
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed dehydrogenative borylation of terminal alkynes with pinacolborane†
Erik A.
Romero
,
Rodolphe
Jazzar
and
Guy
Bertrand
*
UCSD-CNRS Joint Research Chemistry Laboratory (UMI 3555), Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA 92093-0358, USA. E-mail: guybertrand@ucsd.edu
First published on 9th August 2016
Abstract
LCuOTf complexes [L = cyclic (alkyl)(amino)carbenes (CAACs) or N-heterocyclic carbenes (NHCs)] selectively promote the dehydrogenative borylation of C(sp)–H bonds at room temperature. It is shown that σ,π-bis(copper) acetylide and copper hydride complexes are the key catalytic species.
Popularized by the Suzuki–Miyaura reaction, organoboronic esters and acids are now regarded as key building blocks for compounds with applications ranging from material to life sciences. Consequently, numerous methodologies have been developed to access these valuable substrates.1 Following ground-breaking reports by Hartwig et al.,2 and Smith et al.,3 the catalytic dehydrogenative borylation of C(sp3)–H bonds and C(sp2)–H bonds are now well documented.4 In contrast, for C(sp)–H bonds there are only a few reports by Ozerov et al.5 using iridium and palladium complexes supported by pincer ligands, and one by Tsuchimoto et al.6 with zinc triflate, which is only effective with 1,8-naphthalenediaminatoborane as the boron partner. One of the difficulties of the dehydrogenative borylation of terminal alkynes is the competing hydroboration of the triple bond, which affords alkenyl boronic esters.7
We recently reported8 on the role of the X ligand in the mechanism of the LCuX catalyzed azide-alkyne cycloaddition (CuAAC reaction) [L = cyclic (alkyl)(amino)carbene].9,10 Herein we show that these results allow for the rational design of copper catalysts that selectively promote the dehydrogenative borylation of terminal alkynes with pinacolborane.
In the above mentioned study, we found that in the presence of triethylamine, LCuOTf reacts with terminal alkynes to give the catalytically active σ,π-bis(copper) acetylides A,8,11 along with ammonium triflate (Scheme 1). We hypothesized that in dinuclear complexes A, the triple bond is protected which should prevent the classical hydroboration reaction leading to alkenyl boronic esters. Instead, the highly polarized copper–carbon bond could undergo a σ-bond metathesis with pinacolborane (TS) to afford the desired alkynyl boronic ester B, as well as the copper hydride C. The latter should react with triethylammonium triflate to regenerate LCuOTf and triethylamine with the elimination of dihydrogen.
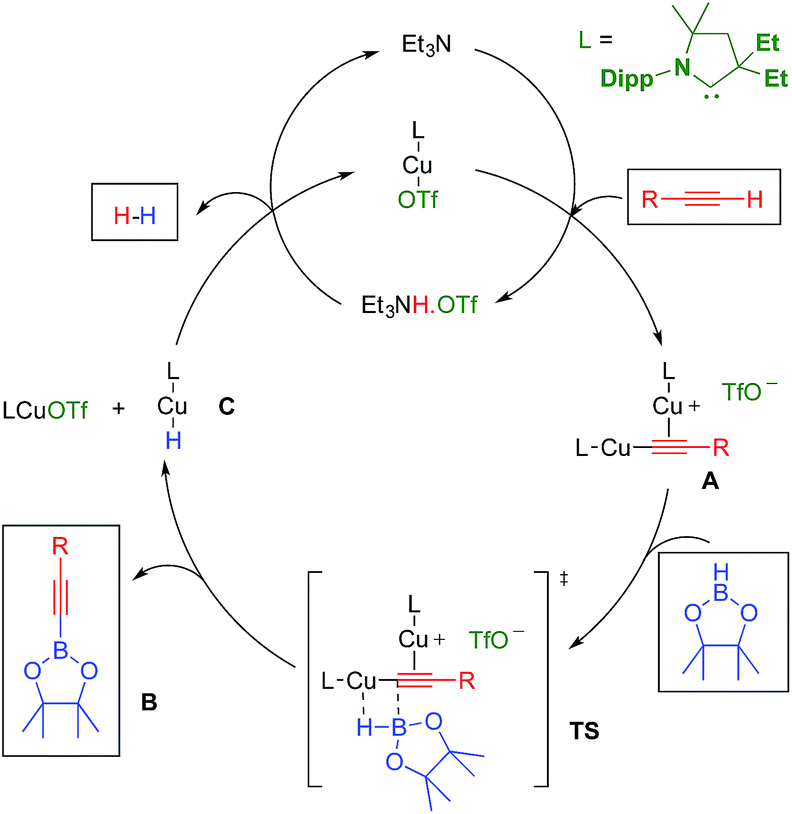 | ||
| Scheme 1 Hypothetical mechanism for the LCuOTf induced dehydrogenative borylation of terminal alkynes. | ||
We first checked that in the absence of catalyst no reaction occurred between p-tolylacetylene 1a and pinacolborane in C6D6 at room temperature for 2 hours (Table 1, entry 1). In the presence of 1 mol% of L1CuOTf, no significant reaction occurred either because of the difficulty of the triflate to deprotonate the alkyne8 (entry 2). In order to promote the deprotonation of the alkyne, 1 mol% of Et3N was added which resulted in immediate hydrogen evolution (as characterized by a singlet at 4.47 ppm in 1H NMR). The major product was the alkynyl boronic ester B1 (48%), but the alkenyl boronic ester D1 (11%), and the styrene derivative E1 (7%) were also formed (entry 3).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Base (mol%) | Solvent | Conc. (M) | 1a (%) | B1 (%) | D1 (%) | E1 (%) |
a Reactions were carried out in a test tube for 2 h at RT under an argon atmosphere using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture (0.69 mmol) of p-tolylacetylene and pinacolborane.
