
Continuous flow synthesis of antimalarials: opportunities for distributed autonomous chemical manufacturing
Joseph L.
Howard
,
Christiane
Schotten
and
Duncan L.
Browne
 *
*
School of Chemistry, Cardiff University, Main Building, Park Place, CF10 3AT UK. E-mail: dlbrowne@cardiff.ac.uk
First published on 3rd April 2017
Abstract
The concept of distributed manufacturing of chemicals is presented and discussed, with specific focus on the context of preparing molecules that can combat the development of geographically-localised resistant strains of infectious pathogens. Specifically we present the case of antimalarial compounds and demonstrate that the flow chemistry community have already designed both a machine capable of distrubuted chemical manufacturing and a module that would be capable of producing artemisin derivatives at the point of use.
Introduction
Distributed manufacturing is an emerging technology whereby the way in which products are manufactured and distributed is turned on its head.1,2 Rather than large scale centralised manufacturing of products, distributed manufacturing features a localised smaller scale manufacturing at the point-of-use. Distributed manufacturing requires the distribution of electronic ‘blue-prints’ to localised manufacturing sites and thus offers benefits in the reduction of transport and logistics costs, both to the economy and the environment. With regards to the distributed manufacturing of chemical entities, the on demand preparation of radiolabelled tracer compounds represents the only significant development in this field.3,4 Such processes use automation to generate the radioactive component before reacting this with a precursor compound to afford the tracer chemical. The tracer is then purified and formulated in an automated fashion prior to use for patient diagnostics. Owing to this automated approach one can imagine a series of automated modules being distributed into the field for the localised and specialised production of chemical entities. Indeed, recent results from MIT have described a machine ‘the size of a large refrigerator’ that is capable of reconfiguration to produce five key drug compounds (Fig. 1).5 We envisage such an approach to the distributed manufacturing of chemical entities as offering great power in situations where the development of resistant strains is an issue; such as antibiotics, agrochemicals and neglected tropical diseases. Such a manufacturing approach would permit easy rotation/interchange of the modules in order to produce different small molecule entities in a rotational manner; analogous to crop rotation regimes. Such modules, which are designed with the capability of preparing particular chemical entities that are bespoke and specific to addressing the localised problem, could then be deployed to the localised manufacturing site so as to combat resistance. The realisation of such an ideal requires consideration of many hurdles, not the least of which is the development of individual modules capable of the synthesis, purification and formulation of active ingredients. Although, even the latter likely pales into insignificance when the hurdle of regulatory control of such a manufacturing approach is considered! | ||
| Fig. 1 Compact, modular system used in different configurations to synthesise five formulated drugs, developed by MIT. From Science, 2016, 352, 61–67. Reprinted with permission from AAAS. | ||
The global antimalarial drug discovery portfolio has significantly improved over the last 15 years.6–9 In part this is attributable to the coordinative work of the Medicines for Malaria Venture (MMV) and the Drugs for Neglected Diseases initiative (DNDi) which were both established in 1999. Amongst others, the Bill and Melinda Gates foundation has made significant financial contribution to aid this work.10 The portfolio now has the opportunity to deliver compounds across a range of modes of action, which will be critical to combatting resistance. A distributed manufacturing approach where modules are designed for successful candidates in this portfolio would enable the rapid rollout of newly approved compounds.
Malaria continues to pose a significant threat to a large number of people throughout the world, claiming hundreds of thousands of lives every year. In 2015 alone, there were over 214 million clinical cases reported and an estimated 438![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths, mostly in very young children.11 However shocking these figures are, they only convey part of the story. The presence of malaria has a detrimental effect on a country's development, with a striking correlation between malaria and poverty having been observed.12 Malaria is therefore a truly tragic disease, but significant recent progress has been made and gives hope that eradication of the parasite will eventually be achieved. Of particular note is the recent discovery of a CRSIPR-cas9 gene drive system, which targets female reproduction in Anopheles gambiae and could suppress mosquito numbers to levels that are not conducive to the effective spreading of malaria.13 Still, the rollout of such a solution is ethically complex.
000 deaths, mostly in very young children.11 However shocking these figures are, they only convey part of the story. The presence of malaria has a detrimental effect on a country's development, with a striking correlation between malaria and poverty having been observed.12 Malaria is therefore a truly tragic disease, but significant recent progress has been made and gives hope that eradication of the parasite will eventually be achieved. Of particular note is the recent discovery of a CRSIPR-cas9 gene drive system, which targets female reproduction in Anopheles gambiae and could suppress mosquito numbers to levels that are not conducive to the effective spreading of malaria.13 Still, the rollout of such a solution is ethically complex.
A major barrier to be overcome in the fight against malaria using a chemical approach is resistance. There is now widespread resistance to all treatments developed in the last century: chloroquine, sulfadoxine–pyrimethamine (SP), amodiaquine and mefloquine.14 Currently, the artemisinin-based combination therapies (ACTs) are used as the first line of treatments. However, resistance is developing to this newest class of antimalarials, which threatens to reverse recent progress.15–18
Another critical advantageous factor that makes the application of distributed manufacturing to addressing tropical diseases an interesting proposal is that of drug quality. Often drugs in tropical climates can be susceptible to increased rates of degradation, criminal production, failed packaging analysis and poor manufacturing.19 A localised, distributed manufacturing approach would at least minimise the degradation of API materials that result from extended and multi-legged transportation routes (which include improper climate control) to the point of use.
Herein we provide an overview of the collective automated flow synthesis routes to antimalarial compounds. Notably, as a community, flow chemists have already collectively designed a module for the synthesis of the artemisinin derived antimalarial compounds and have demonstrated significant advances towards a module for a second endoperoxide; OZ439.
Artemisinin is currently the most effective treatment against the malaria parasite. It is used as the first line of treatment, as recommended by the WHO, in the form of the artemisinin-based combination therapies (ACTs). Global supplies of this vital compound are currently extracted from Artemisia annua (sweet wormwood). However, this source causes the supply to be unstable, leading to dramatic price fluctuations and increased costs for patients.20
There is still no commercially viable total synthesis reported, due to the complexity of the molecular structure. However, there have been several efforts aiming to improve the supply, often employing flow processes.
A common starting point for such efforts is dihydroartemisinic acid (DHAA) or artemisinic acid (AA) (Scheme 1). These compounds can also be extracted from A. annua in higher yields, but AA can also be obtained from glucose using genetically modified yeast.21,22 Therefore, a semisynthetic strategy from these compounds could provide a more reliable global supply of artemisinin. The initial singlet oxygen oxidation of DHAA requires the photochemical generation of singlet oxygen, and photochemical reactions are notoriously challenging to scale up in batch. As such, multiple groups have turned to flow methods for the semisynthesis of artemisinin from DHAA (Scheme 1).
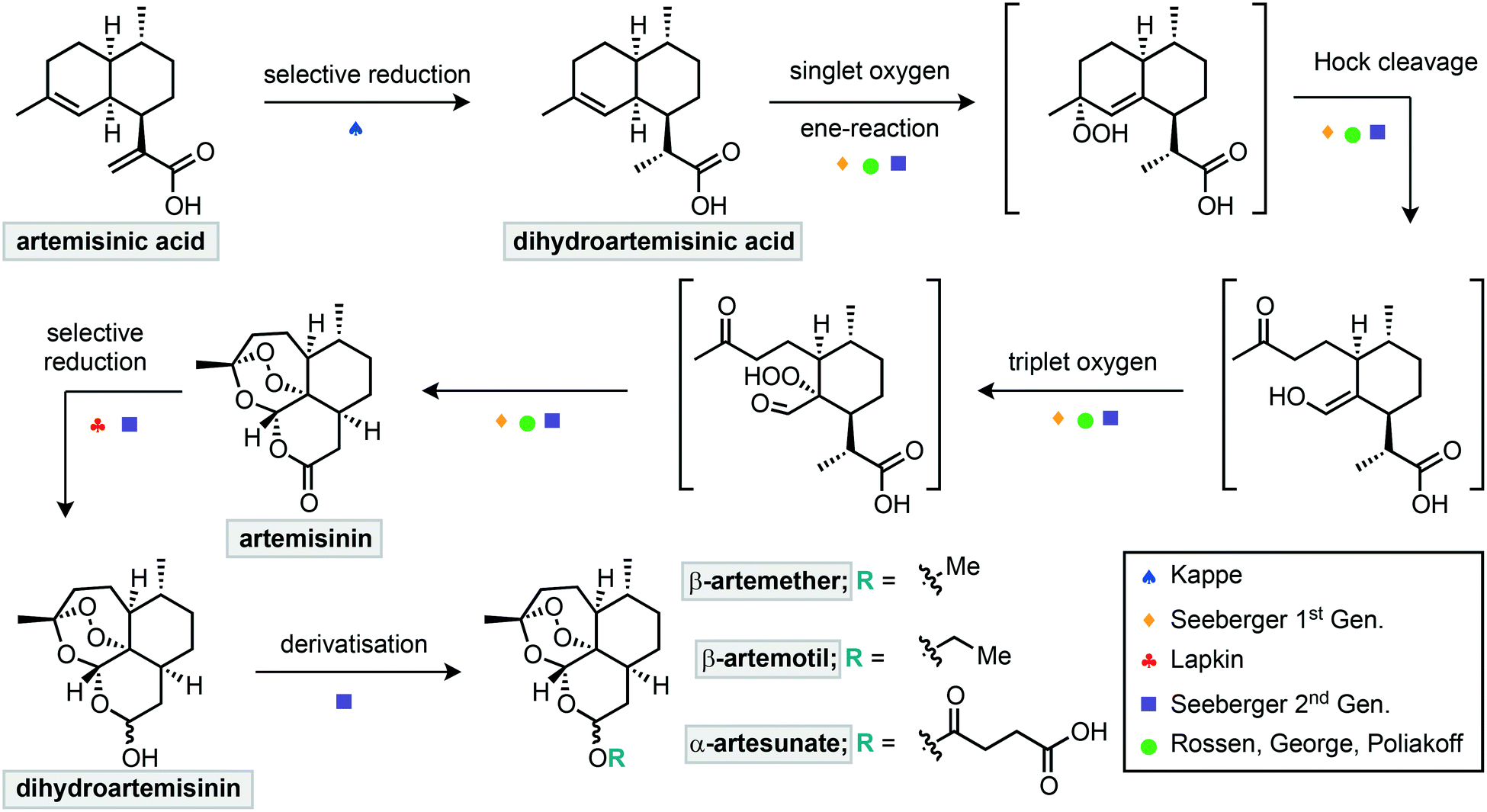 | ||
| Scheme 1 Summary of continuous flow approaches to the semisynthesis of artemisinin and its derivatives. | ||
The first step of this semisynthesis, the selective reduction of artemisinic acid (AA) to dihydroartemisinic acid (DHAA), has been described by Kappe and co-workers (Scheme 2).23 They report the chemo- and diastereoselective reduction of the alkene of the vinylogous acid with a high yield using a diimide reduction. This process avoids the use of catalysts, allowing for simple reaction workup, with side products being water and nitrogen gas. The reactive diimide is generated in situ by oxidation of the readily available hydrazine hydrate using oxygen. The alkene is then reduced by the diimide, liberating nitrogen gas and water (Scheme 2a). In order to avoid reduction of the internal alkene, the authors used a multiple injection strategy. In combination with improved mixing due to the segmented flow conditions, this multiple injection strategy mimics the slow addition of fresh hydrazine using a dropping funnel, driving diimide formation to completion. Using this method, DHAA was obtained with 93% isolated yield on an 8.5 mmol scale with a total reaction time of 37 min.
