
A colorimetric method for rapid and selective quantification of peroxodisulfate, peroxomonosulfate and hydrogen peroxide†
Benjamin J.
Deadman
 a,
Klaus
Hellgardt
b and
King Kuok
(Mimi) Hii
*a
a,
Klaus
Hellgardt
b and
King Kuok
(Mimi) Hii
*a
aDepartment of Chemistry, Imperial College London, South Kensington, London SW7 2AZ, UK. E-mail: mimi.hii@imperial.ac.uk
bDepartment of Chemical Engineering, Imperial College London, South Kensington, London SW7 2AZ, UK. E-mail: k.hellgardt@imperial.ac.uk
First published on 13th June 2017
Abstract
Redox colorimetric tests have been devised for the rapid analysis of the individual components of aqueous mixtures of peroxodisulfate, peroxomonosulfate and hydrogen peroxide; providing a convenient and selective method for the determination of these industrially relevant oxidants, which are known to inter-convert in solution.
Peroxomonosulfate (SO52−) and peroxodisulfate (S2O82−) anions are some of the strongest oxidants with important industrial applications in chemical synthesis, water treatment, pulp and paper, textiles, electronics and metal finishing industries.1 To circumvent the potential hazards associated with handling these reactive oxidants (peroxosulfates are respiratory irritants and sensitizers), our group and others have been developing lab-scale reactors for the on-site, on-demand electrochemical generation of ammonium peroxodisulfate [(NH4)2S2O8] solutions for applications in chemical synthesis.2–4
Aqueous solutions of S2O82− are known to decompose (eqn (1) and (2)) to form SO52− and hydrogen peroxide (H2O2).5 In recent work, we observed that the efficacy and reproducibility of oxidation reactions were highly dependent on the quality of the peroxosulfate solution.6 Consequently, we needed a method that can be used to determine the precise composition of a electrochemically-generated peroxosulfate solution before it can be deployed in a reaction process.
| S2O82− + H+ + H2O → HSO5− + H2SO4 | (1) |
| HSO5− + H2O → H2O2 + HSO4− | (2) |
Analysis of a mixture of these oxidants can be problematic – most redox titrations (e.g. iodometry) are non-selective and provide only the total oxidant content ([Ox]tot). While certain redox titrants can be used for the specific quantification of S2O82−, SO52−, or H2O2 (Table 1), these are tedious to perform, and generate large quantities of aqueous waste containing toxic arsenic, vanadium and manganese salts. Alternatively, quantitative methods based on polarography7 or ion chromatography8,9 had been reported, which will require access to dedicated analytical instruments. Furthermore, analysis of S2O82− by ion chromatography is particularly challenging, requiring special measures to elute the large and highly polar anion.10,11
| Method | Measures | Oxidant selectivity | Readily available equipment | Rapid analysis (<5 min) | Small sample volumes (<100 μL) | Minimal waste (<1 mL) |
|---|---|---|---|---|---|---|
| Iodometric titration15,16 | [Ox]tot | ✗ | ✓ | ✗ | ✗ | – |
| Fe(II) titration17 | [Ox]tot | ✗ | ✓ | ✗ | ✗ | – |
| As(V) back-titration18,19 | [SO52−] | ✓ | ✓ | ✗ | ✗ | ✗ |
| V(IV) back-titration14 | [SO52−] | ✓ | ✓ | ✗ | ✗ | ✗ |
| Ce(IV) titration15 | [H2O2] | ✓ | ✓ | ✗ | ✗ | – |
| Polarography7,17 | [S2O82− + SO52−] | – | ✗ | ✓ | ✗ | – |
| Ion chromatography8–11 | [SO52−], [S2O82−] | ✓ | ✗ | ✗ | ✓ | ✓ |
| TiOSO4 redox colorimetry12,13 | [H2O2] | ✓ | ✓ | ✓ | ✓ | ✓ |
| Redox colorimetry array (this work) | [SO52−], [S2O82−], [H2O2], [Ox]tot | ✓ | ✓ | ✓ | ✓ | ✓ |
Herein, we will report an expedient approach to the rapid analysis of these oxidants by using UV-vis spectroscopy, which can be easily automated for high-throughput reaction analysis using readily accessible laboratory equipment (Fig. 1). Rapid quantification of peroxosulfate ions can be achieved for the first time,12,13 either individually, or as a mixture with H2O2.
