
Carbonate Based Ionic Liquid Synthesis (CBILS®): development of the continuous flow method for preparation of ultra-pure ionic liquids†
Roland S.
Kalb
 *a,
Markus
Damm
a and
Sergey P.
Verevkin
bc
*a,
Markus
Damm
a and
Sergey P.
Verevkin
bc
aProionic GmbH, A-8074 Grambach, Austria. E-mail: roland.kalb@proionic.com; Tel: +43 316 4009 4200
bDepartment of Physical Chemistry, University of Rostock, Dr-Lorenz-Weg 1, 18059 Rostock, Germany
cCompetence Centre CALOR, Faculty of Interdisciplinary Research, University of Rostock, Albert-Einstein Str. 25, 18059, Rostock, Germany
First published on 10th April 2017
Abstract
Carbonate Based Ionic Liquid Synthesis (CBILS®) is one of the most advanced commercialized processes for greener and halide-free technical synthesis of ionic liquids (ILs). To meet the increasing demand for high purity ILs, a continuous flow method with an improved space-time yield was developed. The quality of resulting ILs and IL-intermediates was confirmed to be ultra-high, by traditional analytical methods as well as by extremely sensitive combustion and solution calorimetry techniques.
Ionic liquid (ILs) are amazing liquid materials with highly attractive physicochemical properties, among them are superior thermal stability, low- to non-flammability, high electrical conductivity, nanostructural organization,1 low compressibility and exceptionally low vapor pressure even at elevated temperature.2 These noticeable properties have already been applied in diverse technical processes and products, e.g. ionic compressors, sensors, thermo-fluids, electrolytes, storage of gases, production of chemicals, etc.3 Being a manufacturer of ionic liquids, we recognize the significantly increasing demand for high purity ionic liquids within the last few years, which results from a growing number of ionic liquid-based high-tech applications, e.g. in electronics, energy storage, nanotechnology, dye sensitized solar cells, functional materials, etc.
One of the most advanced commercialized methods for a greener industrial production of high-purity ionic liquids is our so-called Carbonate Based Ionic Liquid Synthesis (CBILS®, a registered trademark of proionic GmbH).4 This entirely halide and waste-free process using carbonic acid esters as quaternization reagents overcomes most of the drawbacks of conventional synthetic approaches and reduces the use of noxious chemicals to a minimum; it is scaled up to the multi-ton level. We recently published a detailed thermodynamic analysis of the CBILS® process, based on quantum-chemical computations, which were validated by comparison with experimental results of 16 typical CBILS® reactions.5
Continuous flow (CF) processes – especially those using microstructured devices – are known to be very effective in organic synthesis. They allow improved and precise heat management, safe handling of aggressive chemicals even under high pressure and temperature, superior mass transfer rates and easier scale up which frequently result in significant process intensification with enhanced yield, selectivity and purity.6 Such improved processes are known for the synthesis and application of ILs as well,7 which encouraged us to evaluate the CBILS® process under CF conditions. For details on the following section, we refer to our ESI.†
The crucial step in the CBILS® process is the formation of the quaternized alkylcarbonate intermediate,5 which is then reacted with Brønsted acids to obtain the final ionic liquid. The alkylcarbonate anion is protonated and hydrolyses under liberation of CO2 and formation of the corresponding alcohol, the Brønsted acid is deprotonated forming the conjugated anion, and the desired ionic liquid is obtained after removal of volatiles in vacuo. For high-tech applications of ultra-pure ionic liquids, imidazolium and pyrrolidinium-based structures are of high importance, that is why we have chosen to investigate the synthesis of the intermediates 1-butyl-1-methylpyrrolidinium methylcarbonate (BMPyr-MC) and 1-ethyl-3-methylimidazolium methylcarbonate (EMIM-MC) under batch and continuous flow conditions.
Scheme 2 and Scheme 1 show the reaction conditions for the synthesis of very pure BMPyr-MC and EMIM-MC investigated in this work, which are obtained as stable solutions in methanol (MeOH) at typical concentrations as being used for commercial products. The starting materials were 1-butylpyrrolidine (BPyr) and 1-ethylimidazole (EI) as nucleophilic bases and dimethyl carbonate (DMC) as the methylating agent. All aforementioned reagents were freshly distilled before use and dried over molecular sieve. The excess of 0.6 equiv. of DMC remains in the reaction mixture and can be easily removed later in vacuo after the final formation of the ionic liquid, as described above. Since BPyr is more reactive than EI, the reaction temperatures are 30 °C higher for the latter to realize residence times of 0.5 to 2 hours. The high amount of 35% to 60% of methanol as solvent is necessary, to suppress the formation of by-products to obtain high qualities; however, this does result in a negative impact on the space-time yields of these reactions.
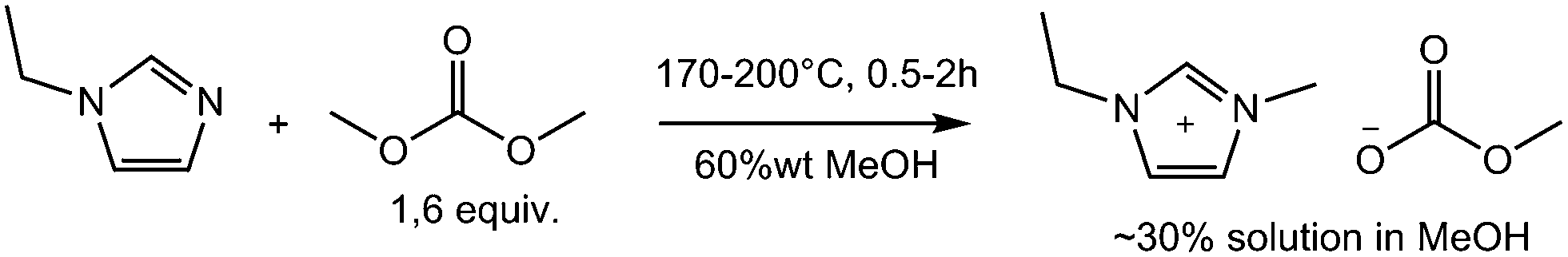 | ||
| Scheme 1 Synthesis of 1-ethyl-3-methylimidazolium methyl-carbonate. | ||
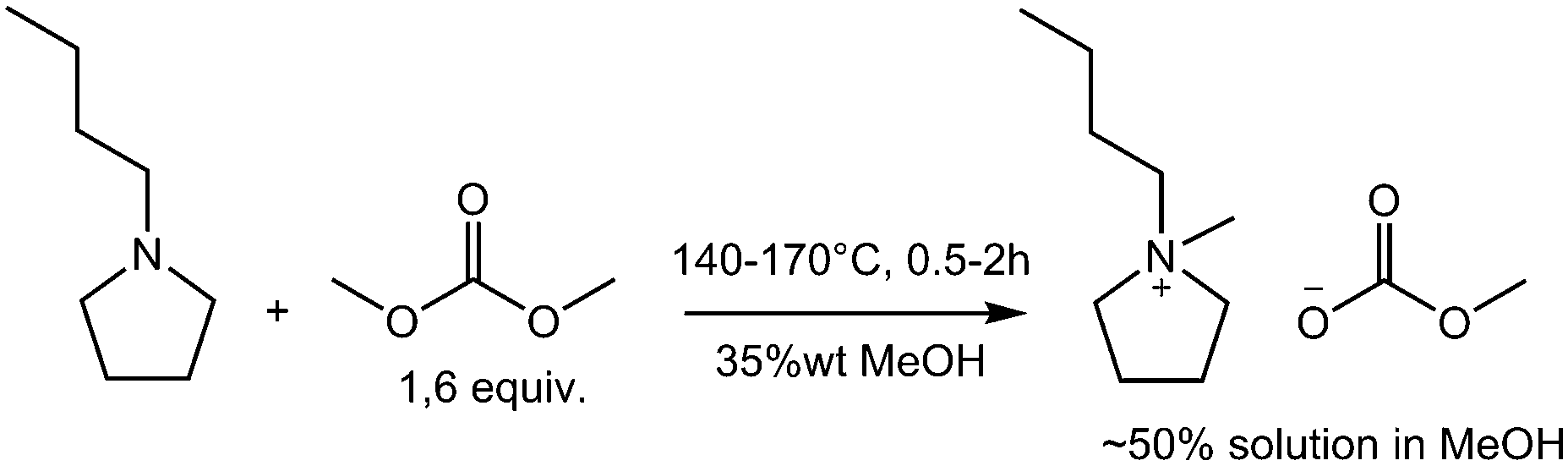 | ||
| Scheme 2 Synthesis of 1-butyl-1-methylpyrrolidinium methyl-carbonate. | ||
Substrate mixtures and product mixtures of both reactions are homogeneous, so the used setup for the continuous flow (CF) experiments was rather simple (Scheme 3). The substrate mixture was pumped using a standard HPLC pump with a pressure indicator at flow rates between 0.1 and 10 ml min−1 and pressures of up to 150 bar through a 1/16 in. stainless steel coil of 38.2 m length, having a reactor volume of 30 ml. The coil was heated inside a standard drying oven with an accuracy of ±1 K. The reaction mixture was cooled down in a second