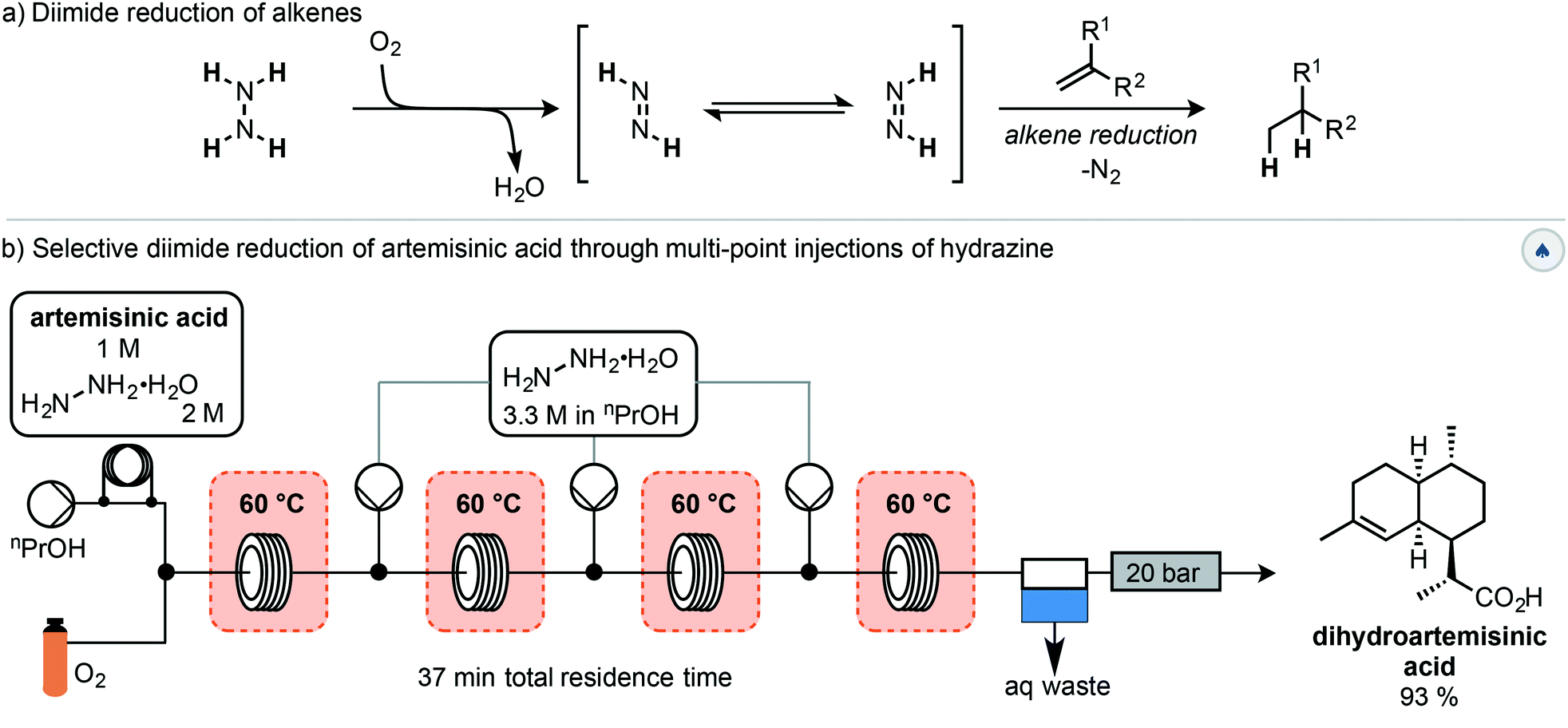 | ||
| Scheme 2 Selective reduction of artemisinic acid in flow using the diimide reduction. | ||
The next step towards the flow of synthesis of artemisinin is the oxidation of DHAA with singlet oxygen in an ene-reaction to yield the allylic hydroperoxide (Scheme 1). This intermediate then undergoes Hock cleavage which results in ring opening to afford a ketone and aldehyde/enol nucleophile. This resulting reactive enol then traps triplet oxygen and triggers a triple cyclisation cascade that affords artemisinin.
In 2012, Seeberger and Lévesque reported the multistep continuous flow synthesis of artemisinin from DHAA.24 Streams of O2 and DHAA with a photosensitizer were combined and passed through a photo-reactor coil where singlet oxygen is generated (Scheme 3). This oxidation is complete after two minutes and after exiting the photochemical reactor, a stream of trifluoroacetic acid (TFA) is introduced to promote the Hock cleavage. After passing through a heated coil for 2.5 minutes, artemisinin was obtained in 39% overall yield from DHAA. This setup is capable of producing 200 g of artemisinin per day and scale up of the photochemical step is relatively straightforward in flow.
