
Towards a sustainable electrochemical activation for recycling CO2: synthesis of bis-O-alkylcarbamates from aliphatic and benzyl diamines†
Gianpiero
Forte
 *a,
Isabella
Chiarotto
a,
Frank
Richter
b,
Vinh
Trieu
b and
Marta
Feroci
*a
*a,
Isabella
Chiarotto
a,
Frank
Richter
b,
Vinh
Trieu
b and
Marta
Feroci
*a
aDipartimento di Scienze di Base e Applicate per l'Ingegneria, Sapienza Università di Roma, Via del Castro Laurenziano 7, 00161 Roma, Italy. E-mail: gianpiero.forte@uniroma1.it; marta.feroci@uniroma1.it
bCovestro Deutschland AG, Kaiser-Wilhelm-Allee 60, 51365 Leverkusen, Germany
First published on 29th August 2017
Abstract
Carboxylation of aliphatic and benzyl diamines with electrochemically activated CO2 led to the synthesis of bis-O-alkyl carbamates in high yields. Reaction conditions, including the geometry of the electrochemical cell, were discussed and optimized. This resulted in a significant reduction of unwanted material associated with the reaction, overcoming the economic and environmental restrictions to its application on large scale.
The compelling need to cut anthropogenic emissions of greenhouse gases,1,2 as part of the endeavor to stop the global warming phenomenon,3,4 has prompted a great deal of effort to reduce the release of carbon dioxide from industrial plants.5,6
In this perspective, the possibility to recycle CO2 from exhaust means not only complying with this requirement, but also accessing a cheap, non-toxic, and readily available carbon feedstock for organic synthesis – finding a replacement for finite fossil resources such as oil and gas, and closing material cycles.7–9 The inert nature of carbon dioxide, however, makes the task particularly challenging, and reactions with CO2 generally require high pressure or metal catalysis.10
Carboxylation of amines (Scheme 1), instead, proceeds spontaneously to form carbamates, and their corresponding esters may serve as intermediates in the industrial manufacture of isocyanates.11
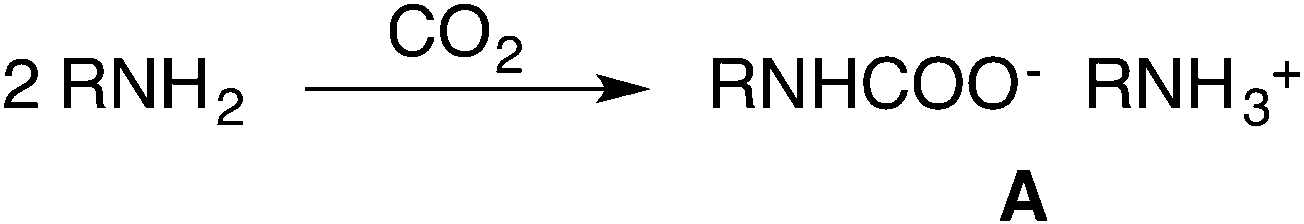 | ||
| Scheme 1 Carboxylation of amines. | ||
Therefore it is not surprising that the synthesis of carbamate esters has been considered an extremely appealing approach to tackle CO2 recycling.12
However, unless additional bases are present, the alkylation of the ammonium carbamate salt A preferably yields N-substituted products.13
In a previous work we demonstrated that,14–18 when reacted with electrochemically activated CO2 and then with an alkylating agent, amines afford carbamate esters in excellent yields without the use of external bases. The formation of N-alkylated products is negligible under these conditions.
Di- and polyisocyanates play an important role in industrial synthesis of polymers, and the lack of literature reports about the carboxylation of diamines is a gap that needs to be filled.
In this communication we assess the formation of bis-carbamate esters from diamines with different structural features (Chart 1), whose corresponding isocyanates are intermediates in the production of polyurethane resins currently on the market. We also report the optimization of the reaction conditions in the perspective of a sustainable application on large scale. Most importantly, we describe a more convenient design of the electrochemical cell, which greatly reduces the amount of waste associated with the activation of carbon dioxide.
