500 MHz, DMSO-d6) δ 6.51 (1H, d, J = 2.4, H-3), 6.48 (1H, d, J = 2.4, H-5), 5.44 (2H, m, H-9′, H-10′), 5.05 (1H, d, J = 10.6, H-1′), 4.45 (1H, td, J = 10.6, 5.6, H-2′), 3.90 (1H, m, H-4′), 3.87 (1H, m, H-6′), 3.83 (3H, s, 4-OCH3), 3.56 (1H, t, J = 7.1, H-5′), 2.30 (1H, dd, J = 12.8, 5.5, H-3′α), 2.11 (1H, m, H-8′), 2.02 (1H, m, H-8′), 1.72 (1H, m, H-7′), 1.66 (1H, m, H-3′β), 1.61 (3H, d, J = 2.1, H-11′), 1.54 (1H, m, H-7′), 1.42 (3H, s, H-1′′), 1.39 (3H, s, H-2′′), 1.33 (3H, s, H-1′′′), 1.32 (3H, s, H-2′′′); 13C NMR (125 MHz, DMSO-d6) δ 167.9 (C, C-12), 166.0 (C, C-4), 163.4 (C, C-2), 143.8 (C, C-6), 130.8 (CH, C-9′), 124.9 (CH, C-10′), 108.2 (C, C-3′′′), 103.8 (CH, C-5), 101.7 (C, C-3′′), 99.8 (C, C-1), 99.7 (CH, C-3), 81.6 (CH, C-5′), 78.4 (CH, C-2′), 78.0 (CH, C-4′), 67.9 (CH, C-6′), 66.8 (CH, C-1′), 55.9 (CH3, 4-OCH3), 36.8 (CH2, C-7′), 33.9 (CH2, C-3′), 28.7 (CH2, C-8′), 27.4 (CH3, C-1′′′), 27.0 (CH3, C-2′′′), 24.5 (CH3, C-1′′), 24.5 (CH3, C-2′′), 17.8 (CH3, C-11′). Positive HRESIMS m/z 463.2335 [M + H]+ (calcd for C25H35O8, 463.2332).
Synthesis of the acetonide derivative 14
Compound 14 was synthesized from compound 2 by using a published method.11 To a suspension of 2 (2.0 mg) in 2,2-dimethoxypropane (0.5 mL) was added TsOH (0.5 mg). The mixture was stirred at room temperature for 4 h, then reaction mixture was diluted with EtOAc and washed with saturated NaHCO3. The organic layer was concentrated under reduced pressure to leave a colorless solid, which was purified by RP-HPLC using MeOH–H2O (95:5) to afford 14 (1.0 mg).
Determination of the absolute configuration at 5′,6′-diol moiety by Snatzke's method
A sample of 3 (0.5 mg) was dissolved in a dry stock solution of the dimolybdenum tetraacetate [Mo2(OAc)4] (0.7 mg) in DMSO (1 mL) and the CD spectrum was recorded immediately after mixing. The CD spectrum was recorded every 10 min until a stationary spectrum was reached (30 min after mixing). The inherent CD from 3 was subtracted to give the induced CD of the complex. The observed sign of the diagnostic band at 318 nm (band IV) was correlated to the absolute configuration at 5′,6′-diol moiety.
Cytotoxicity
The cytotoxicities of compounds 1–13 against the K562, A549 and H460 cell lines were investigated by using the MTT method as previously reported.22
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (81673334).
Notes and references
- P. Jeffries, Can. J. Bot., 1995, 73, 1284–1290 CrossRef.
- R. F. Angawi, D. C. Swenson, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2003, 66, 1259–1262 CrossRef CAS PubMed.
- C. Li, J. B. Gloer, D. T. Wicklow and P. F. Dowd, Org. Lett., 2002, 18, 3095–3098 CrossRef.
- L. E. Schmidt, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2007, 70, 1317–1320 CrossRef CAS PubMed.
- S. H. Shim, J. Baltrusaitis, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2011, 74, 395–401 CrossRef CAS PubMed.
- A. A. Sy, D. C. Swenson, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2008, 71, 415–419 CrossRef CAS PubMed.
- J. L. C. Wright, A. G. Mclnnes, D. G. Smith and L. C. Vining, Can. J. Chem., 1970, 48, 2720 CrossRef.
- J. L. Wee, K. Sundermann, P. Licari and J. Galazzo, J. Nat. Prod., 2006, 69, 1456–1459 CrossRef CAS PubMed.
- K. Dornberger, W. Ihn, M. Ritzau, U. Grafe, B. Schlegel, W. F. Fleck and J. W. Metzger, J. Antibiot., 1995, 48, 977–989 CrossRef CAS PubMed.
- G. M. Hatfield and D. E. Slagle, Lloydia, 1973, 36, 354–356 CAS.
- M. Isaka, A. Yangchum, S. Intamas, K. Kocharin, E. B. G. Jones, P. Kongsaeree and S. Prabpai, Tetrahedron, 2009, 65, 4396–4403 CrossRef CAS.
- M. Nair and S. T. Carey, Tetrahedron Lett., 1980, 21, 2011–2012 CrossRef CAS.
- M. Isaka, C. Suyarnsestakorn and M. Tanticharoen, J. Org. Chem., 2002, 675, 1561–1566 CrossRef.
- L. X. Xu, P. Wu, H. H. Wei, J. H. Xue, X. P. Hu and X. Y. Wei, Tetrahedron Lett., 2013, 5421, 2648–2650 CrossRef.
- K. Matsunami, H. Otsuka and Y. Takeda, Chem. Pharm. Bull., 2010, 58, 438–441 CrossRef CAS PubMed.
- R. D. Burnet and D. N. Kirk, J. Chem. Soc., Perkin Trans. 1, 1981, 1460–1468 RSC.
- Y. Sun, L. Tian, J. Huang, H. Y. Ma, Z. Zheng, A. L. Lv, K. Yasukawa and Y. H. Pei, Org. Lett., 2008, 10, 393–396 CrossRef CAS PubMed.
- G. Dana and H. Danechpajouh, Bull. Soc. Chim. Fr., 1980, 395–399 CAS.
- M. Bock, K. Buntin, R. Muller and A. Kirschning, Angew. Chem., Int. Ed., 2008, 47, 2308–2311 CrossRef CAS PubMed.
- S. Hoshino, M. Okada, T. Awakawa, S. Asamizu, H. Onaka and I. Abe, Org. Lett., 2017, 19, 4992–4995 CrossRef CAS PubMed.
- J. Frelek, Z. Pakulski and A. Zamojski, Tetrahedron: Asymmetry, 1996, 7, 1363–1372 CrossRef CAS.
- Y. Q. Wang, L. Bao, D. L. Liu, X. L. Yang, S. F. Li, H. Gao, X. S. Yao, H. A. Wen and H. W. Liu, Tetrahedron, 2012, 68, 3012–3018 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The NMR spectra of 1–5 and 14. See DOI: 10.1039/c7ra11365j |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab