
Selective continuous flow synthesis of hydroxy lactones from alkenoic acids†
Bruno
Cerra
,
Francesca
Mangiavacchi
,
Claudio
Santi
,
Anna Maria
Lozza
and
Antimo
Gioiello
 *
*
Laboratory of Medicinal and Advanced Synthetic Chemistry (Lab MASC), Department of Pharmaceutical Sciences, University of Perugia, Via del Liceo 1, I-06122 Perugia, Italy. E-mail: antimo.gioiello@unipg.it; Tel: +39 075 5852318
First published on 6th July 2017
Abstract
A novel approach to synthesize hydroxy lactones using flow chemistry is reported. The system could be applied to a variety of functionalized alkenoic acids allowing the simple and eco-friendly generation of lactonic products within one hour (50 min). The reaction was optimized in terms of efficiency, productivity, resources and eco-sustainability using an integrated flow process assisted by in-line work-up and purification. In one example, the method was scaled to deliver 22 mmol of the product.
Introduction
The broad occurrence of lactone rings in natural and bioactive compounds1 and in their precursors2 makes their preparation an important target for synthesis. Diverse approaches have been reported,3 and although major advances have been made, further criteria need to be met to improve the efficiency and versatility, and to provide eco-friendly protocols.To this aim, continuous flow chemistry represents an appealing area of research inspiring the development of green4 and modern methods.5 So far, only two examples have shown the profitable use of flow technology for the preparation of lactones. In particular, Park and collaborators described the palladium-catalyzed diacetoxylation of alkenes using peracetic acid in a microchemical device.6 Albeit the method was designed to provide vicinal diacetoxy compounds, in two cases, the reaction afforded five-membered acetoxy lactones. More recently, the photocatalytic/reductive reaction of in situ generated acyl succinate intermediates was explored to prepare γ-lactones in 51–68% yield and a moderate diastereoselectivity ratio.7 There is, therefore, still room for improvement in terms of substrate scope, yield and selectivity, as well as the flow set-up for integrated downstream operations.
Given that a batch method was recently reported for the bromolactonization of 4-pentenoic acid by using a selenide-based xerogel in the presence of H2O2 and NaBr,8 we were intrigued with the possibility of studying a similar tactic for preparing hydroxy lactones under flow conditions. It is worth noting that, to the best of our knowledge, selenium-mediated catalysis has never been investigated in flow systems. Thus, taking inspiration from the oxidative cycle of glutathione peroxidase (GPx), we sought to employ benzeneperseleninic acid (PhSeO3H, 1) generated in situ from benzeneseleninic acid (PhSeO2H, 2) and H2O2,9 to oxidize alkenoic acids into epoxide intermediates that can readily undergo intramolecular cyclization to form the corresponding hydroxy lactones (Scheme 1).
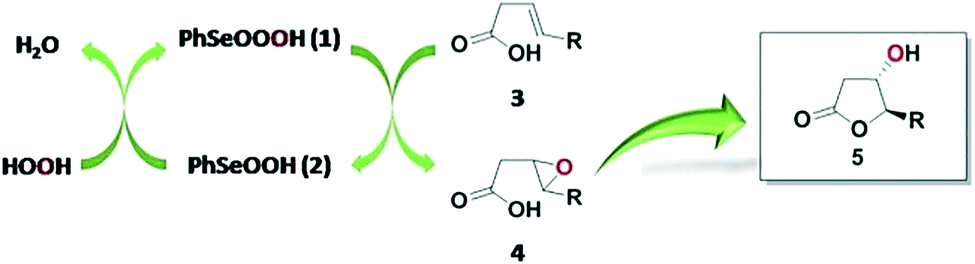 | ||
| Scheme 1 GPx-inspired mechanism induced by selenium-organic reagents employed for the flow synthesis of hydroxy lactones. | ||
Herein, reaction conditions for translating the process into a flow platform are developed to satisfy the specific requirements of continuous flow reactors in a sustainable fashion. Substrate scope and scaling-out are also described, demonstrating the versatility and robustness of the method.
