
Synthesis and molecular weight control of poly(3-hexylthiophene) using electrochemical polymerization in a flow microreactor†
Masatsugu
Mizuno
a,
Hiroyuki
Tateno
a,
Yoshimasa
Matsumura
b and
Mahito
Atobe
 *a
*a
aDepartment of Environment and System Sciences, Yokohama National University, 79-7 Tokiwadai, Hodogaya-ku, Yokohama, Kanagawa 240-8501, Japan. E-mail: atobe@ynu.jp; Fax: +81 45 339 4214; Tel: +81 45 339 4214
bDepartment of Chemistry and Chemical Engineering, Faculty of Engineering, Yamagata University, 4-3-16 Jonan, Yonezawa, Yamagata 992-8510, Japan
First published on 26th July 2017
Abstract
A new approach for the synthesis and molecular weight control of poly(3-hexylthiophene) (P3HT) using electrochemical polymerization in a flow microreactor is described. This synthetic system enabled electrochemical synthesis of soluble P3HT without its deposition using a flow microreactor. Careful selection of the reaction conditions enabled the control of molecular weight and distribution of the synthesized P3HT.
π-Conjugated polymers are used as functional materials in opto-electronic devices due to their lightness, flexibility, and high conductivity.1,2 Molecular weight is closely related to the optical and electronic properties of π-conjugated polymers.3–6 However, controlling the molecular weight of conducting polymers can be difficult due to their insolubility in many solvents. As soluble and processable π-conjugated polymers, poly(3-alkylthiophenes) were first reported by Elsenbaumer in 1986.7 Poly(3-hexylthiophene) (P3HT) is one of the most promising materials for photoelectronics, such as organic solar cells, organic field-effect transistors, and organic EL devices, because P3HT has many useful properties including wide solubility, processability, charge mobility, and environmental stability.8–10 Conventional synthetic methods for molecular-weight-controllable P3HT are based on cross-coupling polycondensation using transition metal catalysts.11,12 However, these synthetic methods have several disadvantages, such as multi-step requirements, long reaction times, difficulty in developing continuous large-scale processes, and use of toxic transition metal catalysts. Therefore, a simple method that can be scaled up and does not require a toxic catalyst is an attractive goal.
Electro-oxidative polymerization of aromatic compounds is useful for preparing the corresponding π-conjugated polymers via anodic oxidative coupling.13 In addition, this polymerization can be conducted without toxic oxidants or catalysts, can be operated under mild conditions, and can be controlled by switching on and off. In the conventional method, electro-oxidative polymerization is conducted in a non-stirred solution using a batch-type reactor. By this method, π-conjugated polymers are usually obtained on the anode surface as a film even in soluble and processable π-conjugated polymer synthesis, because the electron transfer of the monomer and its coupling reaction proceeds near the anode surface and insoluble polymeric products are deposited on it. Therefore, the conventional electro-oxidative polymerization method is suitable for polymer film synthesis, but unsuitable for soluble polymer synthesis and molecular weight control.
To overcome this problem, an electrochemical flow microreactor (illustrated in Fig. 1) was developed for the electrochemical synthesis and molecular weight control of a soluble π-conjugated polymer such as P3HT. This reactor had a simple geometry with working and auxiliary electrodes directly facing each other. Flow microreactors have several advantages such as an extremely high surface-to-volume ratio, short molecular diffusion distance, and precise residence time control in the reactor.14 The precise control of residence time in the reactor, in particular, would enable control of the molecular weight of P3HT. In addition, this flow operation could avoid deposition of P3HT on anode surfaces and allow continuous synthesis of a soluble π-conjugated polymer such as P3HT. On the other hand, the cathodic process in the electrochemical synthesis would be a reduction of protons formed in the anodic process. Hence, the cathodic process would not directly affect the anodic process. Thus, this report describes the work that led to a proof of concept of this process.