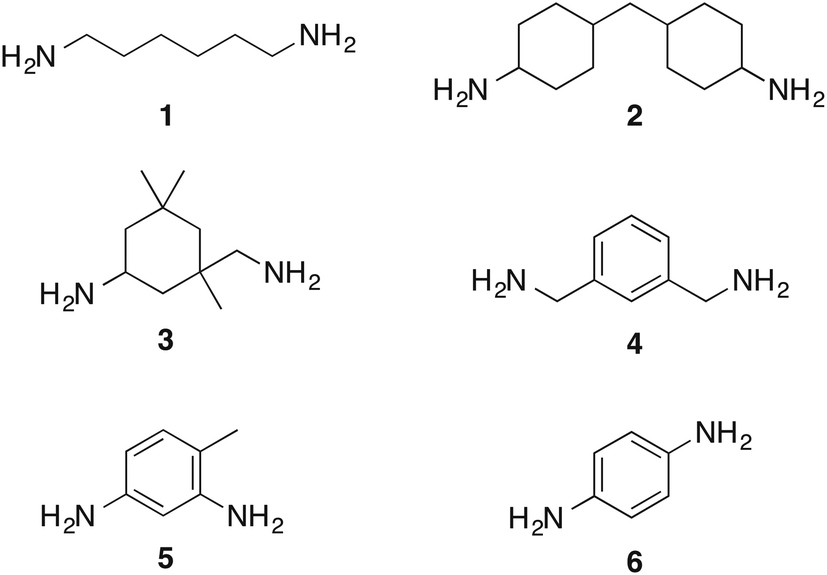 | ||
| Chart 1 Diamines tested in the present study. | ||
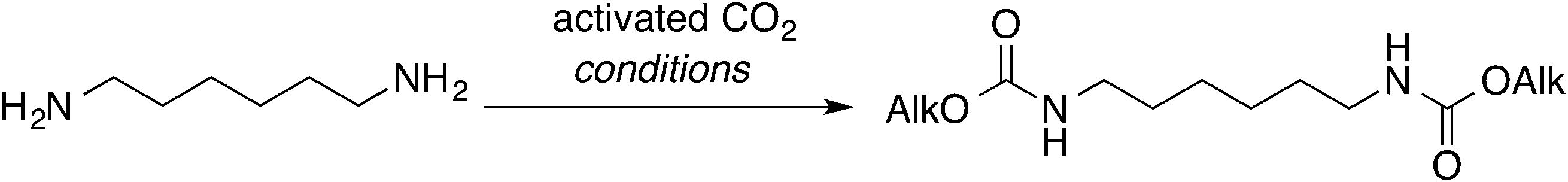
To ascertain whether aliphatic diamines can be effectively carboxylated under the same conditions that we reported for monoamines,14 diamine 1 was chosen as model compound. Cathodic activation of CO2 was achieved on a copper electrode in a two-compartment cell separated by a conductive methylcellulose gel (see ESI†), prior to the addition of 1. Following treatment with ethyl iodide at room temperature, under inert atmosphere, afforded the corresponding bis-carbamate ester 7 in over 90% yield (Scheme 2 and Table 1, entry 1). No trace of partially reacted diamine (i.e. monocarbamate ethyl ester) was observed, the supporting electrolyte being the major impurity in the crude reaction mixture.
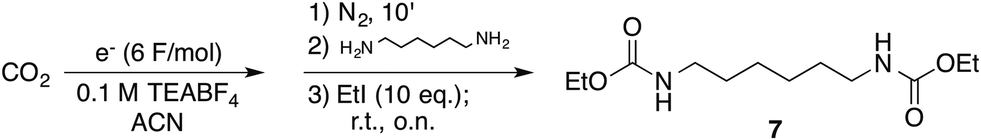 | ||
| Scheme 2 Carboxylation of diamine 1. Carbon dioxide reduced under galvanostatic conditions on a copper cathode in a divided cell (see ESI† for further details). TEABF4: tetraethylammonium tetrafluoroborate. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cell type | Alkylating agent (eq.) | T [°C] | Supporting electrolyte | Bis-carbamate esterb [%] |
| D: divided; U: undivided; H: H-shaped cell. TEABF4: tetraethylammonium tetrafluoroborate. a Hexamethylenediamine, 1 (0.5 mmol), used as model compound for the optimization of the reaction conditions. Copper foil as cathode, glassy carbon bar as anode. CO2 activated in a 0.1 M solution of the supporting electrolyte in ACN. After the consumption of 6 F per mole of diamine, the electrodes were removed and the catholyte treated overnight with the alkylating agent. See ESI for further details. b Isolated yields. c Pt spiral as anode. d 1–5 mol% of TBAI. e 3 hours. f 85% yield on 1.0 mmol scale reaction. | |||||
| 1 | Dc | EtI (10) | 25 | TEABF4 | >90 |
| 2 | U | EtI (10) | 25 | TEABF4 | n.d. |
| 3 | H | EtI (10) | 25 | TEABF4 | >90 |
| 4 | H | EtI (10) | 25 | TEAC | >90 |
| 5 | H | BuI (10) | 25 | TEAC | 82 |
| 6 | H | BuCl (10) | 25 | TEAC | <10 |
| 7 | H | BuCl (10)d | 25 | TEAC | 21% |
| 8 | H | BuCl (10) | 25 | TEAI | 40 |
| 9 | H | BuCl (10) | 50 | TEAC | 64 |
| 10 | H | BuCl (10) | 80e | TEAC | 81 |
| 11 | H | BuCl (6) | 80e | TEAC | 85 |
| 12 | H | BuCl (4) | 80e | TEAC | 82f |
Despite the cleanliness of the reaction, some aspects of this approach set environmental and economic limits to its potential application on industrial scale and needed to be addressed.
Beside the quantity of dimethylformamide and the high concentration of supporting electrolyte necessary for the preparation of methylcellulose gel, its limited lifetime requires frequent replacement.20 This large amount of waste generated may undermine the environmental advantage of recycling CO2.
As much as an undivided cell geometry is desirable, the carboxylation of 1 in such conditions affords only traces of 7 after treatment with ethyl iodide. The alkylated diamine is recovered as major product at the end of the reaction (see ESI† and Table 1, entry 2).
Drastic improvements are achieved with a different cell design, where the two compartments are separated by a simple glass frit without using methylcellulose gel. The electrochemical cell consists of a H-shaped glass vessel fitted with a copper foil as cathode and a glassy carbon bar as anode (Fig. 1 and ESI†). Carbon dioxide was constantly bubbled at the cathode and reduced under galvanostatic conditions until 6 Faradays per mole of diamine were consumed.21 After completion of the electrolysis, the catholyte was transfered into the reaction flask containing 1 without particular precautions, proving that handling under inert atmosphere is not required. Final treatment with ethyl iodide afforded the bis-carbamate ester 7 in very high yields, comparable with those obtained with the previous cell geometry (Table 1, entries 3 and 4).
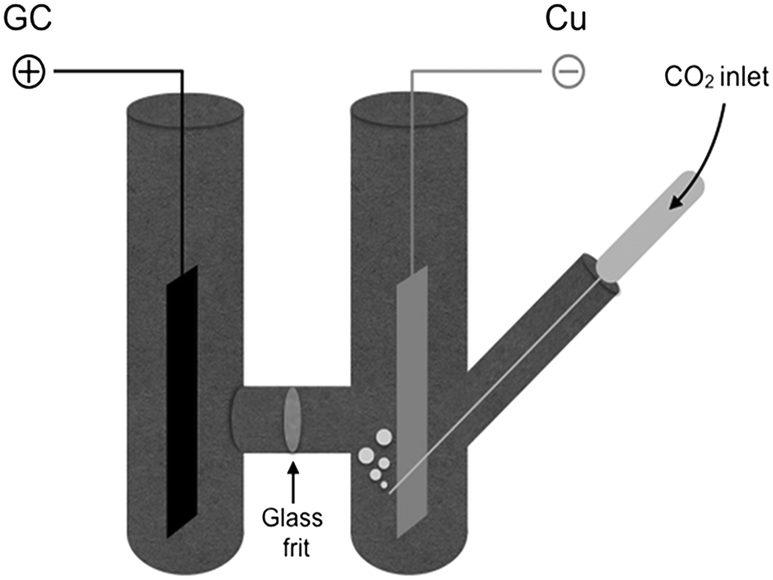 | ||
| Fig. 1 H-cell configuration. The compartments are separated by a glass frit #3. The increase of concentration due to CO2 reduction and anodic oxidation of TEAC causes solvent to migrate towards the catholyte and the anolyte has to be supplemented periodically during the electrolysis. | ||
In the carbon dioxide activation process, the anodic reaction at the expense of the supporting electrolyte must also be considered. Tetraethylammonium tetrafluoroborate (TEABF4) was replaced here by the corresponding chloride (TEAC), whose oxidation leads to the formation of chlorine, less hazardous than BF3 and fluorine generated from TEABF4 in the same process.22
Despite its good reactivity, ethyl iodide is an expensive and potentially harmful reagent which needs to be replaced for future applications on large scale. In this perspective chlorides are preferred to iodides, and their use regenerates TEAC after the alkylation step. On the other hand, ethyl chloride is a gas at room temperature and its use is just as undesirable.