coil of 10 ml volume, submerged in a water bath. To adjust the pressure, a back-pressure regulator with pmax = 175 bar was used. The product was collected in a sealed vessel. For batch experiments, 4 ml screw-capped vials equipped with PTFE coated stir bars, PTFE seals and PEEK screw caps were used, which resist 20 bar pressure and 170 °C; the volume of these vials were filled up with the substrate mixture by 80%. Heating occurred in a massive aluminum block, which was thermostated with an error of ±1 K on a standard hotplate/stirrer under thermal control using an external contact thermometer. Reaction mixtures were characterized using HPLC-UV-CAD (CAD = charged aerosol detector), ion chromatography (IC), Karl-Fisher coulometry (KFC) and alkalimetric or acidimetric titration methods (see the ESI† for details); both compounds EMIM-MC and BMPyr-MC are commercialized products with validated quality control methods.
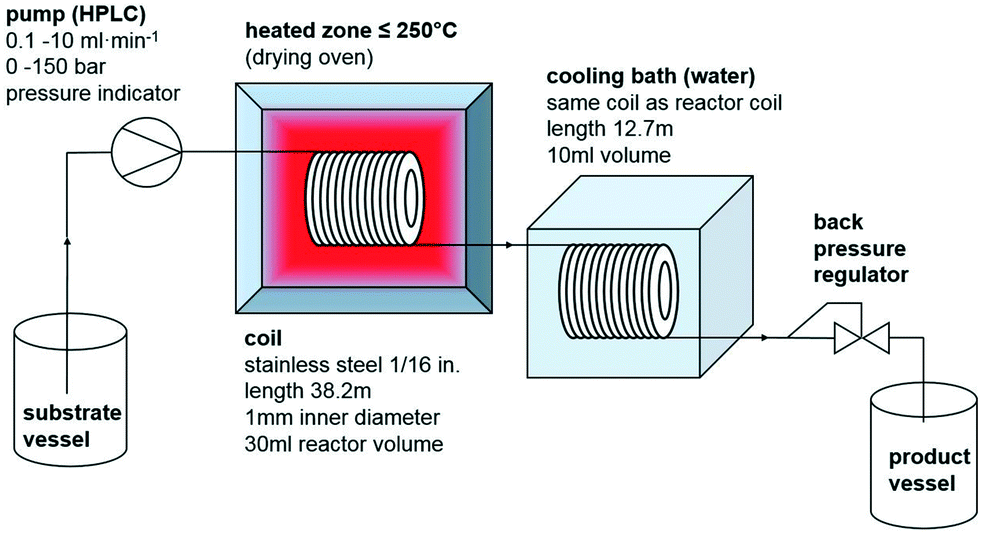 | ||
| Scheme 3 Schematic of the continuous flow setup. | ||
Table 1 shows the results for batch and CF synthesis of BMPyr-MC ∼50% in methanol. In general, it can be seen that CF reactions compared to batch reactions under identical conditions show conversion rates increased by a factor of 1.5 to 2 and space-time yields improved by a factor of 1.3 to 2.6. As we are interested in very high qualities, the residual starting material BPyr at concentrations as low as only a few 100 ppm is highly attractive, as demonstrated in CF reactions no. 9 and 10. Under batch conditions, these low concentrations cannot be reached even after 120 min at 170 °C; compared to reaction no. 9, the residual starting material in no. 6 has a concentration of 1700 ppm at a 4-fold level even after a 4-fold reaction time, and the space-time yield is smaller by a factor of 4.9.