Results and discussion
To realize the translation of the reaction into a flow process, we decided to divide the work into three steps: (a) definition of the model reaction and screening of key experimental parameters, (b) design of a convenient flow set-up, (c) substrate scope and scale-up. Decisions on the best conditions and improvements were made, taking into consideration the efficiency, resources and eco-sustainability.Commercially available (E)-3-pentenoic acid (3a) was selected as the model substrate for initial reaction screenings, while PhSeO2H (2) was employed as the pre-catalyst to form in situ PhSeO3H (1) by reaction with H2O2.9 H2O/acetone (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and EtOAc were the solvents of choice because of their safety profile and ability to solubilize both reagents and products. Reaction screening was performed at room temperature in a modular flow system equipped with loop injection systems, two HPLC pumps, a reactor coil (10 mL), a back pressure regulator (BPR, 100 psi) and a fraction collector (Fig. 1). Thus, a 0.3 M solution of 3a in EtOAc and a solution of 2 (0.1–0.5 equiv.) and H2O2 (30% wt, 5 equiv.) in H2O/acetone were injected into the loops, mixed in a T-piece and flowed through the reactor coil at 25 °C. The crude reaction mixture was collected and analyzed by 1H-NMR analysis for the determination of the reaction yield.
:1, v/v) and EtOAc were the solvents of choice because of their safety profile and ability to solubilize both reagents and products. Reaction screening was performed at room temperature in a modular flow system equipped with loop injection systems, two HPLC pumps, a reactor coil (10 mL), a back pressure regulator (BPR, 100 psi) and a fraction collector (Fig. 1). Thus, a 0.3 M solution of 3a in EtOAc and a solution of 2 (0.1–0.5 equiv.) and H2O2 (30% wt, 5 equiv.) in H2O/acetone were injected into the loops, mixed in a T-piece and flowed through the reactor coil at 25 °C. The crude reaction mixture was collected and analyzed by 1H-NMR analysis for the determination of the reaction yield.
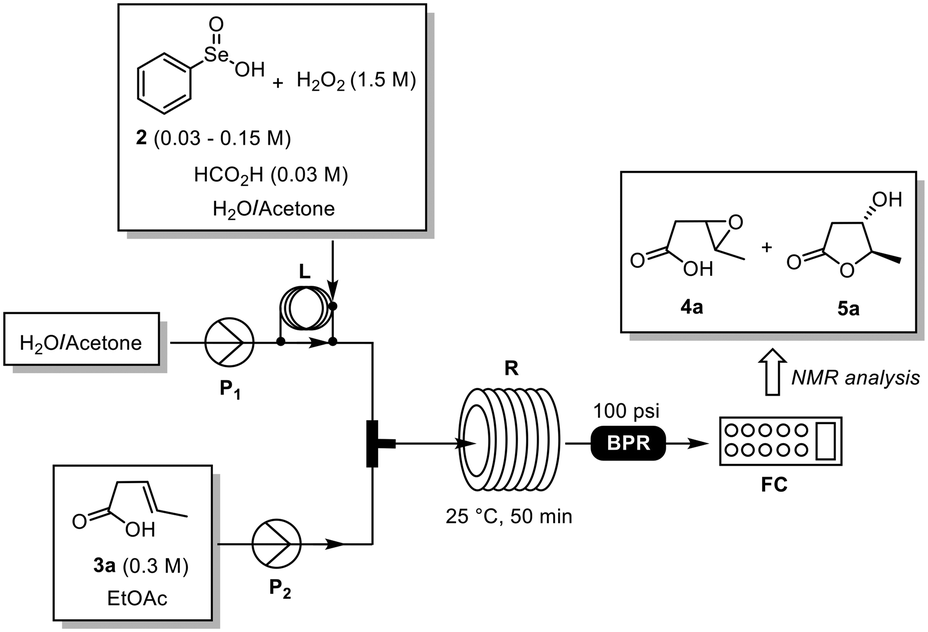 | ||
| Fig. 1 General flow set-up employed during the reaction optimization. BPR: back pressure regulator; FC: fraction collector; L: loop injector; P1–2: pumps; R: 10 mL reactor coil. | ||
Initially we have evaluated the effect of the amount of 2 on the reaction outcome (Table 1). The best result was obtained using 0.5 equiv. of 2 affording 5a in nearly quantitative conversion in 50 min. Intrigued by the high conversion of 3a into the epoxy intermediate 4a (Table 1, entry 2), we then envisaged the possibility of promoting the cyclization of 4a into 5a by acidic catalysis using a volatile acid such as formic acid (10% mol).10 This would allow us to reduce the amount of selenium reagent needed also to convert 4a into 5a. Accordingly, the desired lactone 5a was obtained in 77% yield with a conversion of 90% using 0.2 equiv. of PhSeO2H (2) (Table 1, entry 7). Remarkably, a slight increase of PhSeO2H (2) (0.3 equiv.) afforded 5a in quantitative yield (Table 1, entry 8),11 while higher flow rates had a negative effect on the reaction performance (Table 1, entry 9).