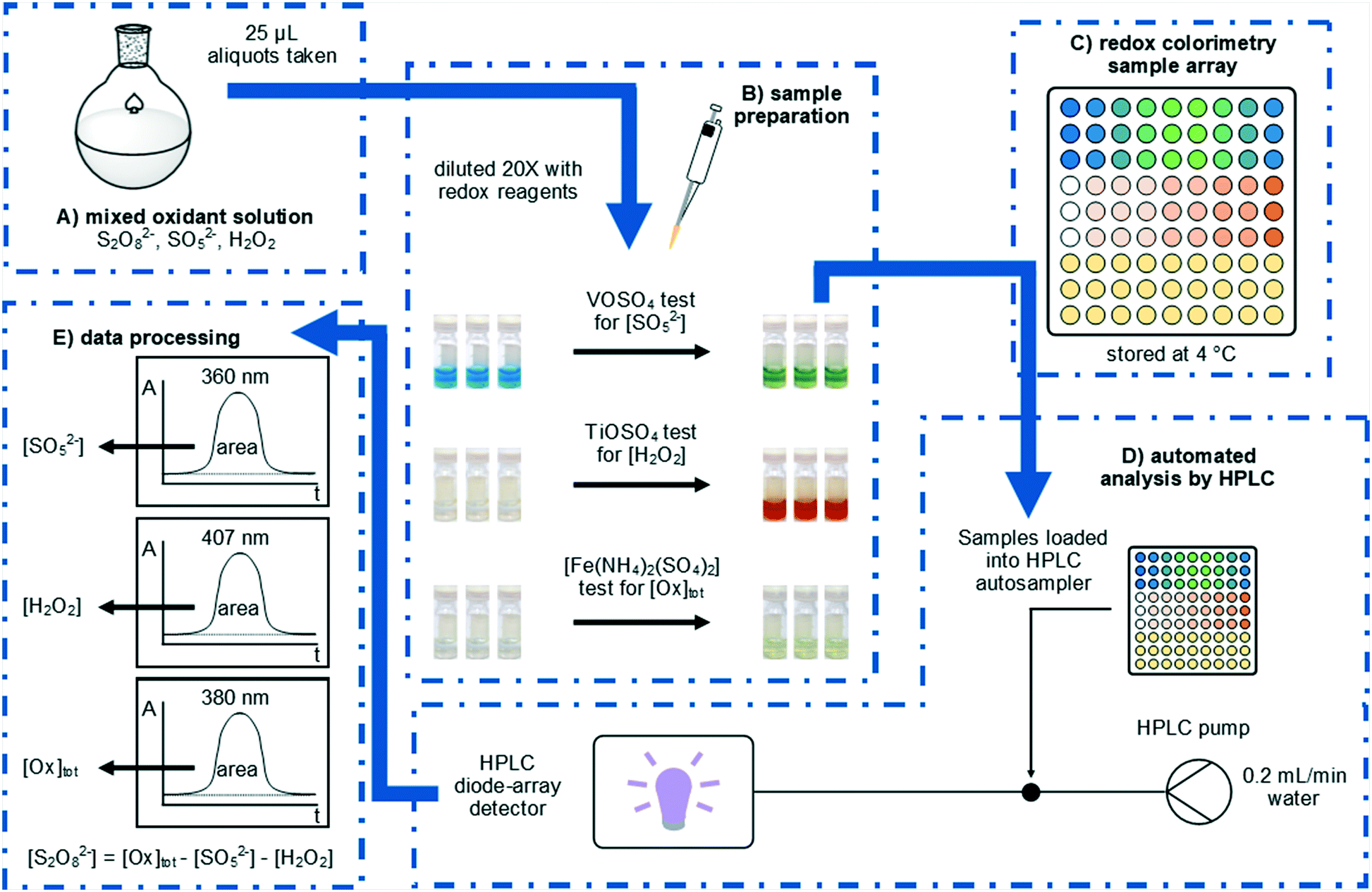 | ||
| Fig. 1 Systematic determination of S2O82−, SO52−, and H2O2 by redox colorimetry. (A) Aliquots were taken from mixed peroxosulfate solutions by autopipette and (B) diluted with redox colorimetry reagents to effect colour changes in response to specific oxidants. (C) Sample arrays were refrigerated before (D) being submitted to automated analysis using an HPLC system with diode array detection. (E) The specific oxidant concentrations were calculated from the peak areas after calibration. | ||
This work was inspired by the observation of colourful transitions during the process of using a VOSO4 titration to determine the concentration of HSO5− in a solution of peroxodisulfate.14 In a typical analysis, a sample (0.5 mL) of the peroxosulfate solution is treated with an excess (20 mL) of the blue VOSO4 solution at room temperature, and the remaining VO2+ then quantified by titration against a standardised KMnO4 solution. The distinct colour changes involved during these processes subsequently prompted us to develop a colorimetric assay, whereby samples of the oxidant (25 μL) were diluted with 0.2 M VOSO4 solution (475 μL) at room temperature. The blue VO2+ ion was oxidised by SO52− to form yellow VO2+ (Fig. 1B); giving rise to a distinct absorption peak at 360 nm which can be used directly in the quantification of the oxidant (eqn (3)).