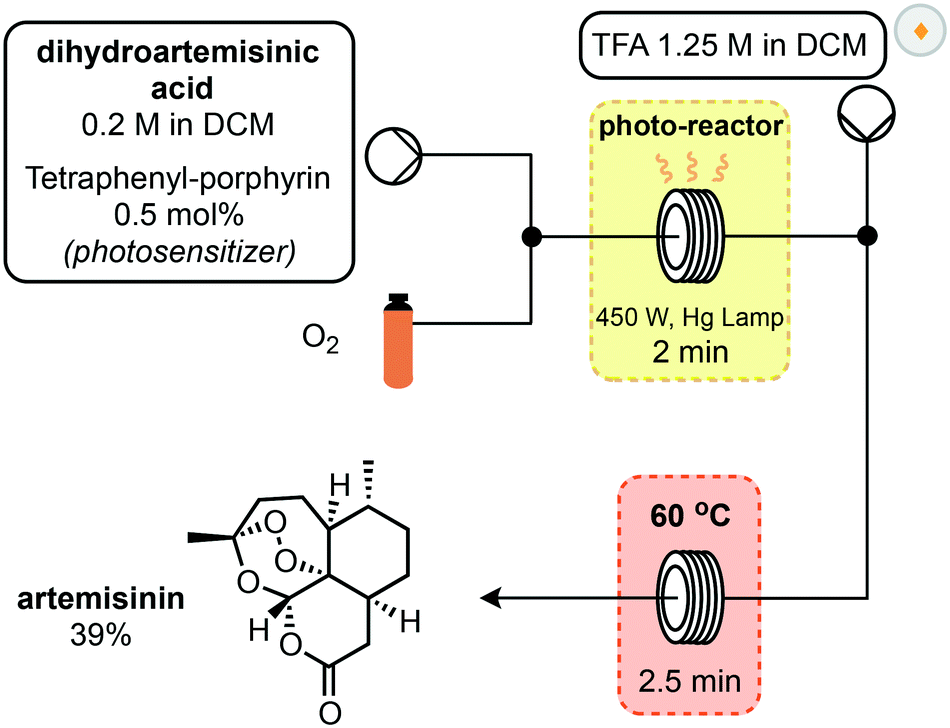 | ||
| Scheme 3 1st generation synthesis of artemisinin from dihydroartemisinic acid. | ||
A similar approach towards artemisinin has been developed by Rossen, Poliakoff, George and co-workers employing a photochemical reactor. In this instance two different strategies were explored for all steps from DHAA to artemisinin (Scheme 4).25 In the first strategy, supercritical carbon dioxide was used as an inexpensive, non-toxic solvent, in which singlet oxygen has a relatively high lifetime. Streams of DHAA, O2 and CO2 are combined and pass into the photo-reactor. The photo-reactor was pre-packed with solid supported tetraphenylporphyrin on Amberlyst 15. This immobilised catalyst serves two purposes; the porphyrin as a photosensitizer for generation of singlet oxygen and Amberlyst resin as a Brønsted acid to promote the Hock cleavage. Passing the reaction mixture through the reactor 4 times was required to achieve full conversion. Subsequent removal of the cosolvent under vacuum afforded artemisinin in 51% yield.
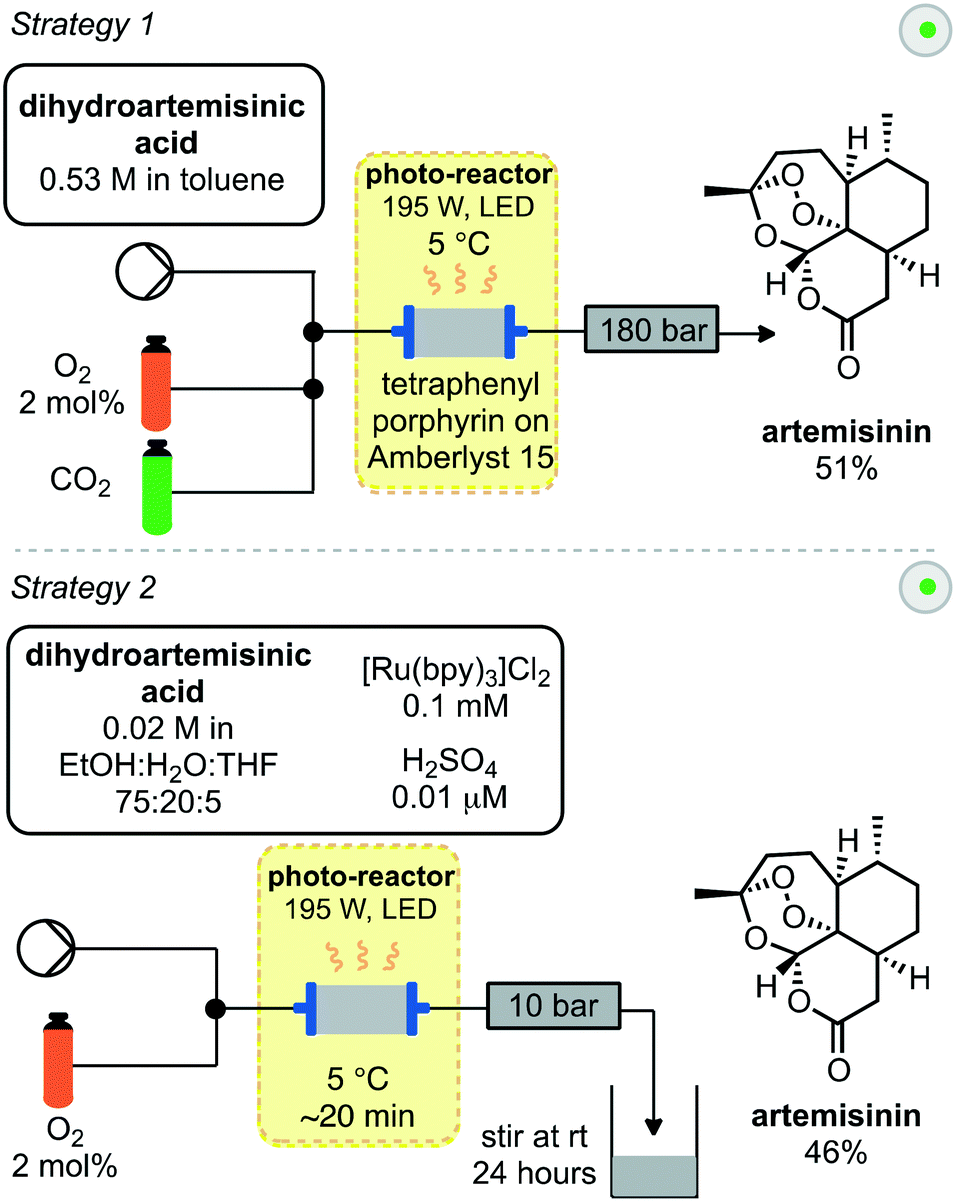 | ||
| Scheme 4 Synthesis of artemisinin from dihydroartemisinic acid in one reactor. | ||
A second strategy (Scheme 4) used a mixture of solvents including water. The photosensitizer was changed to water-soluble [Ru(bpy)3]Cl2 and the acid used was now sulfuric acid. These materials were introduced in combination with the DHAA and combined inline with a stream of O2 before passing into the photo-reactor, which was packed with glass beads to aid mixing. After a residence time of approximately 20 minutes, the output was stirred at room temperature for 24 hours open to air to complete the Hock cleavage and subsequent addition of triplet oxygen. As the organic solvents evaporated, crystals of pure artemisinin were obtained while the acid and catalyst remain in the aqueous solution. After filtration both the acid and catalyst could be recycled and artemisinin was obtained in 46% yield.