| Entry | PhSeO2H (2) (equiv.) | Flow rate (mL min−1) | Conversionb (%) | 4a/5ab |
|---|---|---|---|---|
|
a All reactions were conducted according to Fig. 1. Reagents and conditions: 3a [0.6 mmol, 0.3 M in EtOAc], 2 [0.1–0.5 equiv., 0.06–0.3 mmol, 0.03–0.15 M in H2O/acetone (5:1, v/v)] and H2O2 [30% wt, 5 equiv., 3 mmol, 1.5 M in H2O/acetone (5:1, v/v)], 10 mL coil reactor, 25 °C.
b Determined by 1H-NMR analysis of the crude reaction mixture.
|
||||
| 1 | 0.1 | 0.2 | 85 | 100/0 |
| 2 | 0.2 | 0.2 | 94 | 100/0 |
| 3 | 0.3 | 0.2 | 100 | 55/45 |
| 4 | 0.4 | 0.2 | 100 | 16/84 |
| 5 | 0.5 | 0.2 | 100 | 0/100 |
| 6 | 0.1 + HCO2H | 0.2 | 88 | 80/20 |
| 7 | 0.2 + HCO2H | 0.2 | 90 | 23/77 |
| 8 | 0.3 + HCO2H | 0.2 | 100 | 0/100 |
| 9 | 0.3 + HCO2H | 0.3 | 86 | 0/100 |
Having established the best conditions (Table 1, entry 8), we looked at the downstream section of the process. Besides efficiency, we also sought to simplify as much as possible the flow protocol in order to keep the environmental factor, costs and handling as low as possible. Thus, a continuous liquid–liquid membrane-based separator (S) was employed to separate the aqueous phase from the organic solution (Fig. 2). The organic layer was then directed towards a glass column (C) (Omnifit Labware DIBA HIT column, L × ID 6.6 mm × 100 mm) packed with Amberlyst A21 and silica to catch the selenium-containing reagents and traces of the starting material (Fig. S1, ESI†). The outflow was collected, furnishing 5a in 90% isolated yield with high purity (Table 2, entry 1). Importantly, no traces of selenium and 3a were detected proving the efficiency of the catch system. After reaction, two valves (V1 and V2) ensured the washing of the A21 packed column with an ethanolic solution of NH4OH (5%, v/v), and enabled the recovery of both the catalyst and the unreacted starting material that can be reused for other reactions (Fig. 2).