| No. | Mode | T [°C] | t [min] | BPyr [% wt] | BMPyr-MC [% wt] | Conv. [%] | STY [kg l−1 h−1] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pressure: 50 bar for all CF experiments. Continuous flow (CF), reaction temperature (T) and duration (t), educt 1-butylpyrrolidine (BPyr), product 1-butyl-1-methylpyrrolidinium methylcarbonate (BMPyr-MC), conversion (Conv.), space-time-yield (STY) of the contained pure product. Sum of impurities <0.1% wt. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Batch | 130 | 30 | 22.52 | 13.72 | 26.3 | 0.20 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Batch | 130 | 60 | 14.86 | 25.13 | 49.8 | 0.18 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Batch | 130 | 120 | 7.74 | 38.33 | 74.4 | 0.14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Batch | 170 | 30 | 0.27 | 51.05 | 99.1 | 0.73 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Batch | 170 | 60 | 0.21 | 52.13 | 99.3 | 0.37 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Batch | 170 | 120 | 0.17 | 52.51 | 99.5 | 0.19 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | CF | 130 | 30 | 13.66 | 27.58 | 54.2 | 0.52 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | CF | 130 | 60 | 6.67 | 36.52 | 76.2 | 0.35 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | CF | 170 | 30 | 0.04 | 48.90 | 99.9 | 0.93 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | CF | 170 | 60 | 0.03 | 51.57 | 99.9 | 0.49 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the synthesis of EMIM-MC ∼30% in methanol, results are similar as shown in Table 2. Here conversion rates between CF and batch reactions differ by factors of 2 to 3 and space-time yields by factors of 2 to 4, with CF always being superior. In order to reach very low values of the starting material EI, harsher conditions compared to those of BMPyr-MC had to be applied, which reflects the lower reactivity of EI compared to BPyr. As shown in CF reaction no. 6, only a temperature of 200 °C and a residence time of 120 min led to a residual concentration of EI at an acceptable level of 1600 ppm. Due to the limited pressure and temperature resistance of the used vials, the experiment analogue to no. 6 could not be investigated under batch conditions. A side reaction of DMC, which we observed under the previously described reaction conditions, is its slow decarboxylation into the gases dimethyl ether and CO2 under the basic conditions of the CBILS® process; a very similar reaction with pure DMC is described in the literature to take place with a conversion rate of 4% at 200 °C for 6 h over solid K2CO3 as the catalyst.8 Since DMC is used in excess of 0.6 equiv. and the side products are gases, this does not affect the synthesis of BMPyr-MC and EMIM-MC directly, but the volume of the formed gases reduces the actual available reaction volume in the continuous flow case.
| No. | Mode | T [°C] | t [min] | EI [% wt] | EMIM-MC [% wt] | Conv. [%] | STY [kg l−1 h−1] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pressure: 100 bar for all CF experiments. Continuous flow (CF), reaction temperature (T) and duration (t), educt 1-ethylimidazole (EI), product 1-ethyl-3-methylimidazolium methylcarbonate (EMIM-MC), conversion (Conv.), space-time-yield (STY) of the contained pure product. Sum of impurities <0.1% wt. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Batch | 170 | 30 | 13.52 | 4.89 | 15.7 | 0.065 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Batch | 170 | 60 | 11.59 | 10.33 | 31.5 | 0.069 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Batch | 170 | 120 | 6.31 | 21.34 | 63.6 | 0.071 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | CF | 170 | 30 | 8.92 | 14.05 | 44.9 | 0.25 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | CF | 170 | 60 | 4.82 | 21.56 | 69.8 | 0.19 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | CF | 200 | 120 | 0.16 | 32.83 | 99.1 | 0.15 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In order to suppress this volume loss to a minimum, we investigated the reaction performance under increased pressure. Table 3 shows the results for the EMIM-MC synthesis at a pressure from 20 to 150 bar and temperatures of 170 °C and 200 °C; the residence time has been chosen to be 30 min, which results in unreacted EI levels of some % wt, which is still in the dynamic range of the reaction. It can be seen that space-time yields for 170 °C differ between 0.29 kg l−1 h−1 (100%) for 150 bar and 0.23 kg l−1 h−1 (79%) for 20 bar, which has a relative difference of remarkable 21%. The trend of these changes follows a linear regression with R2 = 0.997, which correlates with the expected compression of the formed gases dimethyl ether and CO2 and subsequent increase of reaction volume. In the case of 200 °C, space-time yields increase only by 2% between 50 and 150 bar, which reflects an almost completed reaction and therefore lower influence of the increased reaction volume and residence time.