b Measured by NMR using 1,4-dioxane as an internal standard. :1 mixture (0.69 mmol) of p-tolylacetylene and pinacolborane.
b Measured by NMR using 1,4-dioxane as an internal standard.
|
||||||||
| 1 | — | — | C6D6 | 1.4 | 100 | 0 | 0 | 0 |
| 2 | L1CuOTf (1) | — | C6D6 | 1.4 | 86 | 0 | 6 | 6 |
| 3 | L1CuOTf (1) | Et3N (1) | C6D6 | 1.4 | 14 | 48 | 11 | 7 |
| 4 | CuOTf | Et3N (1) | C6D6 | 1.4 | 100 | 0 | 0 | 0 |
| 5 | L1CuOTf (1) | Et3N (2) | C6D6 | 1.4 | 1 | 70 | 14 | 12 |
| 6 | L1CuOTf (1) | Et3N (2) | CD2Cl2 | 1.4 | 12 | 42 | 6 | 26 |
| 7 | L1CuOTf (1) | Et3N (2) | THF-d8 | 1.4 | 5 | 64 | 12 | 17 |
| 8 | L1CuOTf (1) | Et3N (2) | CD3CN | 1.4 | 18 | 38 | 0 | 6 |
| 9 | L1CuOTf (1) | iPrNH2 (2) | C6D6 | 1.4 | 67 | 5 | 12 | 10 |
| 10 | L1CuOTf (1) | iPr2NH (2) | C6D6 | 1.4 | 47 | 11 | 13 | 7 |
| 11 | L1CuOTf (1) | iPr2NEt (2) | C6D6 | 1.4 | 10 | 28 | 45 | 3 |
| 12 | L1CuOTf (1) | BnNEt2 (2) | C6D6 | 1.4 | 14 | 53 | 8 | 7 |
| 13 | L1CuOTf (1) | DABCO (2) | C6D6 | 1.4 | 18 | 60 | 1 | 7 |
| 14 | L1CuOTf (0.25) | Et3N (0.5) | C6D6 | 1.4 | 37 | 36 | 15 | 6 |
| 15 | L1CuOTf (0.5) | Et3N (1) | C6D6 | 1.4 | 20 | 54 | 15 | 9 |
| 16 | L1CuOTf (2.5) | Et3N (5) | C6D6 | 1.4 | 4 | 83 | 4 | 7 |
| 17 | L 1 CuOTf (2.5) | Et 3 N (5) | C 6 D 6 | 0.1 | 1 | 98 | 0 | 1 |
| 18 | L2CuOTf (2.5) | Et3N (5) | C6D6 | 0.1 | 0 | 96 | 0 | 4 |
| 19 | L3CuOTf (2.5) | Et3N (5) | C6D6 | 0.1 | 0 | 92 | 0 | 8 |
Note that no reaction occurred under ligand-free conditions (entry 4). Encouraged by these preliminary results, we further optimized the dehydrogenative borylation leading to B1. We found that a two-fold excess of Et3N with respect to (CAAC) CuOTf was beneficial (entry 5). By screening solvents (Entries 5–8) and base additives (Entries 9–13), we identified benzene and Et3N as being most appropriate for the reaction. When we increased the catalyst loading to 2.5 mol% (Entries 14–16), and decreased the concentration of the solution from 1.4 to 0.1 mol L−1 (entry 17) there was quantitative formation of the desired alkynyl boronic ester B1 with excellent selectivity within two hours at room temperature. Notably, substitution of ligand L1 for the more bulky menthyl CAAC L2 or even IPr-NHC12L3 gave comparable results.