| SO52− + 2VO2+ + H2O → SO42− + 2VO2+ + 2H+ | (3) |
In this work, the analysis was automated by utilising an HPLC system fitted with an autosampler: the chromatographic column is removed from the instrument, so that the flow from the injector passes directly to the diode array detector. With a mobile phase consisting of water flowing at 0.2 mL min−1, each sample was monitored for 1 min, with a complete cycle of injection, analysis, and software processing completing within 3 min. The peak areas at selected wavelengths were then used to determine oxidant concentrations, in accordance with Beer–Lambert Law.
This automated process requires minimal user intervention, eliminating the need to employ KMnO4 as an additional titrant (with associated errors), thus greatly improving the workflow as well as accuracy of the analysis. In principle, further increases in the throughput could be realised by use of a microplate photometer (if this is accessible). Another key advantage of the spectroscopic analysis over the titration method is a substantial reduction in the volumes of aqueous manganese and vanadium waste: each analysis generates only 0.5 mL of waste, compared to the 40 to 50 mL generated in each titration analysis. This reduction in volumes greatly extends the number of analyses that can be performed using a single batch of VOSO4 solution (2000 colorimetric analyses, compared to 48 titrations per litre of the titrant).
Calibration of the automated VOSO4 assay provided a linear response (R2 = 0.9993) for SO52− concentrations between 0.20 and 1.00 M, which is a synthetically relevant concentration range required for our research work. For more dilute solutions, treatment of a larger sample (100 μL) with 0.2 M VOSO4 solution (400 μL) allows the quantitation of [SO52−] at concentrations as low as 0.05 M. The sensitivity of the redox colorimetry test for low level [SO52−] analysis can also be further enhanced by increasing the injection volume used in the analysis, and a reduction in the concentration of the VOSO4 reagent solution. For the current method, an injection volume of only 1 μL has been employed to provide a linear response over the concentration range required for our research purposes.
Electrochemically generated peroxosulfate solutions may contain S2O82−, SO52−, and H2O2 and it is this complexity which negates the use of simpler analysis methods which can only determine the total oxidant concentration. To complement our colorimetric test for SO52− we also explored options for the colorimetric measurement of H2O2 in peroxosulfate solutions. The formation of yellow titanic acid has been previously used as a colorimetric test for H2O2 (eqn (4)).12,13 In the present procedure, samples (25 μL) were treated with acidified 0.1 M TiOSO4 solution (475 μL) and the resultant absorbance at 407 nm was measured using the automated HPLC process (Fig. 1B). The calibration curve was linear (R2 = 0.997) for the analysis of H2O2 concentrations between 0.01 and 0.80 M.
| H2O2 + TiO2+ + H2O → H2TiO4 + 2H+ | (4) |
Colorimetric assays for the determination of S2O82− were also investigated. Despite its strong redox potential (E° = 2.01 V), we are not aware of any colorimetric or titration methods which are specific to this oxidant. For peroxosulfate solutions it is assumed that the total oxidant concentration [Ox]tot is equivalent to the sum of [S2O82−], [SO52−] and [H2O2]. Hence, we assume that [S2O82−] can be determined from measurements of [Ox]tot, [SO52−] and [H2O2].