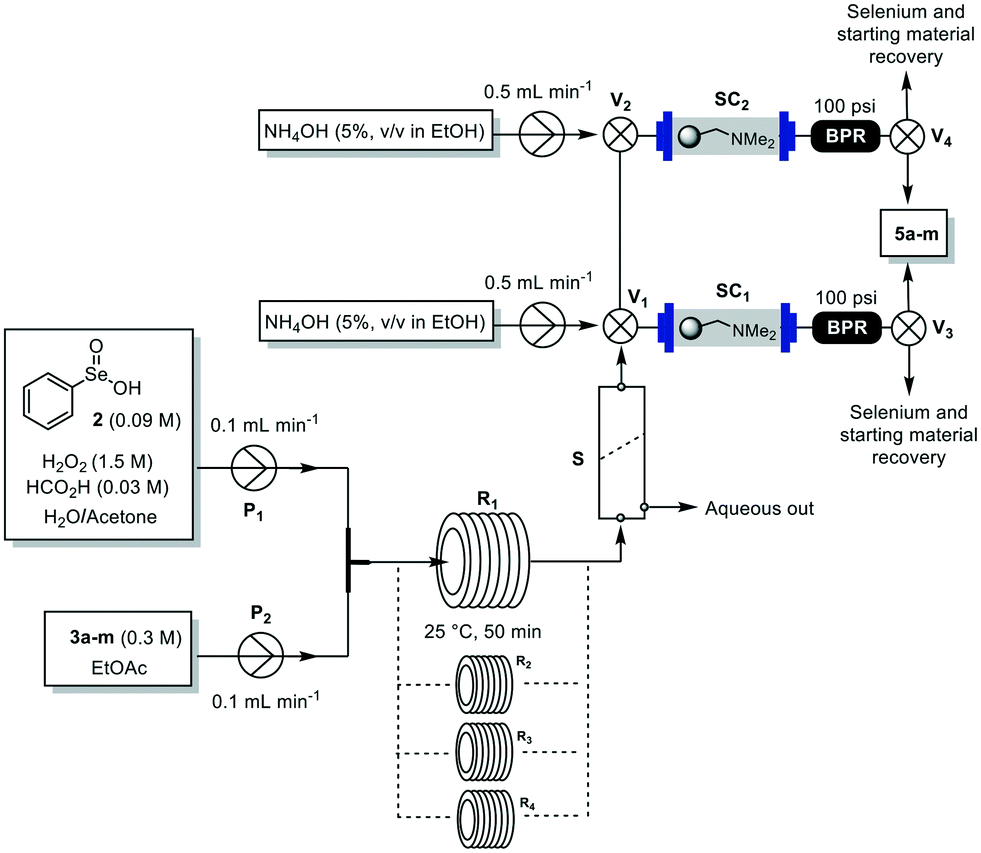 | ||
| Fig. 2 Integrated flow set-up for the synthesis and purification of hydroxy lactones 5a–m. BPR: back pressure regulator; P1–2: pumps; R: 10 mL reactor coil; S: liquid–liquid membrane separator; SC1–2: scavenger column; V1–4: valves. | ||
| Entry | Substrate | Products | Yieldb (%) | d.r.c |
|---|---|---|---|---|
|
a All reactions were conducted according to Fig. 2.
b Isolated yield.
c Diastereomeric ratio (d.r.) was determined by 1H-NMR analysis.
d 4-Hexenoic acid (3h) was used in the commercially available mixture constituted by (E)- and (Z)-isomers in an 8:2 ratio; the E and Z ratio of 3h was maintained during the formation of the corresponding S- and R-product 5h. n.a.: not applicable.
|
||||
| 1 |
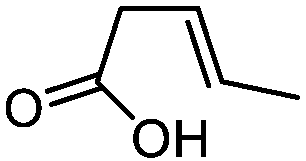 3a
3a
|
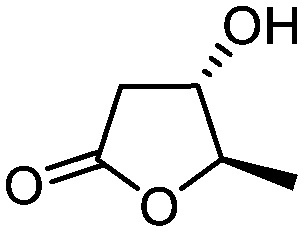 5a
5a
|
90 | >99:1 |
| 2 |
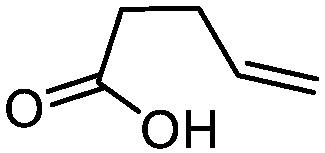 3b
3b
|
 5b
5b
|
80 | n.a. |
| 3 |
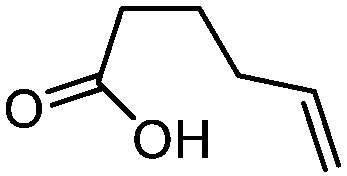 3c
3c
|
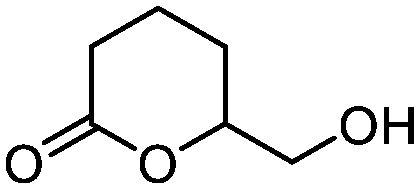 5c
5c
|
74 | n.a. |
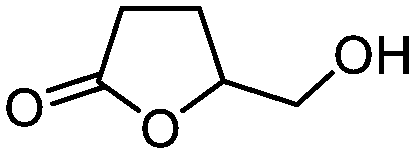
| 4 |
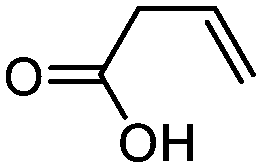 3d
3d
|
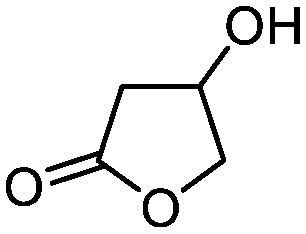 5d
5d
|
79 | n.a. |
| 5 |
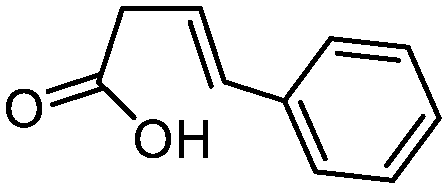 3e
3e
|
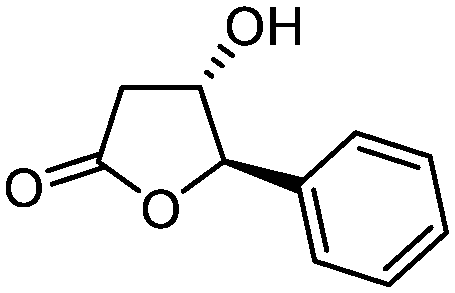 5e
5e
|
75 | 95:5 |
| 6 |
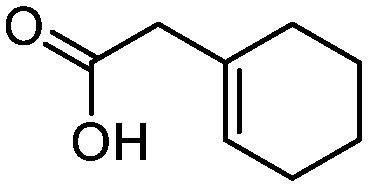 3f
3f
|
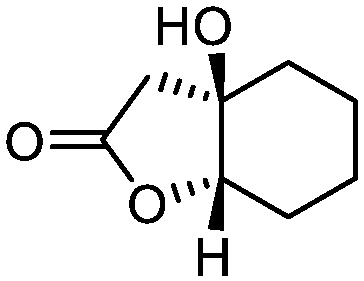 5f
5f
|
92 | >99:1 |
| 7 |
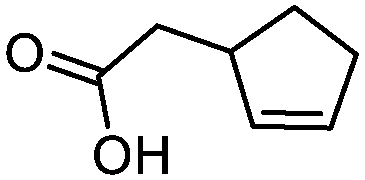 3g
3g
|
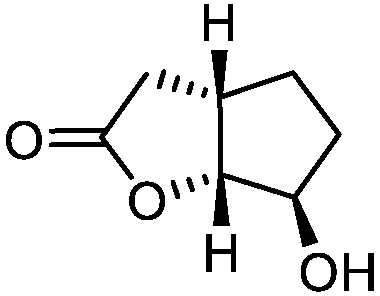 5g
5g
|
75 | >99:1 |
| 8d |