| No. | T [°C] | p [bar] | EI [% wt] | EMIM-MC [% wt] | Conv. [%] | STY [kg l−1 h−1] | STY [%] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Duration: 30 min for all experiments. Reaction temperature (T) and pressure (p); educt 1-ethylimidazole (EI), product 1-ethyl-3-methylimidazolium methylcarbonate (EMIM-MC), conversion (Conv.), space-time-yield (STY) of the contained pure product. Sum of impurities <0.1% wt. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 170 | 20 | 9.03 | 13.09 | 42.8 | 0.23 | 79 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 170 | 50 | 8.70 | 14.13 | 45.6 | 0.25 | 86 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 170 | 100 | 8.22 | 15.17 | 48.8 | 0.27 | 93 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 170 | 150 | 7.61 | 16.46 | 52.8 | 0.29 | 100 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 200 | 50 | 1.96 | 27.55 | 87.9 | 0.49 | 98 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 200 | 100 | 1.64 | 28.16 | 89.9 | 0.50 | 100 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 200 | 150 | 1.49 | 28.06 | 90.7 | 0.50 | 100 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
All reaction products from the systematic experiments listed in Tables 1–3 were analyzed and showed to be very pure with some residual quantities of halides (IC) <5 ppm, water (KFC) <100 ppm and no other impurities detectable by HPLC-UV-CAD or IC (<0.1% wt). However, to demonstrate the applicability of the continuous flow method for practical ionic liquid synthesis, a larger quantity (about 100 g) of EMIM-MC solution in methanol was produced according to the reaction conditions used in no. 6, Table 2, and further reacted with exactly 1.000 equivalent of acetic acid having a purity of 99.96%. Prior to the analytic attestation of the synthesized IL, possible volatiles were removed in vacuo by using thin film evaporator technology under industrially applicable molecular distillation conditions (see the ESI†).9 Then, the resulting 1-ethyl-3-methylimidazolium acetate (EMIM-OAc) was characterized by the methods listed in Table 4 and showed excellent purity at the level of 99.9%, which – to our best knowledge – has never been reached before for this particular IL by other methods.10
| Analyte (method) | Results |
|---|---|
| OAc− calc. as EMIM-OAc (IC) | 99.91 ± 0.05% wt |
| EMIM+ calc. as EMIM-OAc (HPLC) | 99.49 ± 0.07% wt |
| Starting material EI (HPLC) | Not detected (< 500 ppm) |
| F− (IC) | Not detected (< 5 ppm) |
| Cl− (IC) | Not detected (< 5 ppm) |
| H2O (Karl Fisher coulometry) color (acc. to Ph. Eur.) | 304 ± 3 ppm G7 (almost colorless) |
However, the precise determination of impurities in ionic liquids is a challenging task.11 Admittedly, each of the analytic methods applied in this work HPLC-UV-CAD, IC, and KFC is sensitive only to certain kinds of impurities and fails to provide the total amount of possible impurities. For example, the IC method is successful for precise determination of the residual amount of halide anions but it is useless for detection of traces of molecular precursors in the IL. The