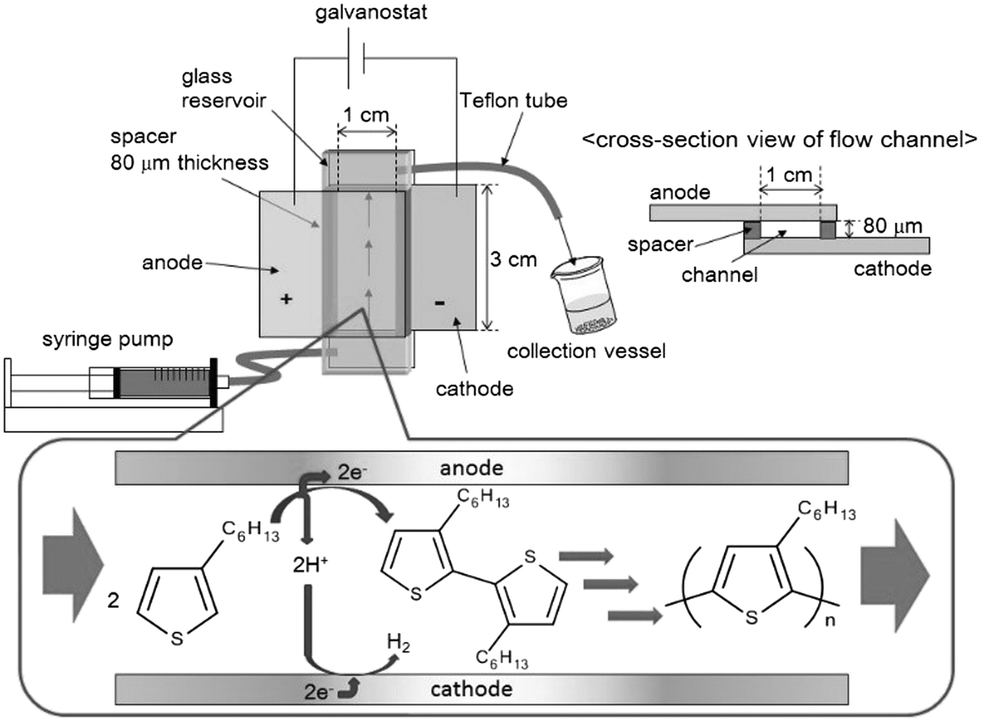 | ||
| Fig. 1 Schematic representation of the electrochemical flow microreactor. | ||
First, the electro-oxidative polymerization of 3-hexylthiophene was conducted using a conventional batch-type reactor and a flow microreactor under the same reaction conditions. As shown in Table 1, monomer conversion was improved by using the flow microreactor, attributed to the better mixing in the flow microreactor. Therefore, the electrode reaction of the monomer in the flow microreactor proceeded more efficiently than that in the batch reactor. In addition, the use of the flow microreactor resulted in a narrower molecular weight distribution. This may be ascribed to the high volume-to-surface ratio of the reactor causing the absence of hot spots and thus better defined polymer products. On the other hand, an insoluble polymer film also was formed on the ITO anode surface after electrochemical polymerization using a batch-reactor (Fig. 2). In contrast, no polymer deposition was found on the anode surface after polymerization using the flow microreactor. These results confirm that the flow microreactor is applicable for electro-oxidative polymerization of soluble polymers with a narrow molecular weight distribution.
| Reactor type | Monomer conversionc (%) | M n | M w | M w/Mnd |
|---|---|---|---|---|
| a Experimental conditions: anode, ITO plate; cathode, stainless plate; current density, 5.0 mA cm−2; electricity, 2.0 F mol−1; solvent, CH3CN; substrate; 10 mM of 3-hexylthiophene; supporting electrolyte, 50 mM of Bu4NClO4. b Electrode distance, 80 μm; flow rate, 0.47 mL min−1. c Determined by HPLC. d Determined by GPC. | ||||
| Batch reactor | 13 | 610 | 2100 | 3.5 |
| Microreactorb | 53 | 550 | 900 | 1.7 |
 | ||
| Fig. 2 Photographic images of the ITO anode surface after electrosynthesis using a (a) batch reactor and a (b) flow microreactor. | ||
During the next stage of investigations, the effect of the reaction conditions on the molecular weight of P3HT synthesized in the flow microreactor was investigated. The anode material is an important factor in controlling the efficiency of the electro-oxidative polymerization. Therefore, to select suitable anode materials for the system, electro-oxidative polymerization of 3-hexylthiophene was conducted using various anode materials such as graphite, ITO, platinum, and glassy carbon. As shown in Table 2, the use of the graphite electrode led to higher monomer conversion and narrower polydispersity compared to the other anode materials. Therefore, the graphite electrode was selected as the anode for this reaction system and used in the following experiments.