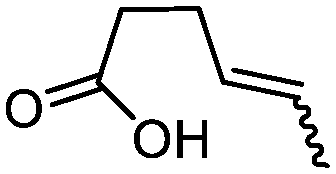 3h
3h
|
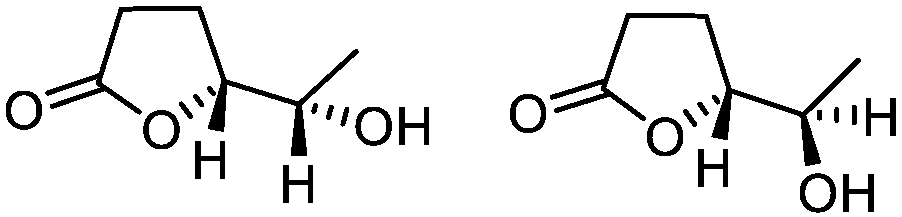 5h
5h
|
78 | 80:20d |
| 9 |
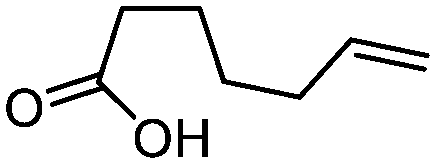 3i
3i
|
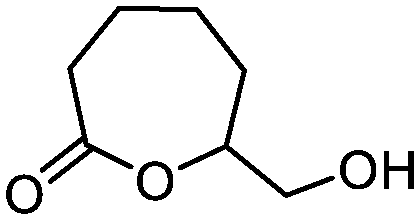 5i
5i
|
67 | n.a. |
| 10 |
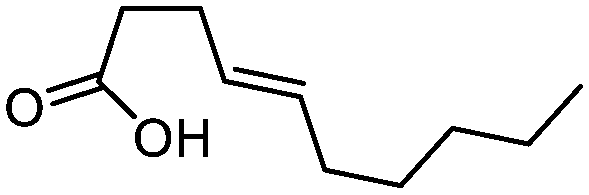 3j
3j
|
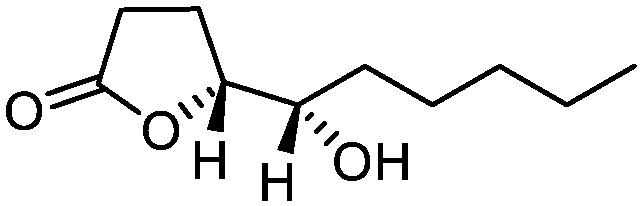 5j
5j
|
82 | >99:1 |
| 11 |
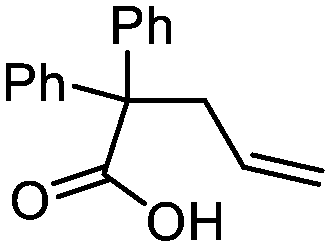 3k
3k
|
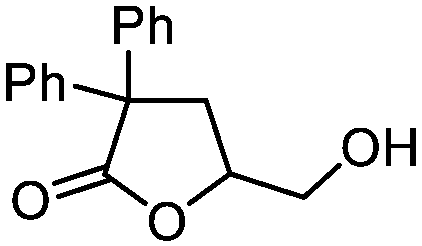 5k
5k
|
78 | n.a. |
| 12 |
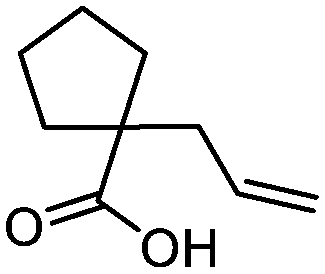 3l
3l
|
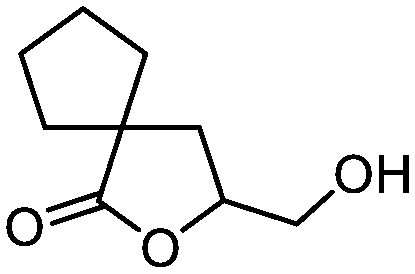 5l
5l
|
77 | n.a. |
| 13 |
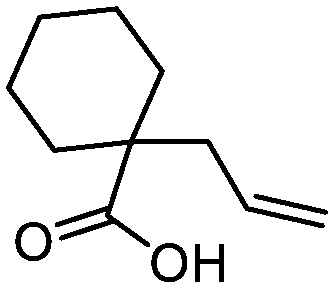 3m
3m
|
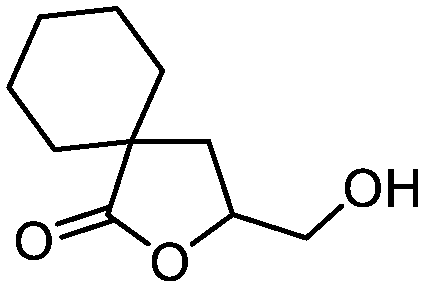 5m
5m
|
76 | n.a. |
We next evaluated the substrate scope of the reaction using different alkenoic acids 3b–m (Table 2). Reactions were performed according to the improved flow set-up depicted in Fig. 2. As a result, the method achieved efficient and versatile furnishing pure hydroxy lactones 5a–m in good to high isolated yields. Both alkyl, cycloalkyl and aromatic substituents, and more constrained substrates were well tolerated.
While the transient epoxyde intermediate is responsible for the exclusive formation of trans stereoisomers, the regiochemistry of the reaction can be explained according to previous reports.12 Indeed, substrates 3a, 3d and 3e (ref. 13) (Table 2, entries 1, 4 and 5) reacted by an anti-Baldwin like mechanism leading to 5-endo products, while 5-exo products were favored over 6-endo analogs (Table 2, entries 2, 8 and 10–13). Analogously, 6-exo ring closure was preferred over the 7-endo path (Table 2, entry 3), and 7-exo ring closure was favored over the 8-endo (Table 2, entry 9). Furthermore, endocyclic compounds 3f and 3g (Table 