
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-induced decarboxylation in a photo-responsive iron-containing complex based on polyoxometalate and oxalato ligands†
Yan
Duan
 a,
João C.
Waerenborgh
b,
Juan M.
Clemente-Juan
a,
Carlos
Giménez-Saiz
*a and
Eugenio
Coronado
*a
a,
João C.
Waerenborgh
b,
Juan M.
Clemente-Juan
a,
Carlos
Giménez-Saiz
*a and
Eugenio
Coronado
*a
aInstituto de Ciencia Molecular (ICMol), Universitat de València, c/Catedrático José Beltrán, 2, 46980 Paterna, Spain. E-mail: carlos.gimenez@uv.es; eugenio.coronado@uv.es
bCentro de Ciências e Tecnologias Nucleares, Instituto Superior Técnico, Universidade de Lisboa, 2695-066 Bobadela LRS, Portugal
First published on 16th August 2016
Abstract
A new photoresponsive molecular polyanion in which two Fe(III) ions are simultaneously coordinated by two [A-α-PW9O34]9− polyoxometalate units and two oxalato ligands has been obtained. When irradiated with UV light its potassium salt, 1, exhibits a remarkable photocoloration effect, attributable to the partial reduction of the POM units to give rise to a mixed-valence species. The photoinduced process is intramolecular and involves electron transfer from the oxalato ligands, which partially decompose releasing CO2, towards the Fe(III) and the POM. This mechanism has been confirmed by DRS, IR, XPS and Mössbauer spectroscopy, magnetism and elemental analysis. An analogous derivative of 1 containing malonato ligands does not exhibit such photoactive behaviour, which is evidence that the oxalate ligand is essential for the photoactivity of 1. To our knowledge, 1 represents the first POM-based compound in which the photocoloration effect does not require the presence of intermolecular short interactions.
Introduction
The study of molecular-based materials whose physical or chemical properties can be tuned by applying external stimuli is attracting considerable interest in chemistry owing to the wide variety of functional materials that one can design.1 As an external stimulus, light has been extensively used in this context. Thus, many examples of photomagnetic materials,2 photochromic materials,3 and electrochromic materials4 have been reported.Polyoxometalates (POMs) are metal–oxo clusters with W, Mo or V in their highest oxidation states which have the ability to accept, step by step, one or more electrons giving rise to mixed-valence coloured species (heteropoly blues or heteropoly browns).5 POMs are being increasingly used as building blocks for the construction of systems with stimuli-responsive behaviour (in particular photoresponsive).6 A common strategy is the combination of POMs with photosensitive groups in order to integrate the smart response in hybrid systems by means of electrostatic interactions, H-bonding, or other intermolecular interactions. Recent examples include photoresponsive systems obtained by combining POM anions with azobenzene-modified cationic surfactants which can reversibly assemble and disassemble in solution with light irradiation,6c or supramolecular systems, obtained by using ionic liquids H-bonded to the surface of fluorescent POMs, that exhibit tunable photoluminescence properties by adjusting the pH of the solution.6g
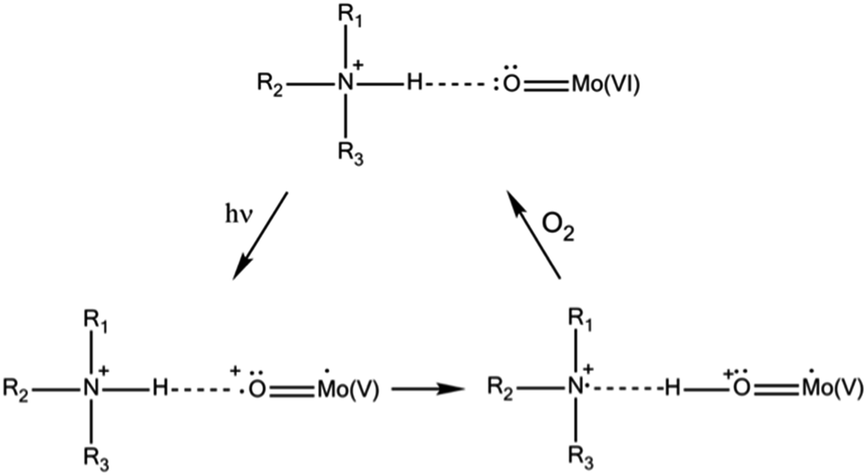
Regarding the solid state, photochromic materials with strong UV-induced colour changes and remarkable coloration contrast can be obtained associating POM anions with organic cations (mainly organoammonium cations (OACs)).6a,b,7 The photoactivity of such systems has been described in terms of a UV-induced photoreduction of the POM, concomitant with an intermolecular electron transfer assisted by proton transfer from the OAC towards the POM (Scheme 1). The effect is reversible in contact with air due to oxidation.
 | ||
| Scheme 1 | ||
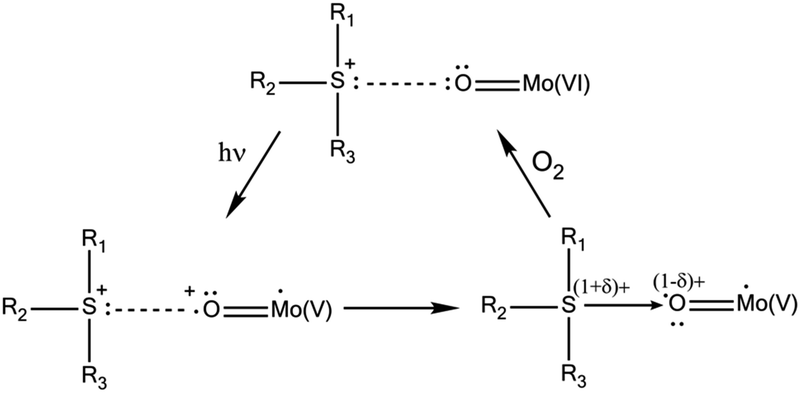
According to this mechanism, the photoactivity in these systems requires the formation of direct H-bonds between POMs and OACs, something that cannot be systematically achieved.8 A way to improve the possibility of H-bond formation has been developed by P. Mialane, R. Dessapt et al., consisting of directly grafting OAC-functionalized bisphosphonate ligands to the inorganic core of POMs.6d The same researchers also proposed a new class of solid-state photochromic hybrids combining sulfonium cations with POMs and so circumvented the dependence on an H-bonding network.6e In this case, the mechanism of the photochromic effect requires the establishment of short S⋯O contacts to allow UV-induced electron transfer from the sulfonium cations to the adjacent POM (Scheme 2). As before, slow bleaching happens in contact with air.
 | ||
| Scheme 2 | ||
On the other hand, some Fe(III) carboxylate complexes exhibit sensitivity to visible and UV light, which makes them photoreactive through ligand to metal charge transfer (LMCT) reactions and, therefore, valuable in the chemistry of environmental systems9 and for laboratory photochemical applications in general (e.g. actinometry).10 A prototypical example of such complexes is the tris(oxalato)ferrate(III) complex whose photoinduced LMCT reaction (reaction (1)) gives rise to the (photo)generation of Fe(II) ions (both in solution and in the solid state), and to a subsequent reductive reaction via CO2˙− radical anions (reaction (3)).11
| [FeIII(C2O4)3]3− + hν → Fe2+ + C2O4˙− + 2(C2O4)2− | (1) |
| C2O4˙− → CO2 + CO2˙− | (2) |
| [FeIII(C2O4)3]3− + CO2˙− → Fe2+ + 3(C2O4)2− + CO2 | (3) |
In this context, we are exploring the possibility of combining Fe(III) carboxylate complexes with POMs in the same molecular unit with the aim of obtaining a new type of photoactive solid state systems which would be independent of the establishment of any kind of inter- or intramolecular short interactions (i.e. H-bonds or S⋯O contacts). This can be realized if both carboxylate and POM act as ligands of a heteroleptic Fe(III) complex.