The scope of the dehydrogenative borylation reaction was then studied at room temperature in a benzene solution (0.1 M) using a stoichiometric mixture of alkyne and borane (1.82 mmol), 5 mol% of Et3N and 2.5 mol% of L1CuOTf (Scheme 2). This methodology is readily applicable to a broad range of terminal alkynes bearing functionalities such as OMe, CN, F, Cl, TMS and CO2Me. It is worth noting that electron-rich terminal alkynes require longer reaction times (12 h instead of 2 h) (B11–15). Alkynyl boronic esters B1–15 were isolated in good to excellent yields via filtration through a short plug of dry neutral alumina using pentane as the eluent. This straightforward protocol allows for gram-scale synthesis, as shown for B3.
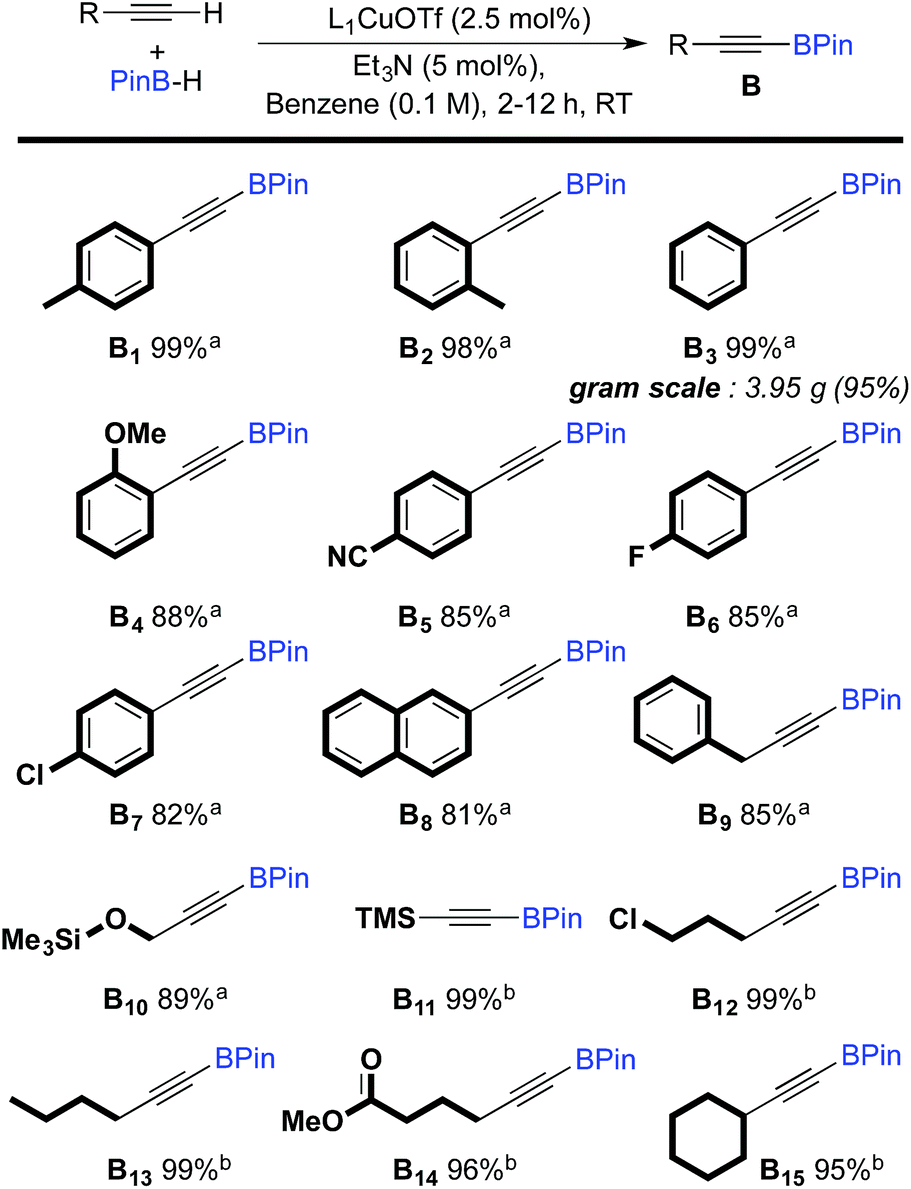 | ||
| Scheme 2 Scope of the dehydrogenative borylation of terminal alkynes. [a] Reaction time 2 h. [b] Reaction time 12 h. | ||
With these results in hand, we performed a set of experiments in order to verify our mechanistic hypothesis. When the 2.5 mol% mononuclear complex F was used, with or without Et3N, no traces of the dehydrogenative borylation product B1 were observed, instead the hydroboration product D1 was quantitatively formed (Fig. 1 and Scheme 3) (see also the kinetic profile in the ESI†). In marked contrast, when 2.5 mol% Et3NH·OTf was added as a proton source, we observed the rapid formation of B1, and the kinetic profile was comparable with those obtained using our standard catalytic conditions (LCuOTf/Et3N) or the bis(copper) acetylide A. Since we already proved that LCuOTf/Et3N reacts with terminal alkynes to afford the dinuclear species A,8 we verified that similarly complex F reacts with Et3NH·OTf to give A. These experiments as a whole strongly suggest that the dinuclear complex A is pivotal in the dehydrogenative process.