2, entries 6 and 7) furnished stereoselectively the corresponding cis-bicyclic lactones 5f and 5g.13 It is interesting to note that no by-products were detected (for instance, the epoxyde) in any of the examples, with water being the only by-product generated during the reaction.
The method was also easily scalable, since we were able to continuously process 22 mmol of 3-hydroxy-γ-butyrolactone (3-BHL) (5d), a useful building block for synthetic transformations and the precursor of cholesterol-reducing drugs such as Crestor™ and Lipitor™, the antibiotic Zyvox™, and the anti-hyperlipidemic Zetia™.1c A continuous flow preparation of 5d employing high pressure hydrogenation of malic acid over a ruthenium-based catalyst has been recently disclosed;14 however, this process employed hazardous conditions, expensive catalyst and purifications. Other chemical and chemo-enzymatic approaches also suffer from several limitations,15 resulting in the high cost of the product. More recently, a biosynthetic pathway to 3-BHL (5d) using acyl-CoA enzymes has been described;16 although fashionable, the efficacy of the route for large scale operations needs to be proven. In our case, we have used the integrated flow system illustrated in Fig. 2 using four reactor coils operating in parallel and an additional A21 packed column for catalyst scavenging. To avoid resin saturation, the column was washed after 3.5 h of continuous pumping (when about half of the reservoir solution of 3d was reacted). Switch to the second column then guaranteed the purification of the rest of the reaction mixture. Under these conditions, 22 mmol of 5d were continuously synthesized in high purity with a productivity of 2.45 mmol h−1 (corresponding to 6 g d−1).