However, as far as we know, the only reported examples of Fe(III) complexes containing both oxalato and POM ligands are given by the family [Fe4(C2O4)4(H2O)2(B-β-XW9O33)2]14− (X = AsIII, SbIII, BiIII), although no photoactivity has been reported for these compounds.12 We report here the synthesis and characterization (X-ray crystal structure, diffuse reflectance, IR evolution upon irradiation, UV in aqueous solution, XPS, Mössbauer spectroscopy, and magnetic properties) of a novel mixed ferrioxalate–POM compound formulated as K15{K⊂[(A-α-PW9O34)2Fe2(C2O4)2]}·29H2O (1) which exhibits fast and evident photocoloration when exposed to direct sunlight or UV irradiation in the solid state. For comparison, we also report the analogous malonate derivative, K15{K⊂[(A-α-PW9O34)2Fe2(C3H2O4)2]}·27H2O (2), which does not exhibit such photoactive behaviour, which is evidence that the oxalate ligand is essential for the photoactivity of 1. We will demonstrate that the photocoloration process involves a simultaneous intramolecular electron transfer from the oxalato ligand to Fe(III) (leading to Fe(II)) and to the POM framework (leading to a reduced mixed-valence species containing W(V)). Such a process is accompanied by the release of CO2.
Experimental section
General methods and materials
57Fe was obtained from CHEMGAS as iron powder with an isotope purity of 96.28% in 57Fe. All other reagents were of high purity grade, obtained from commercial sources, and used without any further purification. The trivacant ligand K9[A-α-PW9O34]·16H2O was prepared according to a literature procedure13 and confirmed by IR spectra. The potassium salts of the tris(oxalato)ferrate(III) and tris(malonato)ferrate(III) anions, K3[Fe(C2O4)3]·3H2O and K3[Fe(C3H2O4)3]·H2O, were synthesized according to published procedures.14 Pure water (ρ > 18 MΩ cm) was used throughout. It was obtained using an Elix-3/Millipore-Q Academic water purification system. IR spectra were recorded with KBr pellets on a Thermo NICOLET-5700 FT-IR spectrophotometer. Microanalysis was carried out with a Philips XL-30 scanning electron microscope coupled with a Philips EDAX Microanalysis system and the carbon content was determined by microanalytical procedures using an EA 1110 CHNS-O elemental analyser from CE Instruments. Thermogravimetric analysis was performed on a Mettler Toledo TGA/SDTA851e analyzer.Diffuse Reflectance Spectroscopy (DRS) of solid samples was performed at room temperature and the data converted to obtain absorption spectra. DRS was carried out on a Jasco V-670 spectrophotometer equipped with an integrated sphere coated with BaSO4 and an internal diameter of 60 mm, where the baseline was recorded using a poly(tetrafluoroethylene) reference. X-ray Photoelectron Spectroscopy (XPS) was performed at the X-ray Spectroscopy Service at the Universidad de Alicante using a K-Alpha X-ray photoelectron spectrometer system (Thermo Scientific). All spectra were collected using Al Kα radiation (1486.6 eV), monochromatized by a twin crystal monochromator, yielding a focused X-ray spot (elliptical in shape with a major axis length of 400 µm) at 3 mA C and 12 kV. The alpha hemispherical analyser was operated in the constant energy mode with survey scan pass energies of 200 eV to measure the whole energy band and 50 eV in a narrow scan to selectively measure particular elements. XPS data were analysed using Avantage software. A smart background function was used to approximate the experimental backgrounds. Charge compensation was achieved with the system flood gun that provides low energy electrons and low energy argon ions from a single source.
Selected IR bands (2% KBr pellet 2500–400 cm−1) (Fig. S1†): 1664.6(m), 1654.8(m), 1637.4(m), 1618.5(m), 1401.0(s), 1284.9(s), 1076.4(m), 1019.8(s), 940.9(m, sh), 885.2(w), 854.5(w), 741.5(m, sh), 658.8(s), 595.3(w), 518.4(m). Anal. calcd (found) for K15{K⊂[(A-α-PW9O34)2Fe2(C2O4)2]}·29H2O: C 0.82 (0.81), Fe 1.90 (1.90), P 1.05 (0.90), W 56.35 (56.85), and K 10.61 (10.24). The TGA curve of 1 (Fig. S2†) shows two distinct weight loss steps (the first one from 25 to ca. 203 °C and the second one from 203 to ca. 450 °C, with a total weight loss of 10.57%), which correspond mainly to the loss of crystal waters and to the decomposition of the oxalato ligands, respectively (calcd 10.56%, see the ESI†).
Selected IR bands (2% KBr pellet 2500–400 cm−1) (Fig. S1†): 2361.8(m), 1598.2(m), 1424.6(s), 1384.4(s), 1074.6(s), 1016.2(s), 937.0(s), 846.3(s), 742.4(m, sh), 666.5(s), 515.2(w). Anal. calcd (found) for K15{K⊂[(A-α-PW9O34)2Fe2(C3H2O4)2]}·27H2O: C 1.22 (1.18), Fe 1.90 (1.91), P 1.05 (0.98), W 56.25 (57.80), and K 10.60 (10.03). The TGA curve of 2 (Fig. S3†) shows three distinct weight loss steps. The first step (25–203 °C) is attributed to the loss of water molecules. The second (203–350 °C) and third steps (350–450 °C) correspond to the decomposition of the malonato ligands. After that, no significant weight loss is detected. The total weight loss observed is 10.66% (calcd 10.44%, see the ESI†).