The determination of [S2O82−] has previously been achieved by iodometry,16 although the titration is not specific to S2O82−. Initial attempts to develop a colorimetric test for [Ox]tot based on the oxidation of aqueous KI proved unsuccessful, as the reaction of I− with S2O82− was found not to be quantitative. In contrast, a uniform response to all three oxidants can be achieved when an acidic solution of [Fe(NH4)2(SO4)2] (Mohr's salt) was used as the reductant (eqn (5)–(7)). In our colorimetric measurement of [Ox]tot, samples (25 μL) were treated with acidified 0.3 M [Fe(NH4)2(SO4)2] solution (475 μL) and the absorbance was measured at 380 nm (Fig. 1B). The calibration curve was linear (R2 = 0.994) for the analysis of total oxidant concentrations between 0.1 and 2.0 M.
| S2O82− + 2Fe2+ → 2SO42− + 2Fe3+ | (5) |
| SO52− + 2Fe2+ + 2H+ → SO42− + 2Fe3+ + H2O | (6) |
| H2O2 + 2Fe2+ + 2H+ → 2H2O + 2Fe3+ | (7) |
Next, the application of the colorimetric redox tests to the study of a mixture of oxidants was demonstrated by the analysis of standardised solutions of 0.99 M (NH4)2S2O8, 1.02 M potassium peroxomonosulfate triple salt [KHSO5·½KHSO4·½K2SO4], and 0.70 M H2O2 (Fig. 2A). As expected, the (NH4)2S2O8 solution only furnished a positive response in the [Fe(NH4)2(SO4)2] total oxidant test, while the [KHSO5·½KHSO4·½K2SO4] solution gave positive responses in both the total oxidant and VOSO4 test. The 0.70 M H2O2 solution gave a positive response using both the total oxidant and TiOSO4 tests; however, it also underwent a reaction with the VOSO4 reagent to produce a dark green colour, giving a measurable absorbance at 360 nm. This colour change is thought to indicate the formation of oxoperoxo vanadium complexes,20 which can limit the applicability of the VOSO4 assay to solutions containing high concentrations of H2O2. Finally, distilled water was subjected to the same redox colorimetry tests in a control experiment, which returned a nil response in every case.
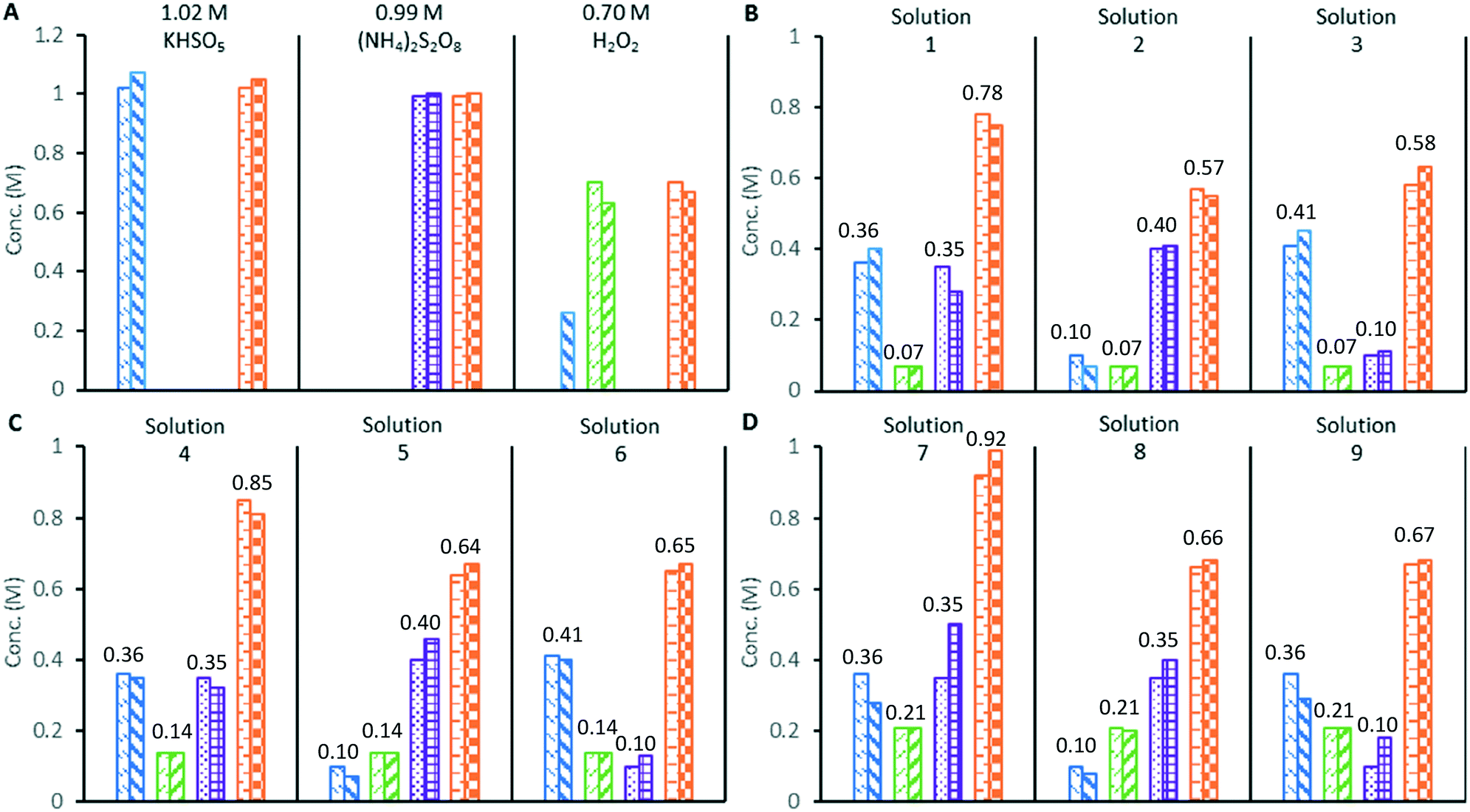 | ||