In the production of isocyanates, the thermal splitting of ethyl carbamates affords ethanol as byproduct. Almost all the industrially relevant processes, however, rely on the cleavage of n-butyl carbamate, as the alcohol evolved is much better separable from the isocyanate formed. Accordingly, we investigated the possibility to use butyl chloride as alkylating agent.23 Carboxylation of 1, followed by overnight treatment with butyl chloride at room temperature, afforded the corresponding bis-O-butylcarbamate in less than 10% yield (Table 1, entry 6). No significant improvements were observed by further increasing the reaction time.
When we carried out the alkylation step in the presence of catalytic amount of tetrabutylammonium iodide (TBAI, 1–5 mol%; Table 1, entry 7; see ESI†) the yield of the product increased to 21%, while 40% yield was obtained using tetraethylammonium iodide (TEAI) as supporting electrolyte (Table 1, entry 8; see ESI†).
Excellent results, instead, are achieved by increasing the temperature during the alkylation step. Heating the crude reaction mixture at 80 °C for 3 hours in the presence of butyl chloride afforded the bis-carbamate ester 8 in 82% yield after purification. Attempts to lower the temperature below 80 °C resulted in a substantial drop in the product yield (Table 1, entries 9 and 10).
Thermal treatment also allowed to decrease the amount of alkylating agent used in the reaction. We found that the equivalents of butyl chloride can be reduced from 10 down to 4 per diamine (i.e. 2 equivalents per amino group) without negatively affecting the outcome of the reaction (Table 1, entries 11 and 12).
As previously mentioned, the major impurity in the crude reaction mixture is the supporting electrolyte. To contain the costs of the process and to further reduce the amount of waste, we recovered the residual salt after the workup. Its use in subsequent reactions did not cause any yield loss.
Under these optimized experimental conditions, the reactivity of diamines with different structures was tested. Aliphatic diamines 2 and 3, and benzyl diamine 4 react smoothly and are converted into their corresponding bis-O-butylcarbamate esters in yields higher than 80% (Table 2). To show that the use of butyl chloride at high temperature is comparable to more reactive alkylating agents, we also report in Table 2 the results of the reactions carried out with butyl iodide at room temperature.
|
|
||||
|---|---|---|---|---|
| Diamine | Product | Bis-carbamate esterb [%] | ||
| BuClc | BuId | EtIe | ||