Results and discussion
Synthetic approach and stability in aqueous solution
Reaction of the potassium salts of [Fe(C2O4)3]3− or [Fe(C3H2O4)3]3− with the trivacant POM [A-α-PW9O34]9− in aqueous solution at 60 °C results in the crystallization of the sandwich-type molecular complexes 1 and 2 which contain two iron atoms, each one coordinated by one oxalato ligand in 1 (or malonato for 2) and two [A-α-PW9O34]9− moieties. The formation of 1 and 2 takes place owing to the lability of the tris(oxalato) and tris(malonato)ferrate(III) complexes, which quickly and readily exchange two dicarboxylate ligands by two [A-α-PW9O34]9− moieties (substitution of the three dicarboxylate ligands is not possible due to the steric effect imposed by the bulky [A-α-PW9O34]9− anion). This is consistent with the impossibility of obtaining analogous POMs containing Cr or Ru using the same reaction conditions as for 1, i.e. using the more kinetically inert tris(oxalato)chromate(III) or tris(oxalato)ruthenate(III) complexes. On the other hand, the reaction of [A-α-PW9O34]9− with the kinetically labile [Co(C2O4)3]3− or [Mn(C2O4)3]3− does not give the analogous POMs of 1 containing Co(III) or Mn(III) due to their strong oxidizing character in aqueous solution.The crystal structures of 1 and 2 (see below) reveal that both POMs enclose a potassium ion in their internal cavity. It seems that K+ ions act as templates for the assembly of two [A-α-PW9O34]9− and two Fe(C2O4)+ (or Fe(C3H2O4)+) moieties to form 1 and 2. We have attempted unsuccessfully to enclose other alkaline ions (Na+, for example) in the central cavity of these POMs. Hence, the presence of K+ ions in the reaction medium seems to be essential for the formation of 1 and 2. This is attributed to the rigidity of the POM, which has a cavity size only suitable to accommodate potassium.
The stability in aqueous solution of 1 and 2 has been studied by UV spectroscopy (Fig. S9†). Both compounds display similar absorption bands in the range 190–400 nm: 1 exhibits a shoulder at 201 nm (204 nm for 2) and a band centred at 257 nm (258 nm for 2). The absorption at higher energy is due to the pπ–dπ charge-transfer transitions of the Ot → W in the polyoxoanion (Ot: terminal oxygen), while the lower energy band can be assigned to the pπ–dπ charge transfer transitions of Ob,c → W in the polyoxoanion21 (Ob,c = bridging oxygen). The evolution of the UV spectra over 24 h reveals that the positions of the absorption bands do not change with time, but they become less intense over time, pointing to a slow decomposition of both compounds in aqueous solution at room temperature. The compounds, however, can be recrystallized from aqueous solution if the process is carried out within a short time using concentrated solutions of 1 or 2 (∼30 min).
Crystal structures
The X-ray structures of 1 and 2 reveal that both POMs contain two [A-α-PW9O34]9− moieties, two iron ions, two oxalato (for 1) or malonato (for 2) ligands, and one potassium cation encapsulated in the central cavity of each POM (Fig. 1). The two iron ions are sandwiched between the trivacant [A-α-PW9O34]9− anions in such a way that each iron atom is coordinated by two oxygen atoms from two edge-sharing tungsten atoms of each trivacant unit, while the two remaining coordination sites are occupied by one chelating oxalato or malonato ligand.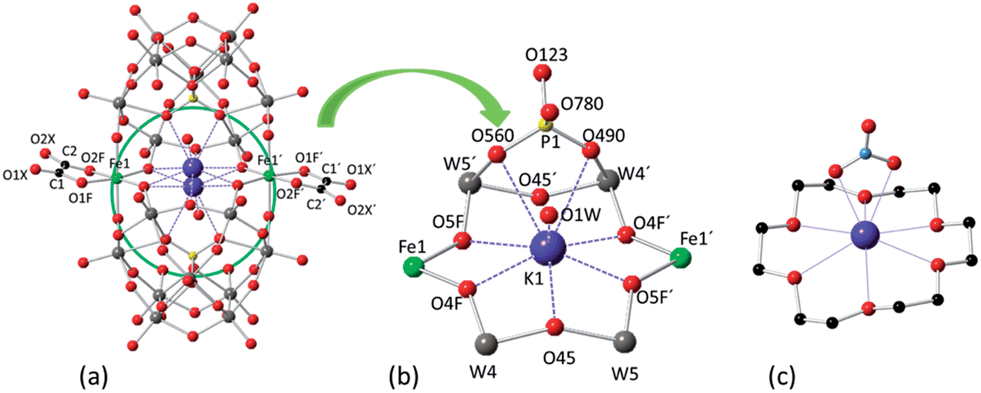 | ||
| Fig. 1 (a) Ball and stick representation of POM 1, showing the disorder of the encapsulated potassium ion in the central cavity of the POM. (b) Detailed view of the coordination of the encapsulated potassium ion (the disorder is not shown). (c) Ball and stick representation showing the coordination of potassium by 18-crown-6 and nitrate in [K⊂(18-crown-6)](NO3)(HNO3).22 The colour code of the spheres is as follows: W (grey), Fe (green), K (violet), P (yellow), O (red) and C (black). K⋯O coordination contacts are shown as dotted lines. | ||
There are many reported examples of sandwich POMs trapping two or more metal ions between two [A-α-PW9O34]9− ligands.23 However, regarding first row 3d-transition metals, the most representative series is [(A-α-PW9O34)2(M(H2O)2)3]n− (MII = Mn, Fe, Co, Ni, Cu, Zn, and MIII = Fe).23a–c The reason why 1 and 2 enclose only two iron atoms and not more lies in the presence of the chelating oxalato or malonato ligands, which occupy two cis positions in the octahedral coordination sphere of the metal ions. This forces the two [A-α-PW9O34]9− units to be slightly side-slipped in order to occupy the other four positions of the octahedral coordination spheres of the iron ions (Fig. 2), as a result giving the impossibility of sandwiching a third iron ion. In the previously reported POM compounds enclosing three octahedrally-coordinated 3d transition metal ions, the absence of a chelating ligand (other than [A-α-PW9O34]9−) allowed the two trivacant units to be placed directly over each other, leaving the remaining trans coordination positions to be occupied by non-chelating ligands.
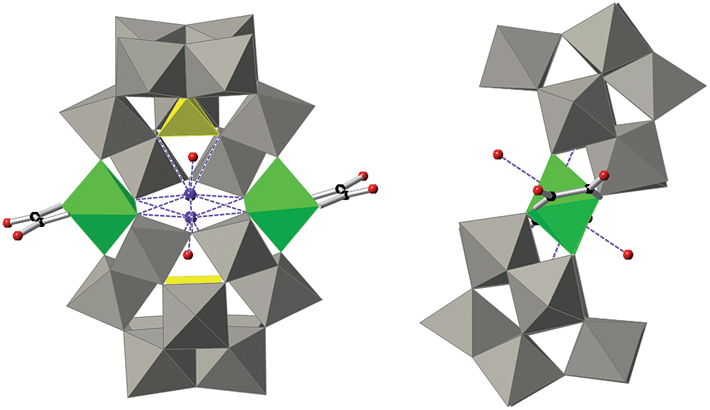 | ||
| Fig. 2 Combined polyhedral/ball-and-stick representations of the POM in 1. Gray octahedra, [WO6]; yellow tetrahedra, [PO4]; green octahedra, [FeO6]; black spheres, C; red spheres, O. | ||
Therefore, the described arrangement of ligands and metal ions in 1 and 2 would give rise to an overall, idealized C2h symmetry (in which the binary axis passes through the two iron atoms). However, the mean planes of the dicarboxylate ligands are tilted in opposite directions with respect to the mean plane formed by the Fe atom and the equatorial O ligands by 18.7(9)° and 22.9(5)° for 1 and 2, respectively (see Fig. 2),‡ reducing the overall symmetry of the POM from C2h to Ci. Bond valence sum calculations24 suggest an oxidation state of +3 for the iron ions in 1 and 2 (this is confirmed by Mössbauer and magnetic measurements, see below and the ESI†) and that all O atoms in the POMs are non-protonated (see Fig. S5†).