Conclusions
We have developed an efficient and eco-friendly strategy for the regio- and diastereo-selective continuous synthesis and purification of hydroxy lactones. To the best of our knowledge, the present study is the only case reported and carried out that uses selenium-mediated catalysis under flow conditions. The results clearly show that our method is robust, reliable and versatile and permits the large scale preparation of important synthons and natural products such as 3-BHL (5d) and 5-hydroxy-4-decanolide (5j), a sex pheromone of the parasitic wasp genus Nasonia. In particular, 3-BHL (5d) was continuously produced (isolated yield: 74%) with high purity without the need of human intervention for work-up and purifications. Overall, this approach was superior to previously reported synthesis3a,9 and more environmentally friendly due to the intrinsic atom economy of the reaction, the use of green solvents and mild reaction conditions, it being safe and amenable for automation, the formation of water as the only by-product of the reaction, and the possibility of recovering and reusing unreacted starting materials and selenium-containing reagents. Current efforts are directed towards the implementation of this method to self-controlled multistep synthesis and bio-enzymatic transformations whose results will be reported in due course.Acknowledgements
Fondazione Cassa di Risparmio di Perugia (2014.01000.021) is gratefully acknowledged for the financial support.Notes and references
- (a) Y.-M. Ham, Y.-J. Ko, S.-M. Song, J. Kim, K.-N. Kim, J.-H. Yun, J.-H. Cho, G. Ahn and W.-J. Yoon, J. Funct. Foods, 2015, 13, 80 CrossRef CAS; (b) N. P. Lopes, M. J. Kato and M. Yoshida, Phytochemistry, 1999, 51, 29 CrossRef CAS; (c) M. K. Wikström and J. A. Berden, Biochim. Biophys. Acta, 1972, 283, 403 CrossRef; (d) S.-H. Lee and O.-J. Park, Appl. Microbiol. Biotechnol., 2009, 84, 817 CrossRef CAS PubMed.
- (a) M. Seitz and O. Reiser, Curr. Opin. Chem. Biol., 2005, 9, 285 CrossRef CAS PubMed; (b) S. Gil, M. Parra, P. Rodriguez and J. Segura, Mini-Rev. Org. Chem., 2009, 6, 345 CrossRef CAS.
- (a) C. Le Floch, E. Le Gall, E. Léonel, T. Martens and T. Cresteil, Bioorg. Med. Chem. Lett., 2011, 21, 7054 CrossRef CAS PubMed; (b) M. Mondal, H. J. Ho, N. J. Peraino, M. A. Gary, K. A. Wheeler and N. J. Kerrigan, J. Org. Chem., 2013, 78, 4587 CrossRef CAS PubMed; (c) C. Shu, M.-Q. Liu, Y.-Z. Sun and L.-W. Ye, Org. Lett., 2012, 14, 4958 CrossRef CAS PubMed; (d) E. Howard and K. A. Woerpel, Tetrahedron, 2009, 65, 6447 CrossRef PubMed; (e) H. S. Park, D. W. Kwon, K. Lee and Y. H. Kim, Tetrahedron Lett., 2008, 49, 1616 CrossRef CAS; (f) G. Kokotos, Org. Lett., 2013, 15, 2406 CrossRef PubMed; (g) O. Anaç, F. S. Güngör and G. Merey, Helv. Chim. Acta, 2006, 89, 1231 CrossRef; (h) O. V. Turova, E. V. Starodubtseva, M. G. Vinogradov, V. A. Ferapontov and M. I. Struchkova, Tetrahedron: Asymmetry, 2009, 20, 2121 CrossRef CAS; (i) A. Comini, C. Forzato, P. Nitti, G. Pitacco and E. Valentin, Tetrahedron: Asymmetry, 2004, 15, 617 CrossRef CAS.
- For recent reviews see: (a) L. Vaccaro, D. Lanari, A. Marrocchi and G. Strappaveccia, Green Chem., 2014, 16, 3680 RSC; (b) C. Wiles and P. Watts, Green Chem., 2014, 16, 55 RSC; (c) S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456 RSC.