| a Reduction of CO2 on a copper cathode (glassy carbon as anode) in H-cell configuration until consumption of 6 F per mole of diamine. b Isolated yields. c 80 °C, 3 h. d Room temperature, over night. e 10 equivalents at room temperature, over night. | ||||
| 1 |
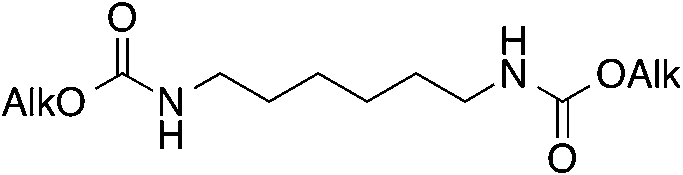 7–8
7–8
|
82 | 82 | >90 |
| 2 |
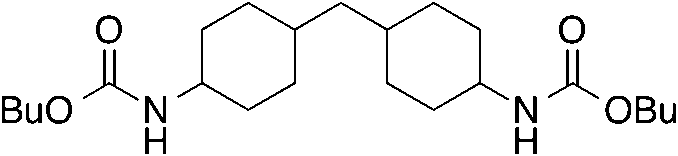 9
9
|
85 | 83 | |
| 3 |
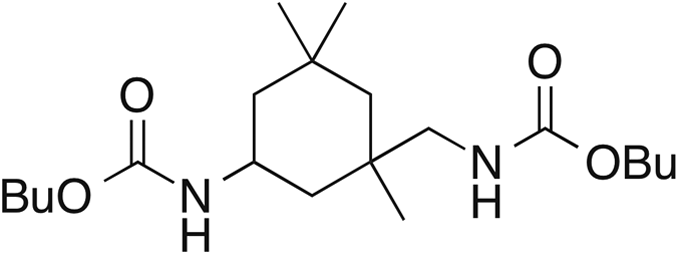 10
10
|
87 | 92 | |
| 4 |
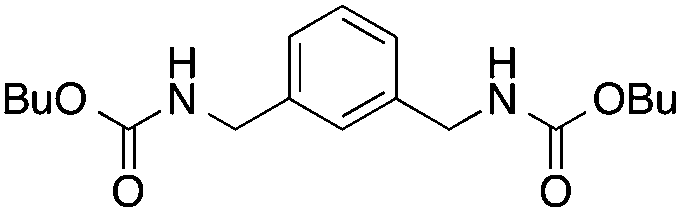 11
11
|
82 | 85 | |
| 5 |
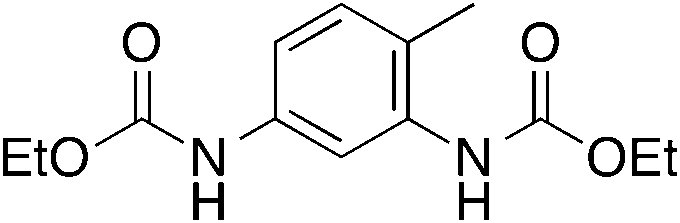 12
12
|
n.d. | 10 | |
| 6 |
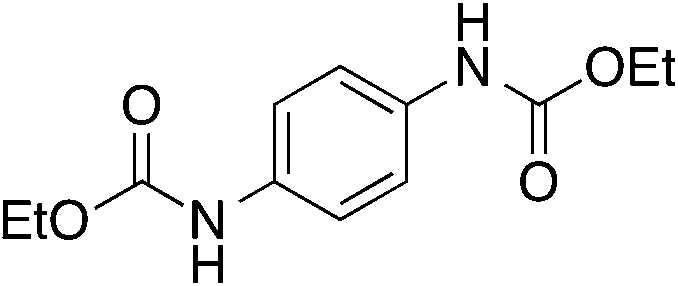 13
13
|
n.d. | 13 | |
Aromatic diamines 5 and 6 are not carboxylated under analogous conditions and mostly unreacted starting material is present in the crude mixture at the end of the reaction. Their bis-carbamate esters 12 and 13 can be isolated in 10% and 13% yield, respectively, only using ethyl iodide as alkylating agent. This highlights how the poor nucleophilicity of these aromatic diamines prevents the formation of corresponding carbamate anions in the first place and is responsible for their lack of reactivity.
Such reduced reactivity of aromatic amines can be exploited for selective carboxylation reactions. Preliminary experiments on the carboxylation of benzylamine in the presence of p-toluidine prove that the butyl carbamate of the former can be obtained in over 60% yield and only traces of the latter (<2%) is converted in the reaction (Scheme 3).
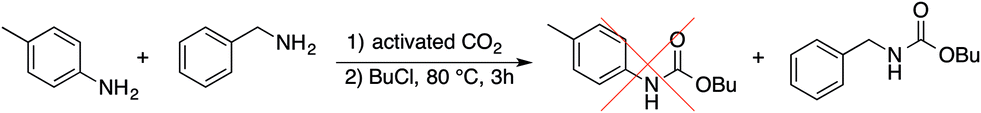 | ||
| Scheme 3 Selective carboxylation of benzylamine in the presence of p-toluidine. | ||
Further experiments are ongoing in our laboratory to selectively carboxylate a single amino group on difunctional substrates, relying on different reactivities. Results will be reported in due course.
Conclusions
In conclusion, aliphatic and benzylic diamines can be converted to industrially relevant bis-carbamate esters in high yields and in a very clean fashion, proving that carboxylation of diamines is a promising approach to recycle carbon dioxide on large scale processes.We demonstrated that activation of CO2 can be successfully carried out in a H-shaped electrochemical cell, where a simple glass frit is required to separate the compartments. This approach avoids the use of methylcellulose gel and large amounts of supporting electrolyte.
Optimization of the alkylation step was achieved by simple thermal treatment. It allows not only to use butyl chloride as alkylating agent, but also to significantly reduce its amount with respect to previous reports. Moreover, TEAC was recovered from the crude reaction mixture and successfully reused as supporting electrolyte.