The side-slipped arrangement of the two [A-α-PW9O34]9− moieties creates an internal cavity in the centre of POMs 1 and 2 which is occupied by a nine-coordinated potassium cation. This internal cavity can be considered as an oxygen pocket formed by an equatorial ring of six µ2-O atoms of the POM (O4F, O5F, O45, O4F′, O5F′ and O45′) which has a chair-like arrangement about the potassium ion, and four other µ3-O atoms of the POM (O490, O560, O490′, O560′), two at each opposite side of the equatorial ring. The six µ2-O atoms either bridge two W atoms (O45 and O45′) or W and Fe atoms (O4F, O5F, O4F′ and O5F′), while the four µ3-O atoms bridge one P and two W atoms. The potassium ion is enclosed in the centre of this oxygen pocket although it is disordered over two equivalent and close positions separated by 1.11 Å (or 1.02 Å) in 1 (or 2). This disorder enables K+ to be coordinated by two of the µ3-O atoms located on one side of the equatorial ring made of the six µ2-O atoms. K⋯µ2-O distances range from 2.595(19) to 2.868(19) Å for 1 (2.559(6) to 2.834(6) Å for 2) and K⋯µ3-O distances are 2.917(19) and 2.943(16) Å in 1 (2.924(6) and 2.957(6) Å in 2). A water molecule completes the ninth coordination of the K+ ion with a K⋯Ow distance of 2.72(3) Å for 1 (2.783(10) Å for 2). This situation is strongly reminiscent of the typical potassium complexes of the 18-crown-6 polyether ligand.25 The rigidity of the internal cavity created by the POM, however, makes it much more selective to K+ coordination than the flexible organic 18-crown-6 polyether, which can also coordinate other smaller or larger alkaline cations.25
Photocoloration effect
When a solid sample of 1 is exposed to direct sunlight or UV irradiation, a colour change from light yellowish-green to black, visible by the naked eye, starts in a matter of seconds (Fig. 3). If this black solid is stored in the dark in the presence of air, the solid reverts to its original colour after about 7 days (no colour reversion takes place under an O2-free atmosphere). | ||
| Fig. 3 (a–h) Photogenerated colour change of 1 after different UV irradiation times at 351 nm (0–20 min). After being irradiated, the compound was stored in the dark for 7 days (i) and then re-irradiated again (j–l). Photographs of individual irradiated crystals of 1 are shown in the ESI (Fig. S13 and S14†). | ||
UV-vis/diffuse absorbance spectra of 1, before and after irradiation with UV light (351 nm) during different irradiation times, are shown in Fig. 4. Before UV irradiation, the sample exhibits two absorption bands in the visible region; a weak, broad absorption band centred at 630 nm and a narrower and stronger band at 460 nm. In addition there are two broad, significant bands at lower wavelengths (ranging between 210–270 and 320–370 nm) attributed to O → W ligand to metal charge transfer (LMCT) bands of the POM (Fig. 4, inset).
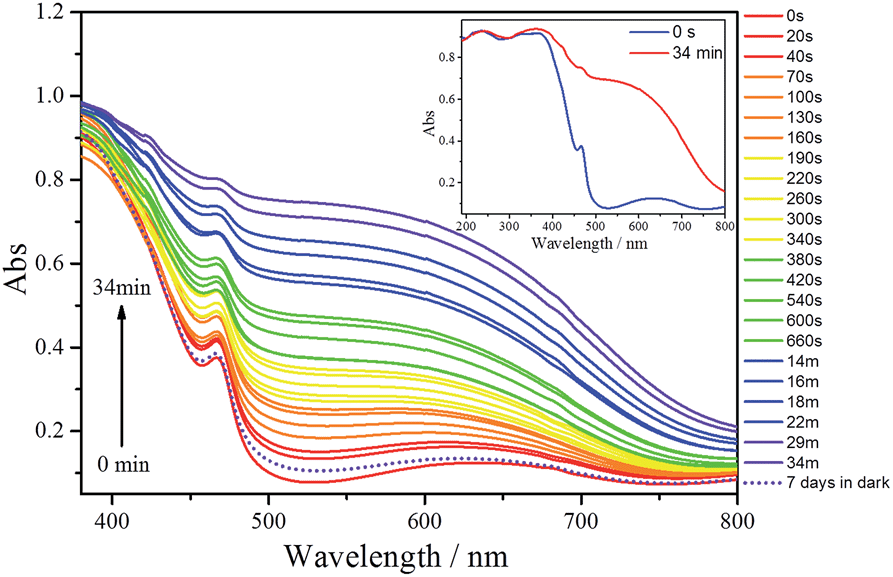 | ||
| Fig. 4 Evolution of the photogenerated absorption of 1 after 0, 0.17, 0.67, 1.17, 1.67, 2.17, 2.67, 3.17, 3.67, 4.33, 5, 5.67, 6.33, 9, 10, 11, 14, 16, 18, 22, 29 and 34 min of UV irradiation (351 nm). After being irradiated for 34 min, the compound was stored in the dark for 7 days and its absorption spectrum recorded (dotted line). Inset: UV-vis/diffuse absorbance spectra of 1 (before UV irradiation and after 34 min of UV irradiation) in the 190–800 nm range. | ||
After irradiation, the colour of the sample turns black, in agreement with the growth of the two absorption bands in the visible range. The broader band in this region is quite comparable with those of other irradiated photochromic POMs,6d,e,26 suggesting that the photoinduced coloration is mainly due to the POM core. This should imply that in 1 the two POM moieties have been reduced as a result of the UV irradiation and contain W(V) species. Therefore, the absorption in the visible range can be attributed to d–d transitions (if the electron is trapped onto a specific W centre) or intervalence charge transfer (IVCT) of W(V) → W(VI) (if it can be delocalized onto several adjacent {WO6}). In addition, the position of the maximum absorption of the broader band undergoes a blue shift with irradiation time from 630 nm to about 550 nm, suggesting that the degree of reduction in the POMs increases with irradiation time. After the UV irradiation was stopped, the sample was stored in the dark at room temperature in the presence of air. Under these conditions, the sample slowly reverts to its original colour. After 7 days, the absorption spectrum is very similar to the original one (dotted line in Fig. 4) meaning that W(V) is reoxidised to W(VI) in the presence of air. The coloration and bleaching process can be repeated at least 6 times if the sample is exposed again to UV light, although the intensity of the black colour decreases after each cycle. In contrast, 2 does not exhibit any light-induced colour change under similar conditions, as shown by its UV-vis/diffuse absorbance spectra (Fig. S12†).