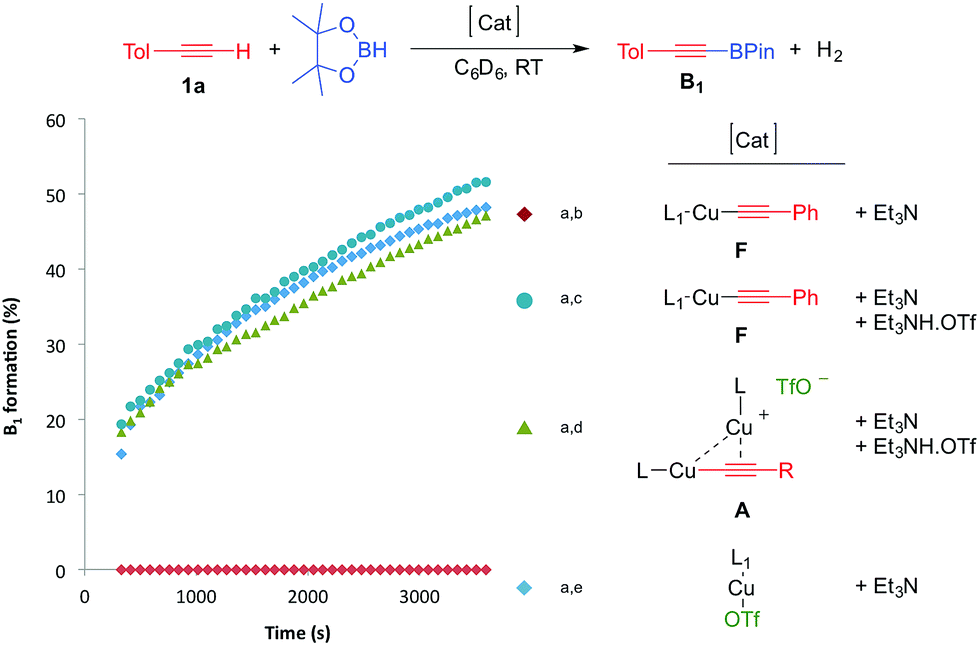 | ||
| Fig. 1 Kinetic profiles of the formation of B1 using various catalytic systems. [a] Reactions were carried out in a J-Young NMR tube at RT under an argon atmosphere using a 1:1 mixture (0.45 mmol) of p-tolylacetylene and pinacolborane in 1 mL of C6D6. [b] 2.5 mol% of F and 5 mol% Et3N; no trace of B1 was observed, instead D1 was obtained quantitatively. [c] 2.5 mol% of F, Et3N and Et3NH·OTf. [d] 2.5 mol% of A, 3.75 mol% Et3N and 1.25 mol% Et3NH·OTf. [e] 2.5 mol% of L1CuOTf and 5 mol% of Et3N. | ||
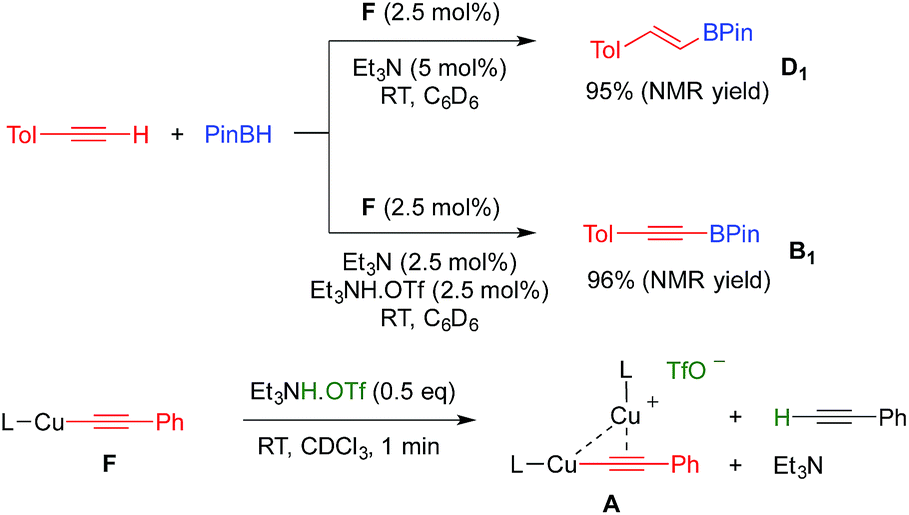 | ||
| Scheme 3 Evidence for the pivotal role of the dinuclear copper complex A. | ||
The other important species in our postulated mechanism is the copper hydride C, a type of complex that has been proposed to play a major role in a number of catalytic transformations.13 While there is still no report of a monomeric mono-ligated Cu hydride,14 a range of dimeric species have been described by us and others.15 An indication of L1CuH formation in this process is the observation of a small amount of styrene derivatives E in our experiments (Table 1). Indeed, copper hydrides are known to undergo 1,2-addition across alkynes to generate copper vinyl complexes, which by protonolysis give alkenes.15 Consistent with this hypothesis, a catalytic experiment using deuterium labelled phenyl acetylene under our optimized conditions, but in CD2Cl2 instead of C6D6 (Table 1, entry 6), afforded B3 and Styrene-D (75% D-incorporation) (Scheme 4).