These examples demonstrate the excellent use of flow processing to overcome some of the difficulties encountered in batch during the synthesis of artemisinin, specifically the productive merger of flow and photochemical methods.26–29 They could lead to a more reliable global supply, and hence reduced cost of this vital medicine. However, artemisinin-based combination therapies (ACTs) also consist of various derivatives of artemisinin. Further derivatisations will be required in the future to combat resistance, which is already becoming a problem, particularly on the Thai–Cambodia border region.
The first step in such derivatisations is reduction of artemisinin to dihydroartemisinin (DHA). A setup for the fast and selective reduction has been reported by Lapkin and co-workers.30 In this setup, a solution of artemisinin is combined with a solution of LiBHEt3 before passing through a reactor for 30 seconds to reduce the lactone (Scheme 5). Upon exiting the reactor, the reaction was quenched by a stream of acetic acid to yield 94% of the reduced compound. The use of a flow setup allowed this exothermic reaction to be conducted safely and without external cooling. Most notably, the endoperoxide moiety remains intact during this reduction process.
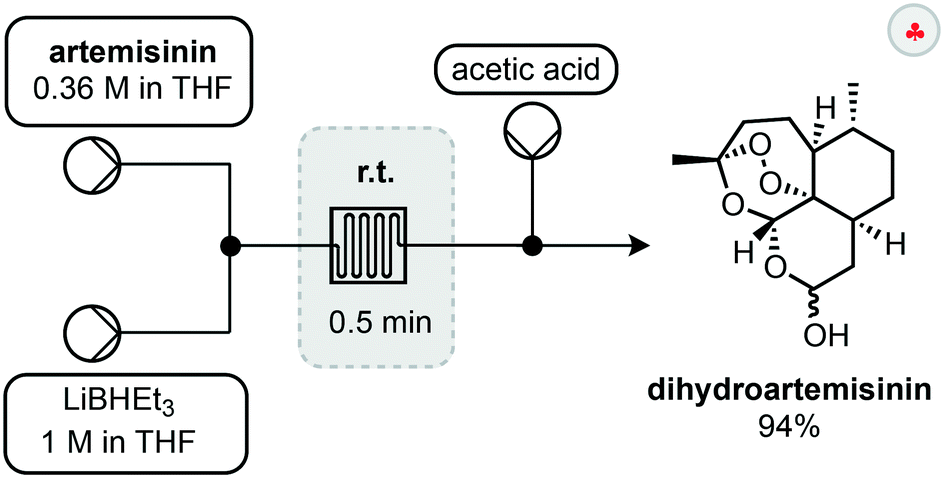 | ||
| Scheme 5 Selective reduction of artemisinin in flow. | ||
Seeberger and co-workers further extended their work to incorporate multistep processing, whereby the initial photochemical process was telescoped into reduction and derivatisation steps to deliver artemisinin derived products.31 Key to the success was the productive merging of the individual steps. In this regard the photosensitizer 9,10-dicyanoanthracene (DCA) was combined with a stream of oxygen and passed through the photo-reactor (Scheme 6). Subsequent passage through a phase separator removed any remaining oxygen before proceeding through a column packed with sodium borohydride to achieve reduction to dihydroartemisinin. This crude material underwent inline liquid–liquid extraction prior to the final derivatisation reactor sequence. The last step consisted of mixing with an appropriate electrophile or latent electrophile, passing through a residence coil of 25 minutes, neutralisation and liquid–liquid extraction to yield three artemisinin derivatives: β-artemether, β-artemotil and α-artesunate in overall yields of 25, 23 and 28% respectively. Each stage of the system was monitored for quality by inline IR spectroscopy.