The coloration kinetics of 1 have been quantified by analysing its reflectivity values, R(t), in the range of 380–800 nm as a function of irradiation time, t (Fig. S10†). Details of the parameters related to the coloration kinetics are given in the ESI (Table S3†). In summary, it has been found that the R525(t) vs. t curves of 1 can be fitted using the pseudo-second order law R525(t) = a/[bt + 1] + [R525(0) − a] (Fig. S11†). The t1/2 value obtained for the photocoloration process (4.25 min) corresponds to a relatively fast photoresponse compared to other POM-based photochromic materials.26c,27
Infrared spectroscopy
The IR spectra of 1 (as a KBr pressed pellet) before and after UV photoirradiation were investigated (Fig. 5). Before irradiation it exhibits the typical bands of the chelating oxalato ligand28 at 1714 cm−1 (νa(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), very weak), 1668 cm−1 (νa(CO), strong), 1384 and 1401 cm−1 (νs(CO) + ν(CC), strong) and 1285 cm−1 (νs(CO) + δ(O–CO), medium), and bands corresponding to the POM framework at 1076 and 1018 cm−1 (νa(P–O), strong), 940 cm−1 (νa(W–Ot), strong), 852 and 800 cm−1 (νa(W–Oa–W), strong) and 742 cm−1 (νa(W–Ob–W), strong). Bands at 659 and 519 cm−1 can be assigned to Fe–O–W and Fe–O–C vibrations. Upon irradiation, the intensity of all oxalato bands decreases gradually as the irradiation time increases, while there are two new bands that appear and increase in intensity with irradiation time, at 2341 and 2364 cm−1. These bands can be attributed to carbon dioxide (R- and P-branches of the νa(CO2) band, respectively) trapped in the KBr pellet.29
O), very weak), 1668 cm−1 (νa(CO), strong), 1384 and 1401 cm−1 (νs(CO) + ν(CC), strong) and 1285 cm−1 (νs(CO) + δ(O–CO), medium), and bands corresponding to the POM framework at 1076 and 1018 cm−1 (νa(P–O), strong), 940 cm−1 (νa(W–Ot), strong), 852 and 800 cm−1 (νa(W–Oa–W), strong) and 742 cm−1 (νa(W–Ob–W), strong). Bands at 659 and 519 cm−1 can be assigned to Fe–O–W and Fe–O–C vibrations. Upon irradiation, the intensity of all oxalato bands decreases gradually as the irradiation time increases, while there are two new bands that appear and increase in intensity with irradiation time, at 2341 and 2364 cm−1. These bands can be attributed to carbon dioxide (R- and P-branches of the νa(CO2) band, respectively) trapped in the KBr pellet.29
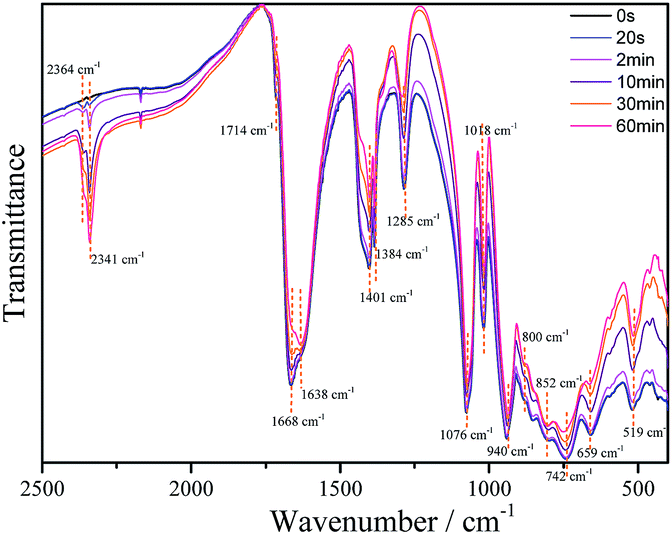 | ||
| Fig. 5 Evolution of the IR spectra of 1 upon different irradiation times of UV light (351 nm). | ||
These results confirm a photoinduced decomposition of the oxalato ligand into carbon dioxide, akin to the photodecomposition of the oxalato ligands in the tris(oxalato)ferrate(III) complex anion.30 During this process, two electrons must be released from each oxalato ligand that decomposes, which may be accepted by the POM framework and/or the iron atoms. The decrease in the intensity of all the POM bands upon irradiation is consistent with a reduction of the POM.26a
XPS spectra
The XPS W4f spectra collected for 1 before irradiation, after UV irradiation, and after being stored in the dark during 7 days are shown in Fig. 6. All three XPS W4f spectra can be resolved into 4f7/2 and 4f5/2 doublets (caused by spin–orbit coupling) and deconvoluted into two couples of peaks (Fig. 6a–c), corresponding to the typical binding energies of W(VI) (centred at 37.9 and 35.7 eV) and W(V) oxidation states (centred at 37.0 and 34.9 eV). As shown in Table 1, W(V) species appear even in the non-irradiated sample (Fig. 6a), suggesting that the photoreduction of the POM occurs in part when the sample is exposed to an X-ray source, as observed in other previously published work.26d,31 Data analysis indicates that after photoirradiation with 351 nm UV light for 3 days under vacuum, the amount of W(V) increases and the amount of W(VI) decreases (Fig. 6b). The concentration of W(V) is 11.0% (Table 1), as determined from the peak area ratio of W(V) to W(VI) (much higher than that observed before irradiation), indicating the reduction of more W(VI) to W(V) under UV irradiation.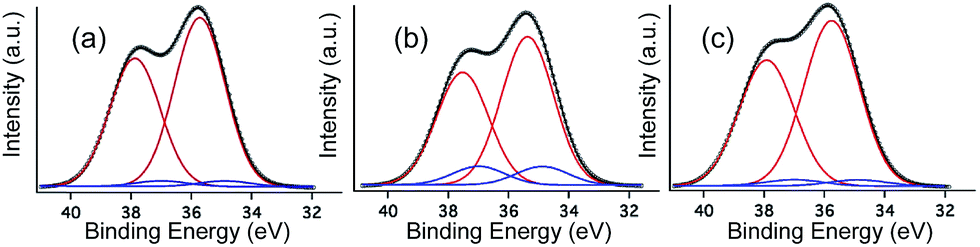 | ||
| Fig. 6 XPS W4f spectra collected for 1 before irradiation (a), after 3 days of UV irradiation (351 nm) (b) and 7 days more in the dark (c). Dotted lines correspond to the experimental data that can be deconvoluted into two pairs of peaks corresponding to W(VI) (red curves) and W(V) (blue curves). Fitting of the experimental data (black curves) has been obtained by summation of the deconvoluted peaks. | ||
| Species | Before irradiation | After irradiation | 7 days in the dark | ||||
|---|---|---|---|---|---|---|---|
| Peak, BE/eV | W(V)% | Peak, BE/eV | W(V)% | Peak, BE/eV | W(V)% | ||
| W4f | W(VI)4f5/2 | 35.7 | 3.0% | 35.4 | 11.0% | 35.7 | 4.0% |
| W(VI)4f7/2 | 37.9 | 37.5 | 37.9 | ||||
| W(V)4f5/2 | 34.9 | 34.9 | 34.9 | ||||
| W(V)4f7/2 | 37.0 | 37.0 | 37.0 | ||||
The XPS spectrum of the photoirradiated sample stored in dark (Fig. 6c) exhibits very similar binding energies of the W4f peaks to those of the non-irradiated sample, and can be deconvoluted into one pair of peaks that corresponds to the typical binding energies of W(VI) (centred at 37.9 and 35.7 eV). The concentration of W(V) in these two samples is also very similar (4.0% compared to 3.0% for the irradiated and the initial samples, respectively).