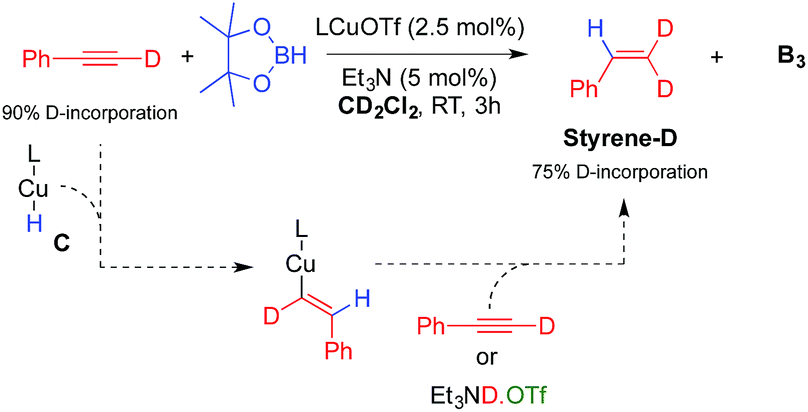 | ||
| Scheme 4 Evidence for the formation of the copper hydride C. | ||
Conclusions
In summary, we have disclosed the first example of a highly selective dehydrogenative borylation of terminal alkynes with pinacolborane, using an inexpensive metal center supported by readily accessible ligands.16 Preliminary mechanistic studies suggest the pivotal role of a σ,π-bis(copper) acetylide A and a copper hydride C.Acknowledgements
The authors gratefully acknowledge financial support from the DOE (DE-FG02-13ER16370).Notes and references
- J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864 CrossRef CAS PubMed; A. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412 RSC.
- H. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995 CrossRef CAS PubMed.
- J. Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka Jr. and M. R. Smith III, Science, 2002, 295, 305 CrossRef CAS PubMed.
- J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC; I. A. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed.
- C. I. Lee, J. Zhou and O. V. Ozerov, J. Am. Chem. Soc., 2013, 135, 3560 CrossRef CAS PubMed; C. I. Lee, N. A. Hirscher, J. Zhou, N. Bhuvanesh and O. V. Ozerov, Organometallics, 2015, 34, 3099 CrossRef; C. I. Lee, W. C. Shih, J. Zhou, J. H. Reibenspies and O. V. Ozerov, Angew. Chem., Int. Ed., 2015, 54, 14003 CrossRef PubMed; C. I. Lee, J. C. DeMott, C. J. Pell, A. Christopher, J. Zhou, N. Bhuvanesh and O. V. Ozerov, Chem. Sci., 2015, 6, 6572 RSC; C. J. Pell and O. V. Ozerov, Inorg. Chem. Front., 2015, 2, 720 RSC.