Fig. 2 Validation of redox colorimetry tests against oxidant mixtures of known composition. A) standardised solutions of single oxidants, B) mixed oxidant solutions with 0.07 M H2O2, C) mixed oxidant solutions with 0.14 M H2O2, D) mixed oxidant solutions with 0.21 M H2O2. Key to bar charts:  [SO52−] by titration, [SO52−] by titration,  [SO52−] by colorimetry, [SO52−] by colorimetry,  [H2O2] by titration, [H2O2] by titration,  [H2O2] by colorimetry, [H2O2] by colorimetry,  [S2O82−] by titration, [S2O82−] by titration,  [S2O82−] by colorimetry, [S2O82−] by colorimetry,  [Ox]tot by titration, [Ox]tot by titration,  [Ox]tot by colorimetry, numbers above bars are the expected concentrations in M. [Ox]tot by colorimetry, numbers above bars are the expected concentrations in M. | ||
The three standardised oxidant solutions were subsequently combined in varying quantities to create 9 test solutions of known compositions, which were analysed in triplicate by redox colorimetry (Fig. 2B–D). Analysis of solutions containing 0.07 or 0.14 M of H2O2 gave oxidant compositions in good agreement with the expected results (solutions 1–6).
Mixed peroxosulfate systems with low levels (<0.2 M) of H2O2 can be characterised by the redox colorimetry assay with an acceptable level of accuracy. However, when the mixed oxidant solutions contained a higher concentration of H2O2 (0.21 M), the formation of the dark green (oxoperoxo) species interfered with the measurement of [SO52−], which was found to be consistently lower (approximately 20%) than expected (solutions 7–9). Since [S2O82−] is derived from the redox colorimetric measurement of the [SO52−], its measurement is also affected by the undesired reaction of VOSO4 with H2O2. In the original redox titration employing VOSO4, the H2O2 was first removed from the sample by a titration against Ce(SO4)2 at 0 °C.14 In this work, addition of a small portion of MnO2 rapidly decomposed H2O2 to O2 in a peroxosulfate solution at room temperature, while the S2O82− and SO52− were unaffected. Hence, redox colorimetric analysis performed before and after quenching with MnO2 allows the concentrations of S2O82− and SO52− to be determined in peroxosulfate solutions containing H2O2 (details in ESI†).