Mössbauer spectroscopy
The Mössbauer spectra of 1-57Fe and 2-57Fe consist of a broad absorption peak (Fig. 7a and S15†), which may be fitted by a quadrupole doublet. Only a broad peak is observed in both spectra because the widths of each line of the doublets are higher than the corresponding quadrupole splittings, QS (Table S4†). The isomer shifts relative to α-Fe at 295 K, IS, are within the range of high spin, S = 5/2, Fe(III) values32 and similar to those of Fe(III) coordinated to oxalato or malonato ligands in other compounds.20,33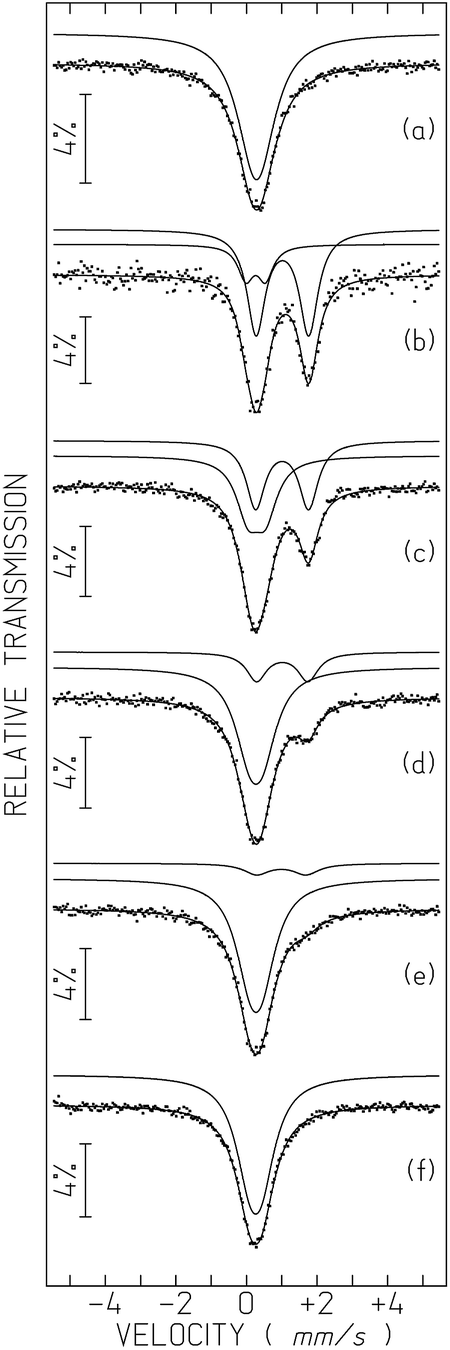 | ||
| Fig. 7 Mössbauer spectra of 1-57Fe sample taken before (a) and after 6 h UV irradiation for tUV ∼ 0.75 h (b), 18 h (c), 42 h (d), 66 h (e) and 12 days (f). tUV, time after irradiation was switched off, is defined in the text. The lines over the experimental points are the sum of two quadrupole doublets shown slightly shifted for clarity (estimated parameters in Table S4†). | ||
After irradiation by UV light (365 nm) the 1-57Fe sample changes colour from light yellowish-green to almost black. After 6 h irradiation time the sample was moved to the Mössbauer spectrometer. At least 45 minutes of data collection was necessary for a reliable analysis (Fig. 7b). The spectrum obtained can only be analysed by two quadrupole doublets. One of the doublets has an IS (Table S4†) typical of high-spin Fe(II), (S = 2),20,32,34 far above the range of the Fe(III) IS values. The second doublet is similar to the Fe(III) signal in the pristine sample but with a higher QS, most likely due to a more distorted Fe(III) environment in the UV-irradiated sample.
Both high-spin Fe(III) and Fe(II) doublets are present in the spectra taken at different times after UV-irradiation (up to 10 days). The relative area of Fe(II) and I(FeII) decreases with increasing time after irradiation (Fig. 7 and S16†). In order to evidence the time dependence of I(FeII), all spectra except for the first one after irradiation, were collected during approximately the same time and tUV was defined as the time elapsed between mid-spectrum collection and the moment when UV irradiation was switched off (which corresponds to tUV = 0). The relative areas estimated for tUV = 0.75 hours suggest that approximately 79% of all the Fe is in the 2+ oxidation state, i.e. I(FeII) ∼79%. The steep decrease of I(FeII) with increasing tUV during the first hours (Table S4, Fig. S16†) suggests that the I(FeII) deduced from the spectra are actually an average of the I(FeII) range of values occurring during the spectrum collection time. This means that immediately after UV irradiation I(FeII) is most likely higher than 79%.
When tUV ∼ 66 hours, I(FeII) reaches 11% ± 2%, but at this stage it decreases very slowly with increasing time. After approximately 10 days the colour of the sample becomes light yellowish-green again but a small shoulder on the Fe(III) absorption peak evidences the presence of an Fe(II) doublet with an estimated I(FeII) ∼4%. Only after 12 days do all traces of Fe(II) disappear from the spectrum (Fig. 7f and S16, Table S4†).
The IS of Fe(III) is consistent with octahedral coordination, in agreement with the structure determined by single-crystal XRD for the pristine compound. The IS of Fe(II) is also consistent with octahedral coordination but the presence of a fraction of Fe(II) in lower coordination, such as penta-coordination, may not be excluded.32,35
The effect of a second UV irradiation of the sample after all Fe returns to the +3 oxidation state, is not the same as the effect of the first irradiation. The sample colour resulting from this second irradiation is not so dark and the Mössbauer spectra (Fig. S17†) taken for similar tUV reveal that I(FeII) is significantly lower in the first days after irradiation (Table S4, Fig. 7, S16 and S17†). The estimated I(FeII) for the spectrum taken during 45 minutes immediately after turning off the UV light is only ∼20%. After 12 days all the Fe is back in the +3 oxidation state.
The IS and the QS of Fe(II) obtained after the second irradiation are significantly different (the IS is smaller and the QS is higher) from those obtained after the first irradiation. The lower IS may be explained if we assume that after the second irradiation Fe(II) is mainly pentacoordinated.32,35 This coordination of Fe(II) would also lead to a more distorted electronic charge distribution around the Fe(II) in agreement with a higher QS.