- T. Tsuchimoto, H. Utsugi, T. Sugiura and S. Horio, Adv. Synth. Catal., 2015, 357, 77 CrossRef CAS.
- R. Barbeyron, E. Benedetti, J. Cossy, J. J. Vasseur, S. Arseniyadis and M. Smietana, Tetrahedron, 2014, 70, 8431 CrossRef CAS.
- L. Jin, D. R. Tolentino, M. Melaimi and G. Bertrand, Sci. Adv., 2015, 1, e1500304 Search PubMed; L. Jin, E. A. Romero, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 15696 CrossRef CAS PubMed.
- For reviews on CAACs, see: M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810 CrossRef CAS PubMed; D. Martin, M. Melaimi, M. Soleilhavoup and G. Bertrand, Organometallics, 2011, 30, 5304 CrossRef PubMed; M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef PubMed.
- For the synthesis of CAACs, see: V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 CrossRef CAS PubMed; R. Jazzar, R. D. Dewhurst, J. B. Bourg, B. Donnadieu, Y. Canac and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 2899 CrossRef PubMed; R. Jazzar, J. B. Bourg, R. D. Dewhurst, B. Donnadieu and G. Bertrand, J. Org. Chem., 2007, 72, 3492 CrossRef PubMed.
- For other mechanistic studies, involving the transient formation of σ,π-bis(copper) acetylides, see: V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210 CrossRef CAS PubMed; F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210 CrossRef PubMed; M. Ahlquist and V. V. Fokin, Organometallics, 2007, 26, 4389 CrossRef; B. F. Straub, Chem. Commun., 2007, 3868 RSC; C. Nolte, P. Mayer and B. F. Straub, Angew. Chem., Int. Ed., 2007, 46, 2101 CrossRef PubMed; A. Makarem, R. Berg, F. Rominger and B. F. Straub, Angew. Chem., Int. Ed., 2015, 54, 7431 CrossRef PubMed; B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457 CrossRef PubMed.
- A. J. Arduengo, R. Krafczyk and R. Schmutzler, Tetrahedron, 1999, 55, 14523 CrossRef CAS.
- For reviews, see: J. D. Egbert, C. S. J. Cazin and S. P. Nolan, Catal. Sci. Technol., 2013, 3, 912 Search PubMed; C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916 CrossRef CAS PubMed; B. H. Lipshutz, Synlett, 2009, 509 CrossRef.
- For an example of diligated copper hydride, see: B. H. Lipshutz and B. A. Frieman, Angew. Chem., Int. Ed., 2005, 44, 6345 CrossRef CAS PubMed.
- G. D. Frey, B. Donnadieu, M. Soleilhavoup and G. Bertrand, Chem.–Asian J., 2011, 6, 402 CrossRef CAS PubMed; N. Cox, H. Dang, A. M. Whittaker and G. Lalic, Tetrahedron, 2014, 70, 4219 CrossRef; M. R. Uehling, R. P. Rucker and G. Lalic, J. Am. Chem. Soc., 2014, 136, 8799 CrossRef PubMed; A. M. Suess, M. R. Uehling, W. Kaminsky and G. Lalic, J. Am. Chem. Soc., 2015, 137, 7747 CrossRef PubMed; N. P. Mankad, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 3369 CrossRef; K. Semba, T. Fujihara, T. H. Xu, J. Terao and Y. Tsuji, Adv. Synth. Catal., 2012, 354, 1542 CrossRef; A. J. Jordan, C. M. Wyss, J. Bacsa and J. P. Sadighi, Organometallics, 2016, 35, 613 CrossRef; A. M. Suess and G. Lalic, Synlett, 2016, 27, 1165 CrossRef; L. R. Collins, I. M. Riddlestone, M. F. Mahon and M. K. Whittlesey, Chem.–Eur. J., 2015, 21, 14075 CrossRef PubMed; C. M. Wyss, B. K. Tate, J. Bacsa, T. G. Gray and J. P. Sadighi, Angew. Chem., Int. Ed., 2013, 52, 12920 CrossRef PubMed.
- CAAC and NHC precursors are commercially available.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02668k |
| This journal is © The Royal Society of Chemistry 2017 |