| Anode material | Monomer conversionb (%) | M n | M w | M w/Mnc |
|---|---|---|---|---|
| a Experimental conditions: anode, graphite, ITO, Pt, or GC plates; cathode, stainless plate; current density, 5.0 mA cm−2; flow rate, 0.47 mL min−1; electricity, 2.0 F mol−1; solvent, CH2Cl2; substrate; 10 mM of 3-hexylthiophene; supporting electrolyte, 50 mM of Bu4NClO4. b Determined by HPLC. c Determined by GPC. | ||||
| Graphite | 61 | 800 | 1100 | 1.4 |
| ITO | 50 | 600 | 1100 | 1.8 |
| Pt | 51 | 540 | 840 | 1.6 |
| GC | 38 | 460 | 770 | 1.7 |
Next, electro-oxidative polymerization was conducted in various electrolytic solvents to select the optimal electrolytic solvent for this synthetic system (Table 3). Monomer conversion with dimethyl sulfoxide was quite low because the oxidation of dimethyl sulfoxide proceeded preferentially due to its low oxidation potential. In contrast, monomer conversion was higher for other solvent systems. However, the use of acetonitrile or nitromethane resulted in insoluble polymers in the reaction mixture. During the operation of continuous reactions using the flow microreactor, no precipitation is desirable, making dichloromethane the best medium for this synthetic system.
| Electrolytic solvent | Monomer conversionb (%) | M n | M w | M w/Mnc |
|---|---|---|---|---|
| a Experimental conditions: anode, graphite plate; cathode, stainless plate; current density, 5.0 mA cm−2; flow rate, 0.47 mL min−1; electricity, 2.0 F mol−1; substrate; 10 mM of 3-hexylthiophene; supporting electrolyte, 50 mM of Bu4NClO4. b Determined by HPLC. c Determined by GPC. | ||||
| DMSO | 8 | 340 | 430 | 1.3 |
| CH3CN | 68 | 950 | 2500 | 2.6 |
| CH3NO2 | 60 | 920 | 2100 | 2.3 |
| CH2Cl2 | 61 | 800 | 1100 | 1.4 |
Supporting electrolytes play an important role in the stability of reaction intermediates in electrochemical syntheses.15,16 Therefore, electrochemical polymerization was preformed using various supporting electrolytes. As shown in Table 4, the molecular weight of the products was strongly influenced by the type of supporting electrolyte used, and the product molecular weight increased with a decrease in the donor number of the anion portion of the electrolyte used in the polymerization. This was attributed to the rate of the coupling reaction between radical-cation intermediates generated from monomers and oligomers accelerated by using a smaller donor number anion because the smaller the donor number of the anion, the fewer the ion-pair interactions with the radical-cation intermediates.
| Supporting electrolyte | Donor numberb | M n | M w | M w/Mnc |
|---|---|---|---|---|
| a Experimental conditions: anode, graphite plate; cathode, stainless plate; current density, 5.0 mA cm−2; flow rate, 0.47 mL min−1; electricity, 2.0 F mol−1; solvent, CH2Cl2; substrate; 10 mM of 3-hexylthiophene; supporting electrolyte conc., 50 mM. b Literature values cited in ref. 17. c Determined by GPC. | ||||
| Bu4NOTf | 16.90 | 500 | 790 | 1.6 |
| Bu4NClO4 | 8.44 | 800 | 1100 | 1.4 |
| Bu4NBF4 | 6.03 | 1200 | 2800 | 2.3 |
| Bu4NPF6 | 2.50 | 1900 | 4700 | 2.4 |
Finally, to precisely control the molecular weight of P3HT, the influence of electricity on the average molecular weight and polydispersity of P3HT in the electrochemical polymerization was investigated. For constant-current electrochemical polymerization in a flow microreactor with fixed channel dimensions, electricity can be controlled by changing the flow rate. As shown in this Table 5, the molecular weight of polymer products increased with electricity (caused by a decrease in the flow rate). These results confirmed that the molecular weight of P3HT could be controlled simply by changing the electricity.