As mentioned above, the QS of Fe(III) increases significantly after the first UV-irradiation, suggesting a more distorted environment of this cation. However, as I(FeII) decreases, QS of Fe(III) also decreases, until it becomes slightly lower than in the pristine sample where I(FeII) is zero. This may suggest that by the end of the first “UV-irradiation-Fe(II) decay” cycle the structure of the sample is no longer identical to the pristine one although all the Fe is in the same oxidation state, +3, and apparently has the same coordination number, 6. Only the ligands of Fe(III) and/or their geometrical arrangement seem to be different. Such a structural modification would be related to the distinct behaviour during the second UV-irradiation, rendering the sample more reluctant to Fe(III) reduction. In summary, Mössbauer spectroscopy clearly shows that UV-irradiation induces a Fe(III) reduction to Fe(II). The variation of hyperfine parameters also suggests structural modifications after the first cycle of UV irradiation and Fe(II) decay back to Fe(III).
Mechanism of the photoinduced coloration and final discussion
In this section we gather the experimental results described in the previous sections with the objective of proposing a mechanism for the photoinduced coloration of 1. When exposed to UV irradiation, the light yellowish-green solid samples of 1 become black in a fast photocoloration process having a t1/2 value of 4.25 min (as indicated by UV-vis/DRS measurements), while 2 does not exhibit any photoinduced colour change. IR spectroscopy clearly indicates that the UV irradiation initiates a photodecomposition of the oxalato ligands of 1 into CO2. This is confirmed by carbon analysis results, which indicate that there is a lesser amount of carbon in the irradiated compound than in the initial one (0.49% vs. 0.81%). Each oxalato ligand that decomposes into CO2 releases two electrons, which presumably are transferred to the iron and/or tungsten atoms of the POM. The DRS measurements suggest that photocoloration is due to the photoreduction of W(VI) (5d0) to W(V) (5d1) and occurs via d–d transitions and/or W(VI)/W(V) intervalence charge transfer in the POM core; XPS measurements confirm the presence of up to 11.0% W(V) in the irradiated compound. On the other hand, Mössbauer measurements confirm the presence of Fe(II) (79%) in the irradiated samples of 1-57Fe and discard it in non-irradiated samples. The magnetic properties of non-irradiated and irradiated samples of 1 are compatible with this scenario (see the ESI†).In view of all the experimental evidence, the photocoloration effect in 1 could arise according to the mechanism proposed in Scheme 3. UV irradiation induces the photodissociation of the coordinated bond between Fe(III) and the oxalato ligand, resulting in a ligand-to-metal charge transfer that yields Fe(II) and a carbon dioxide radical ion (CO2˙−). Concomitantly, the radical ion most likely reacts with the POM yielding reduced W(V) ions, responsible for the intense photocoloration of 1.
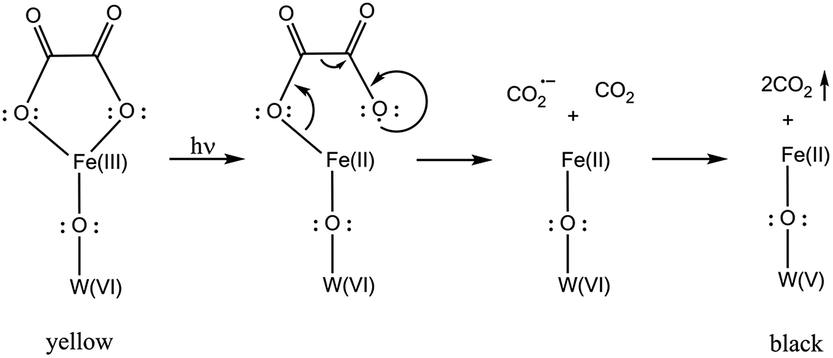 | ||
| Scheme 3 The mechanism depicted here implies a W atom directly connected (through an oxo bridge) to a Fe atom, but can be transposable to other W atoms. | ||
According to the carbon analysis results and the TGA curve of an irradiated sample of 1 (see Fig. S4†), it is possible to deduce the formula K16[(PW9O34)2Fe2(C2O4)x]·nH2O (x = 1.16 and n = 20.6), which clearly reflects the partial loss of oxalato ligands under these conditions of UV irradiation (the decrease in the number of water molecules of crystallization is due to the vacuum conditions employed during the irradiation process). This formula must be regarded in compositional terms only, as it does not reflect either the possible structural modifications of the trivacant [A-α-PW9O34]9− moieties or the change in coordination environment of the Fe atoms that almost certainly happens after the photodecomposition of the oxalato ligands and concomitant reduction of the Fe atoms and POM core (powder diffraction indicates that solid samples of 1 tend to amorphise upon irradiation, see Fig. S8†).
If this irradiated black solid is stored in the dark in the presence of air, it reverts to its original light yellowish-green colour after about 7 days (DRS measurements show that the absorbance of a reversed sample is almost the same as that before irradiation). As no colour reversion takes place under an O2-free atmosphere, it must be assumed that O2 is able to oxidize the W(V) back to W(VI). This is confirmed by XPS measurements of a reversed sample, which indicates that the concentration of W(V) is similar to the value found in the initial, non-irradiated sample. Moreover, Mössbauer measurements indicate also that the Fe(II) ions are oxidized back to Fe(III) after keeping the irradiated samples for 12 days in the dark in the presence of air.
Finally, the process of photoinduced coloration and colour reversion in the presence of air can be repeated at least 6 times for the same sample, although the photoinduced coloration is less intense after every cycle (the fading time keeps approximately constant after each cycle (∼7 days)). This could be anticipated because less amounts of oxalato ligands are expected in the sample after each cycle of UV irradiation. Although the decrease of oxalato ligands after the second irradiation could not be confirmed by carbon analysis (the low percentage of C in the reirradiated samples gave non-reproducible values), it is consistent with the Mössbauer results, which show that a much lower concentration of Fe(II) is attained after the irradiation of a reversed sample with UV light, under the same conditions as the irradiation of the pristine sample (20% after the second irradiation vs. 79% after the first irradiation).
In conclusion, 1 represents a new photoresponsive POM-based system that exhibits a remarkable photocoloration effect (from light yellowish-green to black) in the solid state when irradiated with UV light due to the partial photodecomposition of oxalato ligands and release of CO2, concomitant with a partial reduction of W(VI) and Fe(III) to W(V) and Fe(II). As the photocoloration effect can be repeated several times, this result opens the possibility of obtaining new POM-based materials that incorporate other photoactive Fe(III) carboxylate moieties, which would give rise to different photoresponsive systems with tuneable properties. In addition, we are currently studying the deposition of 1 on different substrates to test its use as chemical actinometer in the solid state. The most widely accepted actinometer is the tris(oxalato)ferrate(III) complex (also called ferrioxalate), due to its wide wavelength range of absorption and high quantum yield.10 Thanks to the POM, 1 exhibits a high contrast photocoloration effect, which is advantageous for its use in solid state actinometry without the need of a post irradiation analytical procedure. Notice that in the standard ferrioxalate actinometry, phenanthroline is needed as a complexation reagent for Fe(II) and subsequent colour development. In our case, this may enable the in situ quantification of the light intensity in a simpler way by monitoring the absorbance of the irradiated solid sample of 1.