- For recent reviews see: (a) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.7b00183 , in press; (b) B. J. Reizman and K. F. Jensen, Acc. Chem. Res., 2016, 49, 1786 CrossRef CAS PubMed; (c) A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J. C. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61 CrossRef CAS PubMed; (d) H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luqueb and T. Noël, Chem. Soc. Rev., 2016, 45, 83 RSC; (e) A. Gioiello, V. Mancino, P. Filipponi, S. Mostarda and B. Cerra, J. Flow Chem., 2016, 6, 167 CrossRef; (f) I. M. Mándity, S. B. Ötvös, G. Szőlősi and F. Fülöp, Chem. Rec., 2016, 16, 1018 CrossRef PubMed; (g) I. M. Mandity, S. B. Oetvoes and F. Fueloep, ChemistryOpen, 2015, 4, 212 CrossRef CAS PubMed; (h) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688 CrossRef CAS PubMed; (i) R. J. Ingham, C. Battilocchio, D. E. Fitzpatrick, E. Sliwinski, J. M. Hawkins and S. V. Ley, Angew. Chem., Int. Ed., 2015, 54, 144 CrossRef CAS PubMed; (j) M. Baumann and I. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194 CrossRef CAS PubMed; (k) Y. Su, N. J. W. Straathof, V. Hessel and T. Noel, Chem. – Eur. J., 2014, 20, 10562 CrossRef CAS PubMed; (l) J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849 RSC.
- J. H. Park, C. Y. Park, H. S. Song, Y. S. Huh, G. H. Kim and C. P. Park, Org. Lett., 2013, 15, 752 CrossRef CAS PubMed.
- M. Fagnoni, F. Bonassi, A. Palmieri, S. Protti, D. Ravelli and R. Ballini, Adv. Synth. Catal., 2014, 356, 753 CrossRef CAS.
- C. M. Gatley, L. M. Muller, M. A. Lang, E. E. Alberto and M. R. Detty, Molecules, 2015, 20, 9616 CrossRef CAS PubMed.
- L. Sancineto, F. Mangiavacchi, C. Tidei, L. Bagnoli, F. Marini, A. Gioiello, J. Scianowski and C. Santi, Asian J. Org. Chem., 2017 DOI:10.1002/ajoc.201700193 , in press.
- Other acids including trifluoroacetic acid, solid and supported p-toluensulfonic acid gave similar results. Hydrochloric acid and acetic acid were less effective and formed by-products.
- When conducted in batch the conversion of 3a to 5a was achieved with a yield of 90% in 8 h using 4 equiv. of H2O2 and 5% mol of diphenyl diselenide (PhSe)2 at room temperature.9 On the gram scale a higher amount of (PhSe)2 or a longer reaction time is however needed to complete the reaction. Moreover, reactions conducted in batch mode requires tedious chromatographic purifications and catalyst recovery is not easily achievable.
- H. Tan and J. H. Expenson, J. Mol. Catal. A: Chem., 1999, 142, 333 CrossRef CAS.
- When prepared in batch mode, 5e and 5g are formed in 85% yield with a lower diastereoselectivity ratio (85:15).9.
- (a) A. M. Rouhi, Chem. Eng. News, 2003, 81, 55 CrossRef; (b) B. S. Kwak, Chim. Oggi, 2003, 21, 23 CAS.
- (a) P. Kumar, A. N. Deshmukh, R. K. Upadhyay and M. K. Gurjar, Tetrahedron: Asymmetry, 2005, 16, 2717 CrossRef CAS; (b) R. I. Hollingsworth, J. Org. Chem., 1999, 64, 7633 CrossRef CAS; (c) A. Nakagawa, H. Idogaki, K. Kato, A. Shinmyo and T. Suzuki, J. Biosci. Bioeng., 2006, 101, 97 CrossRef CAS PubMed; (d) T. Suzuki, H. Idogaki and N. Kasai, Enzyme Microb. Technol., 1999, 24, 13 CrossRef CAS; (e) S. H. Lee, O. J. Park and H. S. Uh, Appl. Microbiol. Biotechnol., 2008, 79, 355 CrossRef CAS PubMed.
- C. H. Martin, H. Dhamankar, H.-C. Tseng, M. J. Sheppard, C. R. Reisch and K. L. J. Prather, Nat. Commun., 2013, 4, 1 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00083a |
| This journal is © The Royal Society of Chemistry 2017 |