HPLC method is developed for quantification of species, which either absorb light in the visible or UV region (UV/VIS-detector) or are significantly less volatile than the eluent (CAD-detector); it is blind towards species that have no chromophore and are too volatile, e.g. methanol and dimethyl carbonate contained in the EMIM-MC intermediate. Moreover, the diversity of the available HPLC detectors (UV/VIS, IR, CAD, MS, etc.) is also adjusted for the detection of a restricted number of impurities responding only to a particular physical or chemical property of the solute (being ideally independent of the mobile phase) and eluating within the time frame of the particular method. Thus, we could pessimistically conclude that even using a broad scope of the analytic techniques for the determination of impurities in ionic liquids, we can overlook a significant amount of impurities due to limitations inherent for each of the methods applied. This conclusion heavily aggravates our main task with the focus on the development of the continuous flow method for the synthesis of ultra-pure ILs. In order to avoid ambiguity with the total amount of impurities in the ultra-pure sample of EMIM-OAc (see Table 4), two additional calorimetric methods have been applied in this work: high-precision combustion calorimetry12 and high precision solution calorimetry.13 The main idea for this double-check of the EMIM-OAc purity is that both classical calorimetric methods are extremely sensitive to the total amount of impurities. Especially sensitive is the combustion calorimetry, where the purity requirement for the sample used is conventionally ≥99.9% wt. Moreover, for this method, the amount of low-molecular weight impurities even at the level of 0.05–0.1% wt can dramatically affect the result. The solution calorimetry is also very sensitive, but for this method, more distracting are the total amounts of residual ionic species and water. The final experimental result from the combustion and the solution calorimetry, which is relevant to the purpose of this study, is the liquid state standard molar enthalpy of formation, 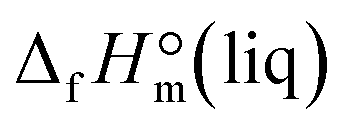 . This thermodynamic property obtained for EMIM-OAc from the two independent calorimetric methods is expected to be essentially the same, provided that the sample taken for the experiments is of impeccable purity as stated in Table 4.
. This thermodynamic property obtained for EMIM-OAc from the two independent calorimetric methods is expected to be essentially the same, provided that the sample taken for the experiments is of impeccable purity as stated in Table 4.
Combustion experiments with ultra-pure EMIM-OAc were performed with an isoperibolic calorimeter with a static bomb and a stirred water bath. The sample was transferred into a polyethylene bulb with a syringe under a nitrogen stream in a glove box. The fine neck of the container was compressed with a pair of special tweezers and was sealed outside the glove box by heating of the neck in close proximity to a glowing wire. Then, the container was placed in a crucible and was burned in oxygen at a pressure 3.04 MPa. The detailed procedure has been described previously.12 The resulting value 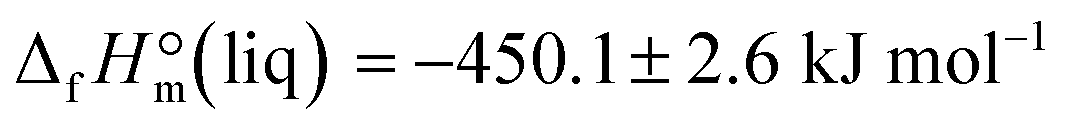 can be now compared with those obtained from the solution calorimetry.