The remarkably low reactivity towards carboxylation observed for aromatic substrates provides a potential feature for the selective formation of carbamates in diamines.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Covestro Deutschland AG, Sapienza Università di Roma and MIUR (Ministero dell'Istruzione, dell'Università e della Ricerca). The authors would like to dedicate this paper to Prof. Achille Inesi on the occasion of his 75th birthday.Notes and references
- M. Meinshausen, Nature, 2009, 458, 1158–1162 CrossRef CAS PubMed.
- C. McGlade and P. Ekins, Nature, 2015, 517, 187–190 CrossRef CAS PubMed.
- Intergovernamental Panel on Climate Change 2001, Climate Change 2001: the Scientific Basis, Cambridge University Press, Cambridge, U.K., 2001 Search PubMed.
- H. D. Matthews, N. P. Gillett, P. A. Stott and K. Zickfeld, Nature, 2009, 459, 829–832 CrossRef CAS PubMed.
- M. Helle and H. Saxén, ISIJ Int., 2015, 55, 2047–2055 CrossRef CAS.
- N. Chang, J. Cleaner Prod., 2015, 103, 40–48 CrossRef CAS.
- K. W. Haider, K. G. McDaniel, J. E. Hayes and J. Shen, (Bayer MaterialScience), Polyether carbonate polyols made via double metal cyanide (dmc) catalysis, US7977501B2, WO2008013731A1, 2008.
- C. Gürtler, J. Hofmann, A. Wolf and S. Grasser, (Bayer MaterialScience), Verfahren zur herstellung von polyethercarbonatpolyolen, WO2012032028A1, 2012.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- M. A. Casadei, S. Cesa, F. Micheletti Moracci, A. Inesi and M. Feroci, J. Org. Chem., 1996, 61, 380–383 CrossRef CAS.
- N. Germain, I. Müller, M. Hanauer, R. A. Paciello, R. Baumann, O. Trapp and T. Schaub, ChemSusChem, 2016, 9, 1586–1590 CrossRef CAS PubMed , and the references reported therein.
- R. H. Heyn, I. Jacobs and R. H. Carr, Adv. Inorg. Chem., 2014, 66, 83–115 CrossRef CAS; D. Reimer, P. Hirapara and S. Das, ChemSusChem, 2016, 9, 1916–1920 CrossRef PubMed.
- W. McGhee, D. Riley, K. Christ, Y. Pan and B. Parnas, J. Org. Chem., 1995, 60, 2820–2830 CrossRef CAS.
- M. A. Casadei, A. Inesi, F. Micheletti Moracci and L. Rossi, Chem. Commun., 1996, 2575–2576 RSC.
- M. A. Casadei, F. Micheletti Moracci, G. Zappia, A. Inesi and L. Rossi, J. Org. Chem., 1997, 62, 6754–6759 CrossRef CAS.
- M. Feroci, M. A. Casadei, M. Orsini, L. Palombi and A. Inesi, J. Org. Chem., 2003, 68, 1548–1551 CrossRef CAS PubMed.
- M. Feroci, M. Orsini, L. Rossi, G. Sotgiu and A. Inesi, J. Org. Chem., 2007, 72, 200–203 CrossRef CAS PubMed.
- M. Feroci, I. Chiarotto, M. Orsini, G. Sotgiu and A. Inesi, Electrochim. Acta, 2011, 56, 5823–5827 CrossRef CAS.
- F. Richter, H. Heckroth, V. Trieu, M. Feroci, G. Forte, A. Inesi and I. Chiarotto, Electrochemical synthesis of dicarbamates, WO2016083475A1, 2016.
- The heat generated from the ion motion during the electrolysis melts the gel. This must be replaced before complete melting, ensuring separation between the compartments of the cell at any time during the experiment.
- Reduction of the amount of current used in the electrolysis leads to a mixture of bis-O-alkylcarbamate ester and N-alkylated monocarbamate ester.
- L. Xiao and K. E. Johnson, J. Electrochem. Soc., 2003, 150, E307–E311 CrossRef CAS.
- H.-W. Michalczak, S. Kohlstruk, M. Kreczinski, G. Grund and R. Lomoelder, Process for the continuous preparation of (cyclo)aliphatic diisocyanates, US20100036154A1, 2010.
Footnote |
| † Electronic supplementary information (ESI) available: Description of electrochemical cells, experimental procedures, characterization of products, copy of 1H and 13C NMR spectra. See DOI: 10.1039/c7re00101k |
| This journal is © The Royal Society of Chemistry 2017 |