Acknowledgements
The present work has been supported by the EU (COST Actions CM1203 Polyoxometalate Chemistry for Molecular Nanoscience (PoCheMon) and CA15128 Molecular Spintronics (MOLSPIN)), the Spanish MINECO (CTQ2014-52758-P, MAT2014-56143-R and Excellence Unit María de Maeztu, MDM-2015-0538), and the Generalitat Valenciana (Prometeo Programme). C2TN/IST authors gratefully acknowledge the Portuguese Foundation for Science and Technology (FCT) support through the UID/Multi/04349/2013 project. We thank José Ma Martínez-Agudo for performing some of the physical measurements. The authors are also grateful to Fernando Coloma for the XPS measurements.Notes and references
-
(a) E. Coronado and G. Mínguez Espallargas, Chem. Soc. Rev., 2013, 42, 1525–1539, 10.1039/C2CS35278H
; (b) S. Biswas, P. Kumari, P. M. Lakhani and B. Ghosh, Eur. J. Pharm. Sci., 2016, 83, 184–202, DOI:10.1016/j.ejps.2015.12.031
-
(a) P. Gütlich, Y. García and T. Woike, Coord. Chem. Rev., 2001, 219, 839–879, DOI:10.1016/S0010-8545(01)00381-2
-
(a) D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349, DOI:10.1002/anie.201500628
-
(a) E. J. Harbron, C. M. Davis, J. K. Campbell, R. M. Allred, M. T. Kovary and N. J. Economou, J. Phys. Chem. C, 2009, 113, 13707–13714, DOI:10.1021/jp9037864
-
(a)
M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag Berlin, Heidelberg, 1983 Search PubMed
-
(a) T. He and J. Yao, Prog. Mater. Sci., 2006, 51, 810–879, DOI:10.1016/j.pmatsci.2005.12.001
- T. Yamase, Chem. Rev., 1998, 98, 307–325, DOI:10.1021/cr9604043
- R. Q. Fang, X. M. Zhang, H. S. Wu and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, M359–M361, DOI:10.1107/S1600536804004647
- L. Deguillaume, M. Leriche, K. Desboeufs, G. Mailhot, C. George and N. Chaumerliac, Chem. Rev., 2005, 105, 3388–3431, DOI:10.1021/cr040649c
- H. J. Kuhn, S. E. Braslavsky and R. Schmidt, Pure Appl. Chem., 2004, 76, 2105–2146, DOI:10.1351/pac200476122105
-
(a) E. L. Simmons and W. W. Wendlandt, Coord. Chem. Rev., 1971, 7, 11–27, DOI:10.1016/S0010-8545(00)80006-5
-
(a) A. Dolbecq, J. Compain, P. Mialane, J. Marrot, E. Rivière and F. Sécheresse, Inorg. Chem., 2008, 47, 3371–3378, DOI:10.1021/ic7024186
-
(a) R. Contant, Can. J. Chem., 1987, 65, 568–573, DOI:10.1139/v87-100
- D. Collison and A. K. Powell, Inorg. Chem., 1990, 29, 4735–4746, DOI:10.1021/ic00348a030
- Agilent Technologies UK Ltd, Oxford, UK, CrysAlis PRO Software system, 2013.
-
G. M. Sheldrick, SHELXTL Version, 2014/7, http://shelx.uni-ac.gwdg.de/SHELX/index.php Search PubMed
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897, DOI:10.1107/S0108767395007367
- R. H. Blessing, J. Appl. Crystallogr., 1997, 30, 421–426, DOI:10.1107/S0021889896014628
- G. J. Long, T. E. Cranshaw and G. Longworth, Moessbauer Eff. Ref. Data J., 1983, 6, 42–49 Search PubMed
- E. Coronado, J. R. Galán-Mascarós, C. Martí-Gastaldo, J. C. Waerenborgh and P. Gaczyński, Inorg. Chem., 2008, 47, 6829–6839, DOI:10.1021/ic800418k
- Y. Wang, X. Sun, S. Li, P. Ma, J. Niu and J. Wang, Cryst. Growth Des., 2015, 15, 2057–2063, DOI:10.1021/cg5012499
- A. N. Chekhlov, Russ. J. Inorg. Chem., 2008, 53, 928–932, DOI:10.1134/S0036023608060193
-
(a) W. H. Knoth, P. J. Domaille and R. D. Farlee, Organometallics, 1985, 4, 62–68, DOI:10.1021/om00120a012
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247, DOI:10.1107/S0108768185002063
- J. W. Steed, Coord. Chem. Rev., 2001, 215, 171–221, DOI:10.1016/S0010-8545(01)00317-4
-
(a) X. A. Zhang, W. J. Wu, Y. H. Man, T. Tian, X. Z Tian and J. F. Wang, Sci. China, Ser. B: Chem., 2007, 50, 318–326, DOI:10.1007/s11426-007-0038-4
- R. Dessapt, M. Collet, V. Coué, M. Bujoli-Doeuff, S. Jobic, C. Lee and M. Whangbo, Inorg. Chem., 2009, 48, 574–580, DOI:10.1021/ic8013865
-
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B: Applications in Coordination, Organometallic and Bioinorganic Chemistry, John Wiley & Sons, 2009 Search PubMed
- G. Keresztury, M. Incze, F. Sóti and L. Imre, Spectrochim. Acta, Part A, 1980, 36, 1007–1008, DOI:10.1016/0584-8539(80)80181-4
-
(a) A. S. Brar and B. S. Randhawa, Bull. Chem. Soc. Jpn., 1981, 54, 3166–3169, DOI:10.1246/bcsj.54.3166
- W. Feng, Y. S. Ding, Y. Liu and R. Lu, Mater. Chem. Phys., 2006, 98, 347–352, DOI:10.1016/j.matchemphys.2005.09.037
-
N. N. Greenwood and T. C. Gibb, Mössbauer Spectroscopy, Chapman and Hall, London, 1971 Search PubMed
- P. S. Bassi, B. S. Randhawa and S. Kaur, Hyperfine Interact., 1986, 28, 745–748, DOI:10.1007/BF02061553
- E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García, J. M. Martínez-Agudo, E. Martínez-Ferrero, J. C. Waerenborgh and M. Almeida, J. Solid State Chem., 2001, 159, 391–402, DOI:10.1006/jssc.2001.9169
- F. Menil, J. Phys. Chem. Solids, 1985, 46, 763–789, DOI:10.1016/0022-3697(85)90001-0
Footnotes |
| † Electronic supplementary information (ESI) available: IR spectra, thermogravimetric analysis, X-ray crystallography, bond valence calculations, X-ray powder diffraction, UV-vis spectra of 1 and 2 in aqueous solution, coloration kinetics, Mössbauer spectroscopy and magnetic properties. ICSD reference 430800. |
| ‡ All six atoms of the oxalato ligand are used to calculate the mean plane, while only the coordinated oxygen atoms and the sp2 carbon atoms of the malonato ligand are used to calculate the mean plane. |
| This journal is © The Royal Society of Chemistry 2017 |