The experimental uncertainty in the colorimetric redox tests was estimated by performing six repeat analyses of the mixed oxidant solutions (1, 4 and 7 from Fig. 2). The uncertainty in [SO52−] and [S2O82−] are both estimated to be ±0.05 M, while [H2O2] could be determined to within ±0.01 M. The uncertainty in [Ox]tot is estimated to be ±0.02 M (note: these uncertainty estimates apply to the measurement of peroxosulfate solutions with [H2O2] < 0.2 M).
Samples treated with the VOSO4 and TiOSO4 reagents should be analysed immediately or stored at reduced temperatures (3–4 °C). In our ongoing research into applications of peroxosulfates, we have observed that the [SO52−] and [H2O2] measured by redox colorimetry will increase over a period of 1–2 h when samples are stored in a reasonably warm laboratory (>25 °C). This could be due to the interconversion of the oxidants (eqn (1) and (2)), or slow reaction of off-target oxidants with the redox reagent. We have found that refrigeration of the treated samples stabilises them for at least 4 h, during which time any change in the measured oxidant concentrations is within the limits of uncertainty.
In an application of these redox colorimetric tests we have followed the decomposition of S2O82− into SO52− and H2O2 in an electrochemically-generated acidic peroxosulfate solution (Fig. 3): the solution (containing 0.42 M S2O82− and 0.55 M SO52−) was heated at 50 °C and aliquots were analysed by redox colorimetry. The expected conversion of S2O82− into SO52− was observed to progress over 3 h with the formation of only 0.03 M of H2O2. After 3 h, [SO52−] reached a maxima and proceeded to decrease, with a concomitant increase in [H2O2]. After 25 h, the solution contained a mixture of 0.59 M SO52− and 0.38 M H2O2 (determined using MnO2 to quench H2O2). This application demonstrates the utility of these assays for characterising the specific concentrations of oxidants in peroxosulfate solutions.
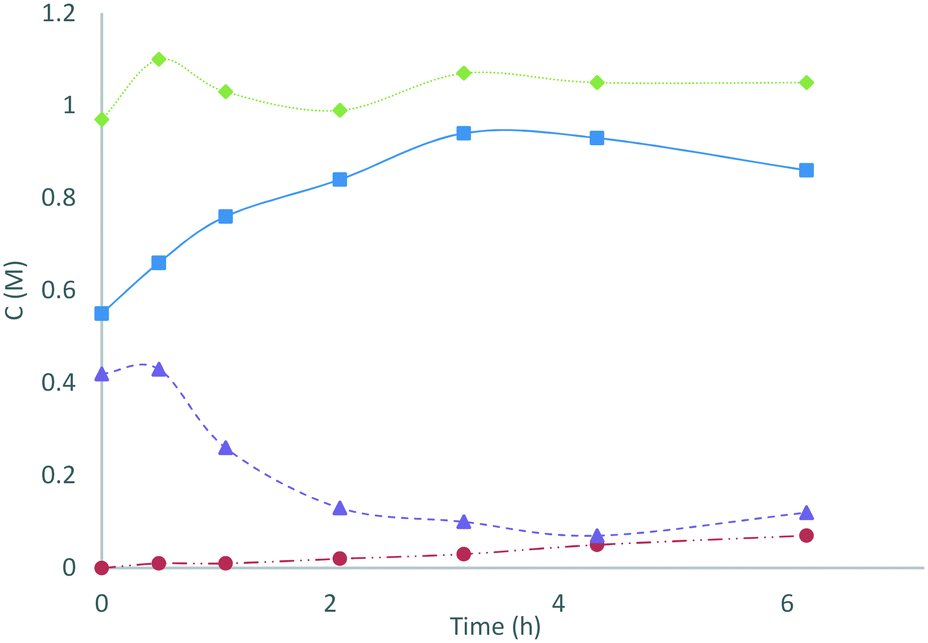 | ||
Fig. 3 Changes in the oxidant composition of an electrochemically generated acidic peroxosulfate solution heated at 50 °C were monitored using the colorimetric redox assays. Key to chart:  [SO52−], [SO52−],  [H2O2], [H2O2],  [Ox.]tot, [Ox.]tot,  [S2O82−]. [S2O82−]. | ||
Conclusions
To summarise, we have developed novel colorimetric redox assays for the selective determination of S2O82−, SO52− and H2O2 where they co-exist in solution. These assays are now being routinely employed in our ongoing investigation into synthetic applications of electrochemically generated solutions of peroxosulfates. In this application we have automated the analysis of redox colorimetry samples, which were collected manually by autopipette in this study. Going forward, automated liquid handling and sampling technologies can be utilised to achieve an uninterrupted workflow; from sample collection to analysis and processing (refer to ESI† for design details of an automated sampling system). Implementation of these technologies in future work will make redox colorimetry an attractive tool for characterisation and quality control of processes involving peroxosulfates.Acknowledgements
The authors thank Dr K. N. Loponov (University of Loughborough) for the suggestion of using an HPLC system for measurement of colorimetric assays. This work is supported by the award of a research grant by the Engineering and Physical Sciences Research Council (EPSRC), U.K., titled: Manufacturing in Flow: Controlled Multiphase Reactions on Demand (CoMRaDe, EP/L012278/1).References
- H. Jakob, S. Leininger, T. Lehmann, S. Jacobi and S. Gutewort, Ullmann's Encycl. Ind. Chem., 2007, vol. 26, pp. 293–324 Search PubMed.