can be now compared with those obtained from the solution calorimetry.
The molar enthalpy of solution of the ultra-pure EMIM-OAc was measured with a commercial LKB 8700-2 isoperibol solution calorimeter. Dissolution of EMIM-OAc was carried out using the ampoule technique. A cylindrical glass ampoule was filled with the sample (0.01 to 0.05 g), sealed, weighed (±0.01 mg), inserted in a sample holder, and finally immersed into the solvent. After thermal equilibration with the solvent, the ampoule was broken and the temperature change in the cell was recorded. The standard molar enthalpy of dissolution ΔsolHm was derived from five experiments. The process of dissolution of EMIM-OAc in water is ascribed to the following reaction:
| EMIM − OAc(liq) + aq = [EMIM]+(aq) + [OAc]−(aq) |
The enthalpy of this reaction is defined as the enthalpy of solution, and the value ΔsolHm = −(50.0 ± 0.2) kJ mol−1 was measured in this work by using solution calorimetry. According to Hess's Law, this enthalpy of solution is also calculated over the enthalpies of formation of the reaction participants:
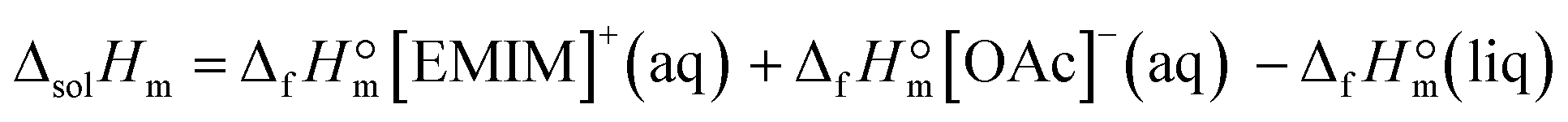
From the latter equation, the enthalpy of formation of EMIM-OAc can be derived with the known enthalpies of formation, 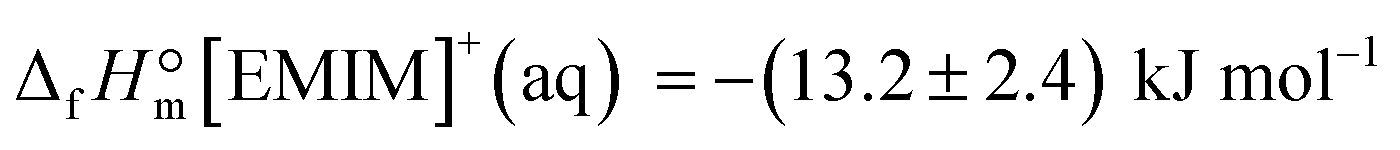 of the cation14 and the
of the cation14 and the 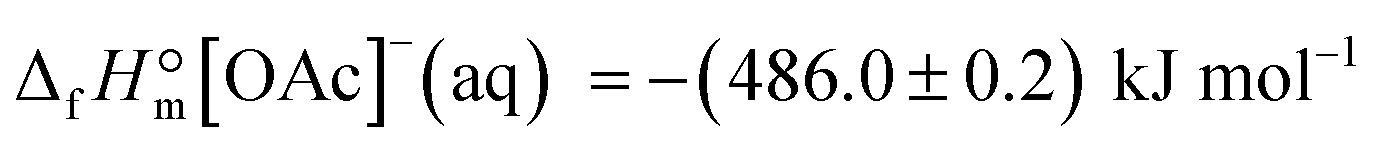 of the acetate anion.15 The resulting enthalpy of formation of EMIM-OAc
of the acetate anion.15 The resulting enthalpy of formation of EMIM-OAc 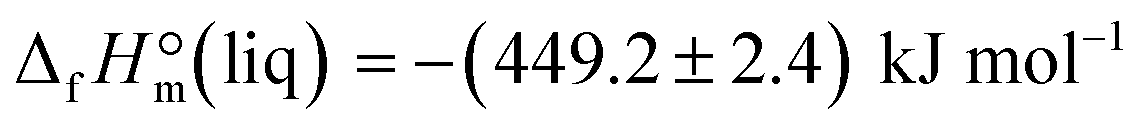 derived from the solution calorimetry experiments has been in most excellent agreement with that
derived from the solution calorimetry experiments has been in most excellent agreement with that 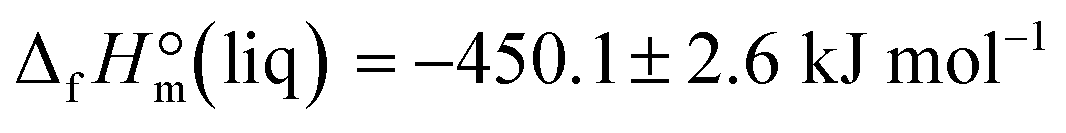 measured by the combustion calorimetry. This remarkable agreement is the convincing validation for the highest level of purity, achieved for the sample of EMIM-OAc, by using the continuous flow method for the synthesis of the ultra-pure ILs.
measured by the combustion calorimetry. This remarkable agreement is the convincing validation for the highest level of purity, achieved for the sample of EMIM-OAc, by using the continuous flow method for the synthesis of the ultra-pure ILs.
In conclusion, we have successfully shown, on the important examples of 1-butyl-1-methylpyrrolidinium methylcarbonate and 1-ethyl-3-methylimidazolium methylcarbonate, that the continuous flow synthesis according to the CBILS® process is a versatile tool to produce ultra-pure ionic liquids with significantly increased space-time yields compared to batch processes. An exemplified synthesis of the well-known ionic liquid 1-ethyl-3-methylimidazolium acetate from continuous flow generated EMIM-MC resulted in the purest sample reported in the literature so far.