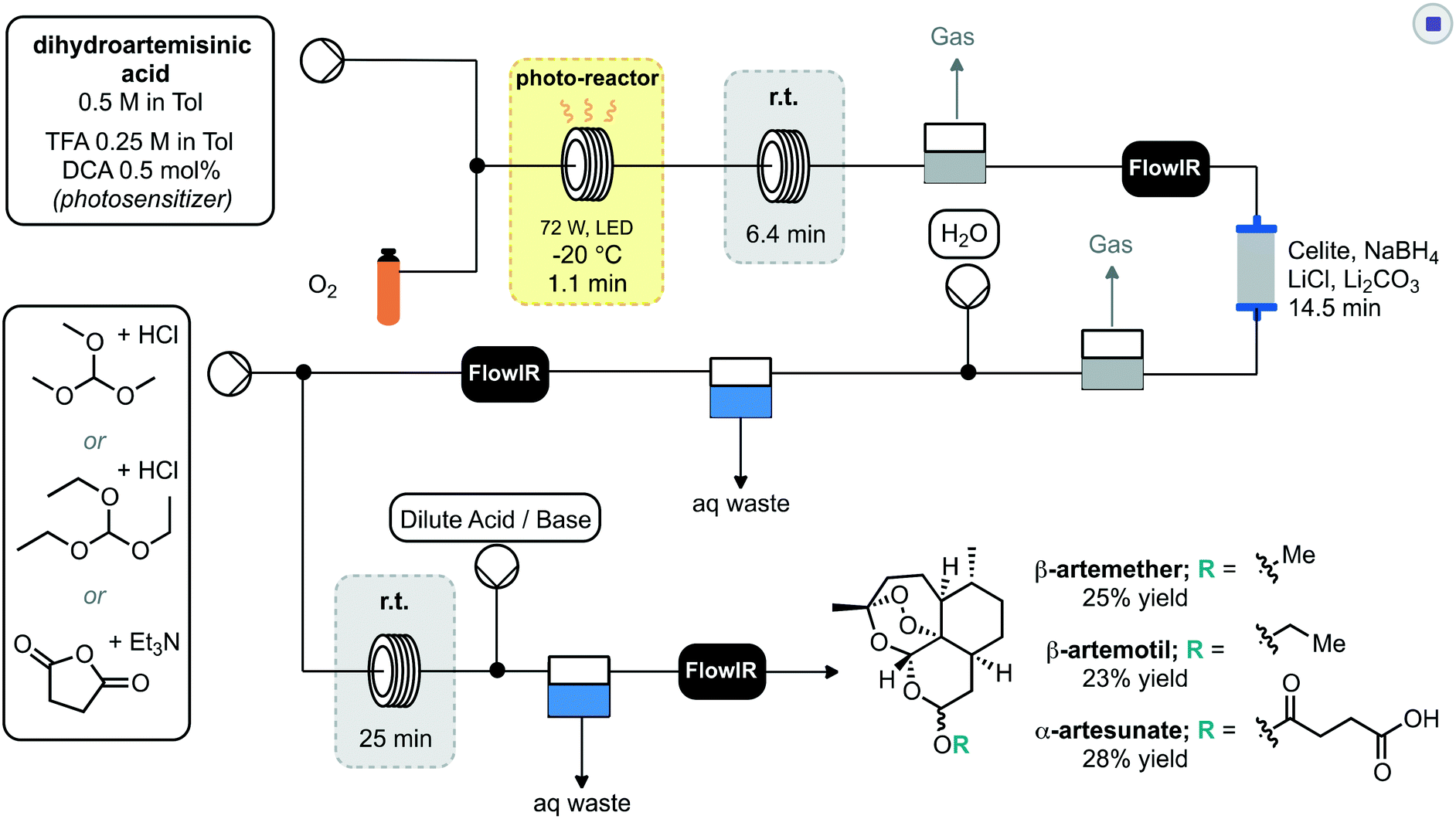 | ||
| Scheme 6 2nd generation synthesis of artemisinin from dihydroartemisinic acid and subsequent derivatisation using one setup. | ||
This system demonstrates the facile nature by which different products can be made using one flow setup. This feature is essential for distributed manufacturing systems where new modules can be “bolted on” when required in order to synthesise different derivatives in different locations.
As resistance continues to be a problem, it is necessary for new compounds to be discovered and manufactured continually. One compound showing potent antimalarial properties is OZ-439 (artefenomel). An important property is that one single, oral dose can cause complete eradication of the parasite in humans. It has undergone a phase 2a clinical trial, with an estimated parasite elimination half-life of 46–62 hours and no serious side effects.32 However, the original synthesis starts from the relatively costly 4-(4-hydroxyphenyl) cyclohexan-1-one (USD 10.60 per g, Sigma-Aldrich). Ley and co-workers have demonstrated the use of flow techniques in a synthetic pathway starting from biphenol (USD 3.20 per g, Sigma-Aldrich).33 Initially, the biphenol was reduced by passage through a heated column of Pd/C with H2 for 3 minutes to furnish the ketone in 58% yield (Scheme 7). The resulting phenol was protected using acetic anhydride in a reactor coil held at room temperature with a residence time of 40 minutes and quantitative yield. A stream of this protected phenol and oxime was then combined with a stream of ozone to synthesise the key 1,2,4-trioxolane motif in 70% yield via the Griesbaum co-ozonolysis. Having achieved a reliable setup for continuous synthesis of this intermediate at a rate of 1.9 g h−1, the remaining steps were performed in batch. Subsequent deprotection using sodium hydroxide generated the phenolate in the presence of electrophilic 4-(chloroacetyl)-morphine. After the nucleophilic substitution, the amide was reduced using a Zn catalyst in the presence of triethoxysilane to afford OZ439 in 86% isolated yield.