- J. Zhu, K. K. Hii and K. Hellgardt, ACS Sustainable Chem. Eng., 2016, 4, 2027–2036 CrossRef CAS.
- K. K. Hii, K. Hellgardt, G. H. Kelsall, J. B. Brazier and L. A. Adrio, GB Pat., P62616GB, 2015 Search PubMed.
- P. C. B. Page, F. Marken, C. Williamson, Y. Chan, B. R. Buckley and D. Bethell, Adv. Synth. Catal., 2008, 350, 1149–1154 CrossRef CAS.
- D. A. House, Chem. Rev., 1962, 62, 185–203 CrossRef CAS.
- K. N. Loponov, B. J. Deadman, J. Zhu, C. Reilly, R. G. Holdich, K. K. Hii and K. Hellgardt, Chem. Eng. J., 2017 DOI:10.1016/j.cej.2017.05.017.
- I. M. Kolthoff and I. K. Miller, J. Am. Chem. Soc., 1951, 73, 3055–3059 CrossRef CAS.
- J. R. Davis, J. C. Baygents and J. Farrell, J. Appl. Electrochem., 2014, 44, 841–848 CrossRef CAS.
- J. Davis, J. C. Baygents and J. Farrell, Electrochim. Acta, 2014, 150, 68–74 CrossRef CAS.
- N. E. Khan and Y. G. Adewuyi, J. Chromatogr. A, 2011, 1218, 392–397 CrossRef CAS PubMed.
- Z. Huang, C. Ni, F. Wang, Z. Zhu, Q. Subhani, M. Wang and Y. Zhu, J. Sep. Sci., 2014, 37, 198–203 CrossRef CAS PubMed.
- G. Eisenberg, Ind. Eng. Chem., Anal. Ed., 1943, 15, 327–328 CrossRef CAS.
- D. B. Graves, Peroxide (H2O2) quantification, http://www.graveslab.org/lab-resources/procedures/peroxide-h2o2-quantification, (accessed August 2016).
- A. J. Berry, Analyst, 1933, 58, 464–467 RSC.
- G. H. Jeffery, J. Bassett, J. Mendham and R. C. Denney, Vogel's textbook of quantitative chemical analysis, Longman Scientific & Technical, London, 5th edn, 1989 Search PubMed.
- N. Wahba, M. F. El Asmar and M. M. El Sadr, Anal. Chem., 1959, 31, 1870–1871 CrossRef CAS.
- I. M. Kolthoff, L. S. Guss, D. R. May and A. I. Medalia, J. Polym. Sci., 1946, 1, 340–352 CrossRef CAS.
- L. J. Csanyi and F. Solymosi, Fresenius' Z. Anal. Chem., 1954, 142, 423–426 CrossRef CAS.
- K. Gleu, Z. Anorg. Allg. Chem., 1931, 195, 61–74 CrossRef CAS.
- P. Schwendt and M. Pisárčik, Spectrochim. Acta, Part A, 1990, 46, 397–399 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00050b |
| This journal is © The Royal Society of Chemistry 2017 |