A 1000-fold scale up experiment at proionic GmbH recently resulted in perfectly transferable reaction parameters and large space-time yield. A simple tube reactor with a heated zone of 30 L was operated at 195 °C and a backpressure of 100 bar, and a space-time yield of 0.14 kg l−1 h−1 of very pure EMIM-MC was obtained, which correlates with a productivity of 4.2 kg h−1. A similar batch process operated in a 200 L pressure autoclave at 150 °C and a resulting pressure of 16 bar delivered a space-time yield of only 0.0046 kg l−1 h−1 and a productivity of 0.93 kg h−1 of EMIM-MC with a somewhat worse quality. Due to the limited pressure maximum in the autoclave, a direct comparison between batch and CF at identical temperatures was not possible; however, the 30-fold difference in space-time yields indicates the advantage of the CF process. From an investment point of view, it is clear that the technical production of CBILS® ionic liquids based on the continuous flow process will be cheaper compared to that of the batch process, while realizing the same productivity. As a result, we expect to commercialize ultra-pure ionic liquids on a larger scale in the nearest future.
This work has been supported by the German Science Foundation (DFG) in frame of the priority program SPP 1708 “Material Synthesis Near Room Temperature”.
Notes and references
- T. L. Greaves and C. J. Drummond, Chem. Soc. Rev., 2013, 42, 1096 RSC.
- (a) M. J. Earle, J. M. S. S. Esperança, M. A. Gilea, J. N. Canongia Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 83 CrossRef PubMed; (b) J. M. S. S. Esperanca, J. N. Canongia Lopes, M. Tariq, L. M. N. B. F. Santos, J. W. Magee and L. P. N. Rebelo, J. Chem. Eng. Data, 2010, 55, 3 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- (a) R. S. Kalb, W. Wesner, R. Hermann, M. Kotschan, M. Schelch and W. Staber, Patents WO2005021484, EP1658262B1, US8075803B2, 2005 Search PubMed; (b) R. S. Kalb, Patents WO2008052860, EP2079705B1, 2008 Search PubMed; (c) R. S. Kalb, Patents WO2008052863, EP2079707B1, 2008 Search PubMed; (d) R. S. Kalb, Patent Application WO2008052861, 2008 Search PubMed.
- R. S. Kalb, E. N. Stepurko, V. N. Emel'yanenko and S. P. Verevkin, Phys. Chem. Chem. Phys., 2016, 18, 31904 RSC.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688 CrossRef CAS PubMed.
- E. García-Verdugo, B. Altava, M. I. Burguete, P. Lozano and S. V. Luis, Green Chem., 2015, 17, 2693 RSC.
- M. Selva, M. Fabris and A. Perosa, Green Chem., 2011, 13, 863 RSC.
- K. C. D. Hickman, in Chemical Engineers' Handbook, ed. R. H. Perry and C. H. Chilton, McGraw-Hill Book Co., Inc., New York, 5th edn, 1973, section 13 Search PubMed.
- H. Rodríguez, G. Gurau, J. D. Holbrey and R. D. Rogers, Chem. Commun., 2011, 47, 3222 RSC.
- A. Stark, P. Behrend, O. Braun, A. Müller, J. Ranke, B. Ondruschka and B. Jastorff, Green Chem., 2008, 10, 1152 RSC.
- V. N. Emel'yanenko, S. P. Verevkin and A. Heintz, J. Am. Chem. Soc., 2007, 129, 3930 CrossRef PubMed.
- A. V. Yermalayeu, D. H. Zaitsau, V. N. Emel'yanenko and S. P. Verevkin, J. Solution Chem., 2015, 44, 754 CrossRef CAS.
- D. H. Zaitsau, A. V. Yermalayeu, V. N. Emel'yanenko, S. Butler, T. Schubert and S. P. Verevkin, J. Phys. Chem. B, 2016, 120, 7949 CrossRef CAS PubMed.
- CODATA Key Values for Thermodynamics, ed. J. D. Cox, D. D. Wagman and V. A. Medvedev, Hemisphere, New York, 1989 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on experimental procedures and calculations. See DOI: 10.1039/c7re00028f |
| This journal is © The Royal Society of Chemistry 2017 |