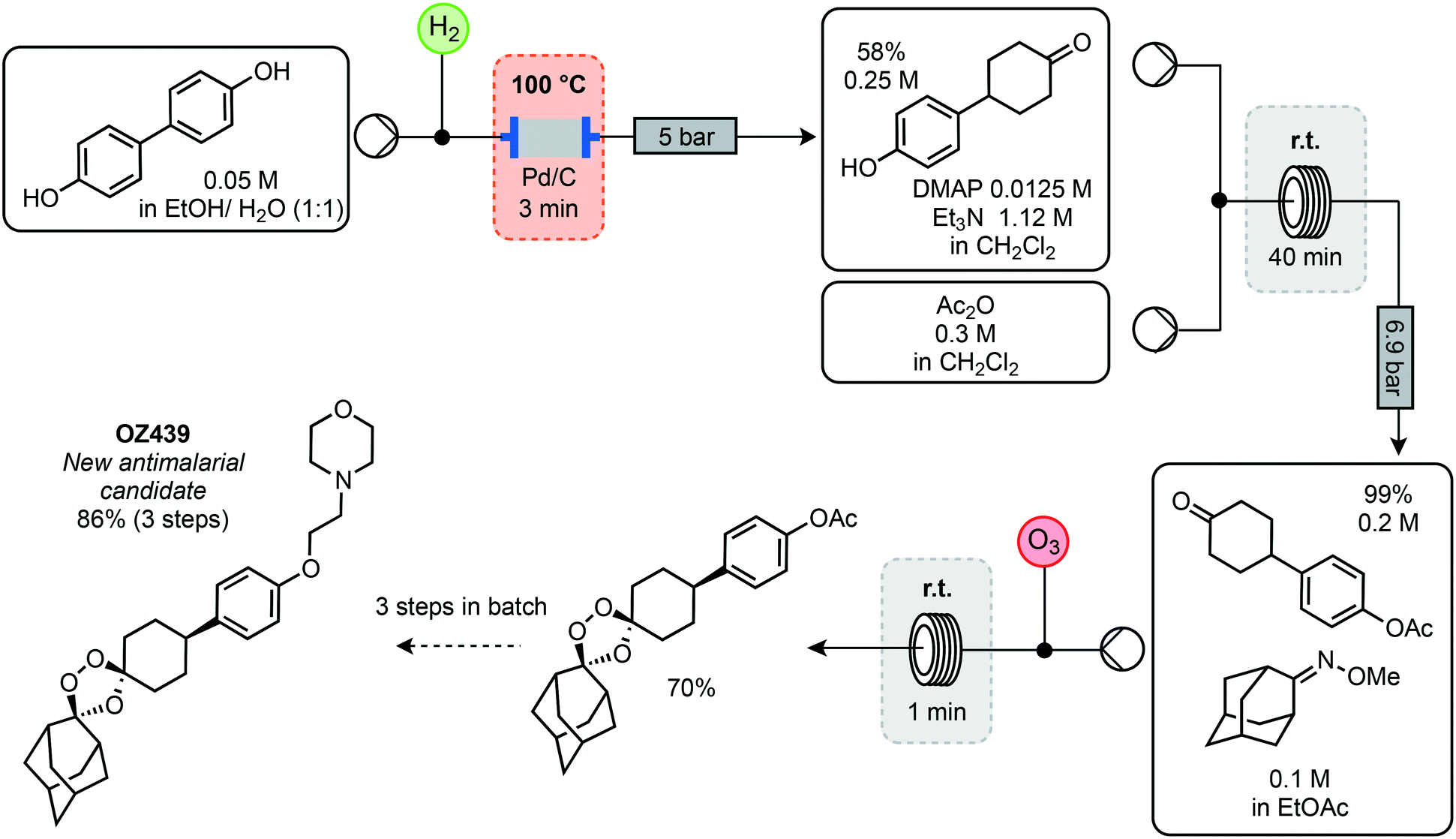 | ||
| Scheme 7 Cost effective flow synthesis of new antimalarial candidate OZ439 from biphenol in flow. | ||
Conclusion and outlook
In conclusion this mini-review/essay has highlighted the potential for the distributed manufacturing of chemicals, with specific focus on combating the development of geographically-localised resistant strains of infectious pathogens. Specifically we present the case of antimalarial compounds and summarise the contributions towards the continuous synthesis of artemisinin and derivatives as well as work towards related endoperoxides. Whilst it is clear that regulation and management of both the devices and chemical products derived from localised manufacturing would be a key critical consideration to help drive this forward, the power of the ongoing combination of chemists and chemical engineers working together to solve problems relating to human healthcare and wellbeing is critical. Indeed, a significant hurdle, which still remains to be more frequently considered by the flow chemistry community and is critical to the idea of distributed manufacturing of drug compounds, is the focus on the downstream purification of products, including formulation. Such processes have been reported, but not in the context of antimalarials and would need to consider solvent switching, liquid–liquid extraction, catalyst recovery or recycling, in-line analytics, product purification, isolation and formulation. If distributed manufacturing is the appropriate solution then such considerations would also have to be proven as well as the synthetic chemistry.Acknowledgements
D. L. B. is grateful to Prof. Simon Croft and Dr Vanessa Yardley of the London School of Hygiene and Tropical Medicine for insightful discussions.References
- H. Kühnle, Distributed manufacturing: paradigm, concepts, solutions and examples, Springer Science & Business Media, 2009 Search PubMed.
- M. Kreiger and J. M. Pearce, ACS Sustainable Chem. Eng., 2013, 1, 1511–1519 CrossRef CAS.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- A. M. Elizarov, Lab Chip, 2009, 9, 1326 RSC.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- T. N. C. Wells, R. H. van Huijsduijnen and W. C. Van Voorhis, Nat. Rev. Drug Discovery, 2015, 14, 424–442 CrossRef CAS PubMed.
- M. Schlitzer, Arch. Pharm., 2008, 341, 149–163 CrossRef CAS PubMed.
- M. A. Biamonte, J. Wanner and K. G. Le Roch, Bioorg. Med. Chem. Lett., 2013, 23, 2829–2843 CrossRef CAS PubMed.
- M. Mishra, V. K. Mishra, V. Kashaw, A. K. Iyer and S. K. Kashaw, Eur. J. Med. Chem., 2017, 125, 1300–1320 CrossRef CAS PubMed.