| Electricity/F mol−1 | Flow rate/mL min−1 | M n | M w | M w/Mnb |
|---|---|---|---|---|
| a Experimental conditions: anode, graphite plate; cathode, stainless plate; current density, 5.0 mA cm−2; solvent, CH2Cl2; substrate; 10 mM of 3-hexylthiophene; supporting electrolyte, 50 mM of Bu4NPF6. b Determined by GPC. | ||||
| 0.5 | 1.88 | 2100 | 4100 | 1.9 |
| 1.0 | 0.94 | 2500 | 5000 | 2.0 |
| 2.0 | 0.47 | 3100 | 7600 | 2.4 |
Conclusions
Electrochemical synthesis and molecular weight control of P3HT were demonstrated using a flow microreactor. The electrochemical flow microreactor allowed effective synthesis of a soluble π-conjugated polymer such as P3HT without polymer deposition. The molecular weight distribution of P3HT synthesized in the microreactor was much narrower than that produced in a conventional batch reactor. In addition, the molecular weight of P3HT could be controlled by selecting the reaction conditions for the electrochemical polymerization. This method has the major advantage of not requiring sensitive, expensive, or toxic reagents. In addition, the reaction could be conducted using single flow-through operations under very mild conditions. These promising results provide a method for efficient and environmentally benign π-conjugated polymer syntheses.This work was financially supported by a Grant-in-Aid for Scientific Research on Innovative Areas (No. 2707: Middle Molecular Strategy).
References
- K. Kaneto, K. Yoshino and Y. Inuishi, Jpn. J. Appl. Phys., 1982, 21, L567 CrossRef.
- G. E. Wnek, J. C. W. Chien, F. E. Karasz and C. P. Lillya, Polymer, 1979, 20, 1441 CrossRef CAS.
- K. Meerholz and J. Heinze, Synth. Met., 1993, 57, 5040 CrossRef CAS.
- S. V. Meille, V. Romita and T. Caronna, Macromolecules, 1997, 30, 7898 CrossRef CAS.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu and J. M. J. Frechet, Adv. Mater., 2003, 15, 1519 CrossRef CAS.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu, J. M. J. Frechet and M. F. Toney, Macromolecules, 2005, 38, 3312 CrossRef CAS.
- K.-Y. Jen, G. G. Miller and R. L. Elsenbaumer, J. Chem. Soc., Chem. Commun., 1986, 1346 RSC.
- R. D. McCullough, Adv. Mater., 1998, 10, 93 CrossRef CAS.
- A. Zen, J. Pflaum, S. Hirschmann, W. Zhuang, F. Jaiser, U. Asawapirom, J. P. Rabe, U. Scherf and D. Neher, Adv. Funct. Mater., 2004, 14, 757 CrossRef CAS.
- P. Schilinsky, U. Asawapirom, U. Scherf, M. Biele and C. J. Brabec, Chem. Mater., 2005, 17, 2175 CrossRef CAS.
- M. C. Iovu, E. E. Sheina, R. R. Gil and R. D. McCullough, Macromolecules, 2005, 38, 8649 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 17542 CrossRef CAS PubMed.
- R. J. Waltman and J. Bargon, Can. J. Chem., 1986, 64, 76 CrossRef CAS.
- B. P. Mason, K. E. Price, J. L. Steinbacher, A. R. Bogdan and D. T. McQuade, Chem. Rev., 2007, 107, 2300 CrossRef CAS PubMed.
- R. Yamaguchi, B. Hatano, T. Nakayasu and S. Kozima, Tetrahedron Lett., 1997, 38, 403 CrossRef CAS.
- J. Yoshida, S. Suga, S. Suzuki, N. Kinomura, A. Yamamoto and K. Fujiwara, J. Am. Chem. Soc., 1999, 121, 9546 CrossRef CAS.
- W. Linert, A. Camard, M. Armand and C. Michot, Coord. Chem. Rev., 2002, 226, 137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00089h |
| This journal is © The Royal Society of Chemistry 2017 |