- P. L. Alonso, G. Brown, M. Arevalo-Herrera, F. Binka, C. Chitnis, F. Collins, O. K. Doumbo, B. Greenwood, B. F. Hall, M. M. Levine, K. Mendis, R. D. Newman, C. V. Plowe, M. H. Rodríguez, R. Sinden, L. Slutsker and M. Tanner, PLoS Med., 2011, 8(1), e1000406 Search PubMed.
- WHO, World Malaria Report 2015, http://www.who.int/malaria/publications/world-malaria-report-2015/report/en/accessed 10/01/17 Search PubMed.
- J. D. Sachs and P. Malaney, Nature, 2002, 415, 680–685 CrossRef CAS PubMed.
- A. Hammond, R. Galizi, K. Kyrou, A. Simoni, C. Siniscalchi, D. Katsanos, M. Gribble, D. Baker, E. Marois, S. Russell, A. Burt, N. Windbichler, A. Crisanti and T. Nolan, Nat. Biotechnol., 2015, 34, 78–83 CrossRef PubMed.
- L. Cui, S. Mharakurwa, D. Ndiaye, P. K. Rathod and P. J. Rosenthal, Am. J. Trop. Med. Hyg., 2015, 93, 57–68 CrossRef CAS PubMed.
- A. P. Phyo, S. Nkhoma, K. Stepniewska, E. A. Ashley, S. Nair, R. McGready, C. L. Moo, S. Al-Saai, A. M. Dondorp, K. M. Lwin, P. Singhasivanon, N. P. J. Day, N. J. White, T. J. C. Anderson and F. Nosten, Lancet, 2012, 379, 1960–1966 CrossRef.
- I. Herlekar, Curr. Sci., 2014, 106, 345 Search PubMed.
- J. Straimer, N. F. Gnädig, B. Witkowski, C. Amaratunga, V. Duru, A. P. Ramadani, M. Dacheux, N. Khim, L. Zhang, S. Lam, P. D. Gregory, F. D. Urnov, O. Mercereau-Puijalon, F. Benoit-Vical, R. M. Fairhurst, D. Ménard and D. A. Fidock, Science, 2014, 2624, 428–431 Search PubMed.
- A. M. Dondorp, F. Nosten, P. Yi, D. Das, A. P. Phyo, J. Tarning, K. M. Lwin, F. Ariey, W. Hanpithakpong, S. J. Lee, P. Ringwald, K. Silamut, M. Imwong, K. Chotivanich, P. Lim, T. Herdman, S. S. An, S. Yeung, P. Singhasivanon, N. P. J. Day, N. Lindegardh, D. Socheat and N. J. White, N. Engl. J. Med., 2009, 361, 455–467 CrossRef CAS PubMed.
- G. M. L. Nayyar, J. G. Breman, P. N. Newton and J. Herrington, Lancet Infect. Dis., 2012, 12, 488–496 CrossRef PubMed.
- R. Van Noorden, Nature, 2010, 466, 672–673 CrossRef CAS PubMed.
- D. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Y. Chang, S. T. Withers, Y. Shiba, R. Sarpong and J. D. Keasling, Nature, 2006, 440, 3–6 CrossRef PubMed.
- For a chemoenzymatic synthesis of dihydroartemisinic aldehyde see: M. Demiray, X. Tang, T. Wirth, J. A. Faraldos and R. K. Allemann, Angew. Chem., Int. Ed., 2017, 56, 4347–4350 CrossRef CAS PubMed.
- B. Pieber, T. Glasnov and C. O. Kappe, Chem. – Eur. J., 2015, 21, 4368–4376 CrossRef CAS PubMed.
- F. Lévesque and P. H. Seeberger, Angew. Chem., Int. Ed., 2012, 51, 1706–1709 CrossRef PubMed.
- Z. Amara, J. F. B. Bellamy, R. Horvath, S. J. Miller, A. Beeby, A. Burgard, K. Rossen, M. Poliakoff and M. W. George, Nat. Chem., 2015, 7, 489–495 CrossRef CAS PubMed.
- L. D. Elliott, J. P. Knowles, P. J. Koovits, K. G. Maskill, M. J. Ralph, G. Lejeune, L. J. Edwards, R. I. Robinson, I. R. Clemens, B. Cox, D. D. Pascoe, G. Koch, M. Eberle, M. B. Berry and K. I. Booker-Milburn, Chem. – Eur. J., 2014, 20, 15226–15232 CrossRef CAS PubMed.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- Y. Su, N. J. W. Straathof, V. Hessel and T. Noël, Chem. – Eur. J., 2014, 20, 10562–10589 CrossRef CAS PubMed.
- J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025–2052 CrossRef CAS PubMed.
- X. Fan, V. Sans, P. Yaseneva, D. D. Plaza, J. Williams and A. Lapkin, Org. Process Res. Dev., 2012, 16, 1039–1042 CrossRef CAS.
- K. Gilmore, D. Kopetzki, J. W. Lee, Z. Horváth, D. T. McQuade, A. Seidel-Morgenstern, P. H. Seeberger, Z. Horvath, D. T. McQuade, A. Seidel-Morgenstern and P. H. Seeberger, Chem. Commun., 2014, 50, 12652–12655 RSC.
- A. P. Phyo, P. Jittamala, F. H. Nosten, S. Pukrittayakamee, M. Imwong, N. J. White, S. Duparc, F. Macintyre, M. Baker and J. J. Möhrle, Lancet Infect. Dis., 2015, 61–69 Search PubMed.
- S. H. Lau, A. Galván, R. R. Merchant, C. Battilocchio, J. A. Souto, M. B. Berry and S. V. Ley, Org. Lett., 2015, 17, 3218–3221 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |