
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unified synthesis of mono/bis-arylated phenols via RhIII-catalyzed dehydrogenative coupling†
Qian
Wu
a,
Ying
Chen
a,
Dingyuan
Yan
b,
Muyue
Zhang
b,
Yi
Lu
b,
Wei-Yin
Sun
b and
Jing
Zhao
 *ab
*ab
aShenzhen Key Lab of Nano-Micro Material Research, School of Chemical Biology and Biotechnology, Shenzhen Graduate School of Peking University, Shenzhen, 518055, China. E-mail: jingzhao@nju.edu.cn
bState Key Laboratory of Coordination Chemistry, Institute of Chemistry and BioMedical Sciences, School of Chemistry and Chemical Engineering, Collaborative Innovation Center of Chemistry for Life Sciences, Nanjing University, Nanjing 210093, China
First published on 3rd August 2016
Abstract
2,6-Bis-arylated phenols are rarely reported and are synthetically challenging. Directed C–H functionalization reactions, using a directing group (DG), might provide a convenient solution to their synthesis. However, this strategy usually results in partial cleavage of the directing group, preventing further/second C–H activation cascades. Herein we report a general strategy that allows for the precise control of the oxidation pathways so that directing groups can be either preserved or cleaved. We found that N-phenoxyacetamides could undergo ortho-arylation reactions with or without an external oxidant, yielding products with different oxidation states, notably the rare bis-arylated phenols. Notably, a unique rhodacycle intermediate was isolated, characterized by X-ray crystallography, and confirmed to be an active catalyst. Switching between internal and external oxidation could be a general strategy in diverse directed C–H functionalization reactions to realize bis-functionalized products.
Introduction
Transition metal-catalysed C–H activation reactions directed by a coordinative group have become one of the most efficient and straightforward synthetic strategies for the direct functionalization of inert C–H bonds.1 The role of a DG extends beyond a simple anchor for the selective cleavage of a neighbouring C–H bond. DGs can often undergo further in situ condensation reactions that yield products of great structural diversity.2 More recently, DGs containing an oxidative N–O or N–N bond were introduced to replace the required external oxidant to render the C–H functionalization reactions redox-neutral.2d,3 Among them, oxyacetamide (O–NHAc) is one of the most versatile functionalities for directed C–H functionalization reaction cascades (Fig. 1a). The reactions involving this unique DG can be classified into two types: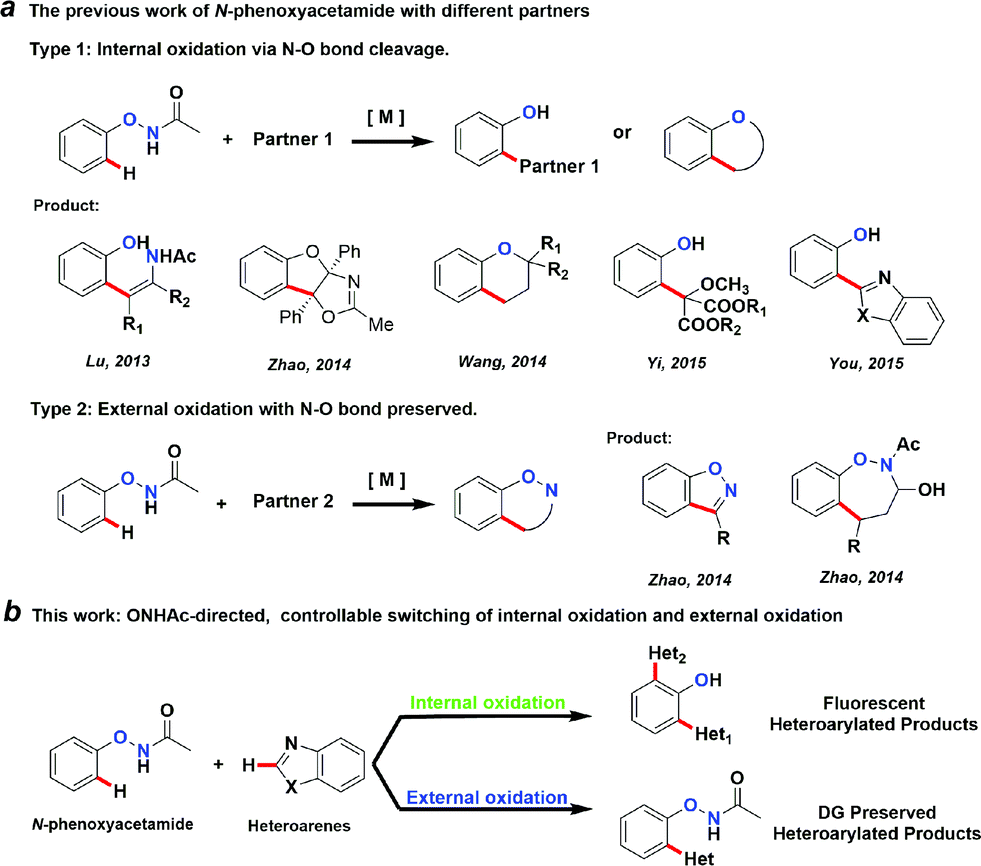 | ||
| Fig. 1 O–NHAc group-directed C–H activation reactions. | ||
Type 1: internal oxidation with N–O bond cleavage. Lu’s group first reported O–NHAc as a superb directing group for redox-neutral olefination reactions of phenol derivatives by coupling with alkynes3l and alkenes.3n Subsequently, our group reported a number of reaction cascades using rhodium catalysis in which complexed heterocyclic scaffolds were synthesized in one step from N-phenoxyacetamides and alkynes with up to a quadruple cascade.2d Wang and Yi made impressive progress on O–NHAc-directed C–H reactions using cyclopropenes3t and carbenoids3x as coupling partners, respectively. In these reactions, the N–O bond of the DG was cleaved and serves as an internal oxidant, leading to the corresponding phenol products. Notably, You’s group reported a pioneering C–H/C–H cross-coupling between N-phenoxyacetamides and heteroarenes through a traceless directing strategy to synthesize the highly functionalized 2-(2-hydroxyphenyl)azoles, which are novel optoelectronic materials.3v
Type 2: external oxidation with preservation of the N–O bond. In contrast to type 1 reactions, we recently described a series of oxidative C–H functionalization reactions. In the presence of a stoichiometric external oxidant, N-phenoxyacetamides could react with aldehydes or α,β-unsaturated aldehydes using palladium4 and rhodium5 catalysis. In these reactions, the N–O bond of the DG remained intact after the reaction and was retained in the products.
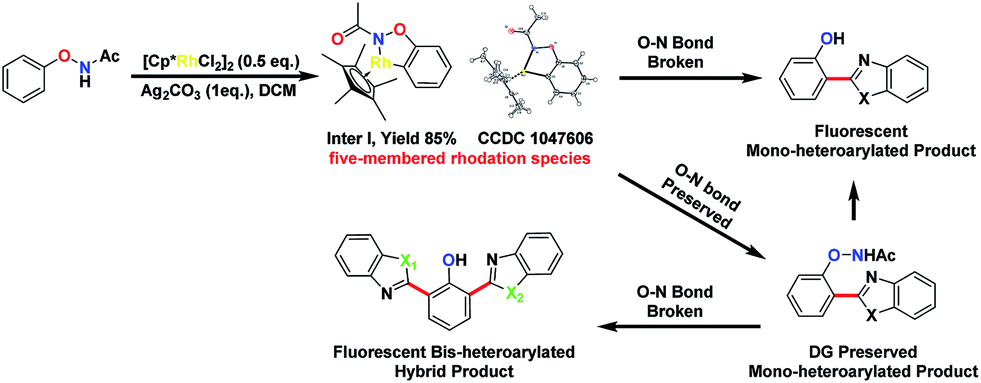
We thought that the unification of type 1 and type 2 into a single reaction would allow general access to products with different oxidation states with controlling the cleavage of the N–O bond. For example, based on an isolated rhodacycle intermediate Inter I6 from our previous studies,5 the O–NHAc-directed cross dehydrogenative coupling reactions with simple heteroarenes would be an attractive strategy to access diverse heteroarylated phenol scaffolds (Scheme 1).1b,d,7–9
 | ||
| Scheme 1 Proposed transformations based on an isolated organometallic intermediate. | ||
Heteroarylated phenols, such as 2-(2-hydroxyphenyl)benzothiazole (HBT) and 2-(2-hydroxyphenyl)benzoxazole (HBO), possess high fluorescence quantum yields and a large Stokes shift due to the excited-state intramolecular proton transfer (ESIPT) effect, and are widely used in various fluorescent probes and related fields.10 If the redox activity of the N–O bond could be tuned using a proper external oxidant, switching between type 1 and 2 reactions could be enabled, and up to three coupling products could be obtained selectively in a unified fashion (Scheme 1). Based on this design, we set out to explore the combination of different reaction parameters from phenoxyacetamides to achieve a unified strategy.
Results and discussion
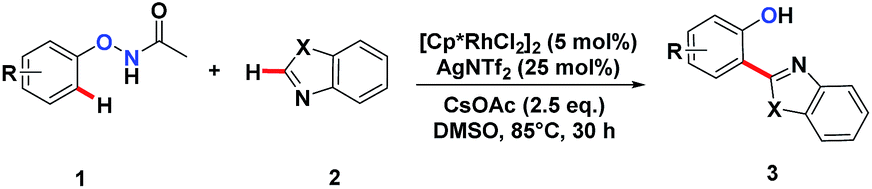
We embarked on our design by investigating a reaction between N-phenoxyacetamide (1a) and benzothiazole (2a). As expected, the desired ortho-heteroarylated phenol (3aa) was obtained as the main product in the absence of any external oxidant. After careful condition optimization, [Cp*RhCl2]2 (5 mol%), AgNTf2 (25 mol%), and CsOAc (2.5 eq.) in 1 mL DMSO at 85 °C for 30 h under an N2 atmosphere was found as the best reaction conditions for 3aa, affording an 85% isolated yield. A non-coordinating counter ion (SbF6, NTf2, CO3, OTf) was essential for the catalytic activity of Rh.3d,k,t,11A series of substituted N-phenoxyacetamides were examined for substrate scope (Table 1). For methyl-substituted N-phenoxyacetamides, the para-methyl substrate 1b gave the corresponding phenol product 3ba in 80% yield. The meta-methyl analogue 1c afforded a 72% yield, and the ortho-methyl derivative 1d yielded 76% of the product. The yield for meta-OMe N-phenoxyacetamide 1j was noticeably less. No obvious electronic effect was observed. Substrates bearing either electron-donating (phenyl and methoxy) or electron-withdrawing groups (ester), delivered their corresponding heteroaryl phenols in comparable yields.

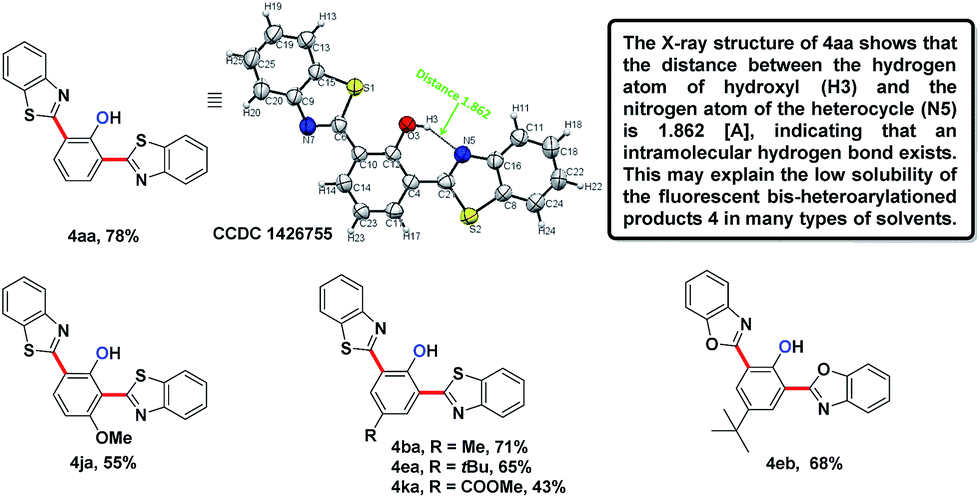
In the reaction of the mono-heteroarylated product 3aa, a different product with a stronger fluorescence was isolated in a small quantity. This double arylation product was determined as the bis-heteroarylated phenol by single crystal X-ray crystallography and it became predominant when a silver salt was introduced as the external oxidant along with 2.5 eq. of benzothiazole. Treating 1a and benzothiazole with [Cp*RhCl2]2 (5 mol%) and PivOH (3 eq.) in THF at room temperature for 20 h afforded 4aa in 78% isolated yield. Highly fluorescent bis-heteroarylated phenols were obtained in moderate to high yields under the optimized reaction conditions (Table 2). Para- and meta-substituents were well tolerated (4ba, 4ea, 4ka and 4ja). In addition, benzoxazoles reacted with comparable efficiency (4eb).

Interestingly, subjecting product 3aa and benzothiazole to various Rh catalysis conditions did not yield 4aa, with or without silver oxidants. This result suggested that 3aa was not one of the intermediates leading to 4aa. The formation of the bis-heteroarylated phenol arose from the type 2 product B (Fig. 2). Based on this result, we decided to target this type 2 product. Despite previous reports that the N–O bond was always broken in the presence of a silver oxidant, we were excited to isolate a small amount of the mono-heteroarylated N-phenoxyacetamide 5aa (8% yield) by replacing PivOH with excess triethylamine (10 eq.). The structure was unambiguously confirmed by NMR, HRMS and single crystal X-ray crystallography. Further condition optimization revealed that [Cp*RhCl2]2 (5 mol%), AgF (3 eq.) and benzothiazole (2 eq.) in a solvent mixture of iPrOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N,N-diisopropylethylamine (DIPEA):H2O = 1:1:0.1 at room temperature for 18 h afforded 5aa in a 20% yield. The starting material N-phenoxyacetamide 1aa remained largely intact. Adding AgF in small portions (2 eq. upon mixing and 1 eq. every six hours for 4 eq. total) improved the yield of 5aa to 68%.
:N,N-diisopropylethylamine (DIPEA):H2O = 1:1:0.1 at room temperature for 18 h afforded 5aa in a 20% yield. The starting material N-phenoxyacetamide 1aa remained largely intact. Adding AgF in small portions (2 eq. upon mixing and 1 eq. every six hours for 4 eq. total) improved the yield of 5aa to 68%.
The substrate scope for this type 2 heteroarylation reaction was explored using the optimized conditions. para-Substituted N-aryloxyacetamides afforded the corresponding products in moderate yields (5ba, 5ea, 5ga, 5ha, 5ia, 5ka in Table 3). The yield for the para-COOMe substrate was particularly high (72%), suggesting that an electron-withdrawing group favoured the type 2 reaction. A lower yield was observed for those carrying a meta-substituent. meta-Methyl and meta-methoxy substrates gave the desired products in 30% and 35% yield, respectively (5ca, 5ja). Both substituted benzothiazoles and benzoxazoles worked equally well (5ac, 5kd).


To explore the formation of bis-heteroarylated phenols, phenol 5aa was subjected to the rhodium catalyst in the presence of an external oxidant AgF (2 eq.) and 4aa was obtained nearly quantitatively (Scheme 2, eqn (1)). Encouraged by this result, we attempted to synthesize more interesting hybrid bis-heteroarylated phenols. Gratifyingly, the hybrid product 6a was obtained in high yield (Scheme 2, eqn (2)). This type 2–type 1 sequence enhanced the structural diversity of these fluorescent bis-heteroarylated phenols and provided a general strategy for devising better fluorescent probes.
 | ||
| Scheme 2 Route to novel fluorescent bis-heteroarylated hybrid products. | ||
To understand the mechanism of the O–NHAc-directed C–H activation reactions, we obtained the five-membered rhodation intermediate Inter I in 85% yield. The structure was confirmed by NMR spectroscopy, HRMS, and X-ray crystallography (Fig. S1†). When Inter I was used as the catalyst, our three reaction conditions led to three expected heteroarylated products, suggesting the rhodation species Inter I was the active intermediate (Scheme 3).
 | ||
| Scheme 3 The confirmation of catalytically active species Inter I. | ||
You’s group had demonstrated that the reaction might start from the cyclometalation of N-aryloxyacetamide rather than the heteroarene by ortho-deuterium labelling experiments. You’s group reported that the KIE value was 1.04 for the N-phenoxyacetamide substrate, while the KIE value was 2.89 for the benzoxazole substrate, suggesting that the rate-limiting step might involve the C–H bond breaking of heteroarenes.3v,12 Thus, we proposed a mechanism with two pathways of internal oxidation and external oxidation to afford different products (Fig. S1†). First, after the generation of the real [RhIII] catalyst by the anion exchange of [NTf2]−, a five-membered rhodation species Inter I formed, which was demonstrated as an active intermediate. Second, the heteroarene was inserted to give a heteroaryl–RhIII–phenyl intermediate, which would undergo reductive elimination to a RhI complex (Inter III in Fig. S1†). Third, two pathways are possible depending on the presence of the external oxidants: (1) in the absence of the external oxidants, the RhI complex Inter III would undergo an internally oxidizing pathway to form a RhIII complex (Inter IV in Fig. S1†) with O–N bond cleavage.3l,n Protonation would afford the mono-arylated product 3. (2) In the presence of the external oxidants, the RhI complex Inter III could undergo an external oxidation pathway to form the O–N bond-preserved mono-arylated product 5. As the DG was retained in product 5, it could subsequently react with another heteroarene to afford the bis-arylated products 4.
Application
The fluorescent properties of mono- and bis-heteroarylated phenols were evaluated (Fig. 2). Considering the solvent effect on ESIPT, a series of common organic solvents was screened, and dichloromethane was chosen for measurement (Fig. S6†).13 The fluorescence spectra of mono-substituted HBTs showed a strong ESIPT emission band in the region of 480–540 nm. Both the λmax and the intensity of the absorption were affected by the substituents. A methyl group in the para- and meta-positions caused a bathochromic shift (∼15 nm), while the ortho-counterpart showed no obvious change. Halogen substituents (–F, Cl, Br) led to increased fluorescence intensity with small red shifts (∼10 nm). Products with an extra phenyl group resulted in the highest red shift (∼25 nm), and the ester group caused the highest blue shift. By contrast, the bis-substituted products demonstrated significant bathochromic shifts with strong yellow fluorescence.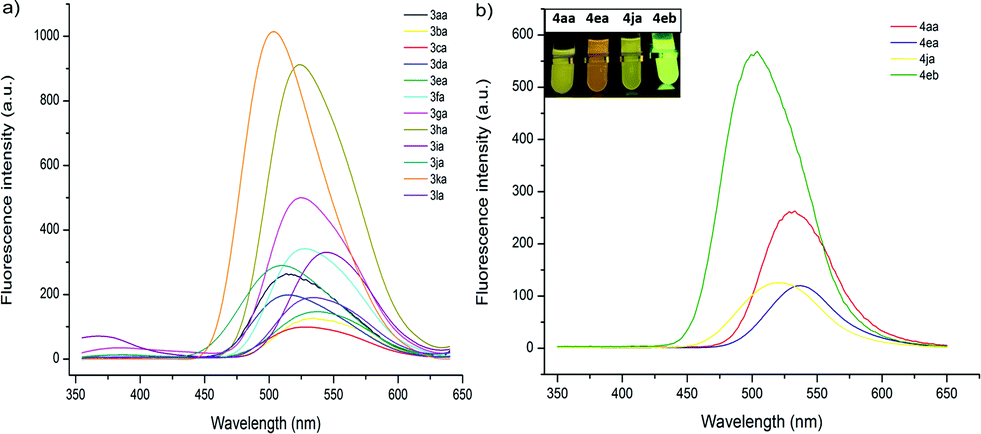 | ||
| Fig. 2 Fluorescence properties of 3 and 4. (a) Fluorescence spectra of mono-substituted HBTs in DCM (2 × 10−6 mol L−1, λex = 330 nm). (b) Fluorescence spectra of bis-substituted products in DCM (2 × 10−6 mol L−1, λex = 360 nm). | ||
Considering that the fluorescence of the DG-preserved products 5 was effectively blocked due to the O–N bond, a small molecule that can cleave the O–N bond would have great potential for developing fluorescent probes.
Conclusions
In summary, we developed a unified strategy for cross dehydrogenative coupling reactions between arenes and heteroarenes. Internal and external oxidation could be controlled using N–O bond cleavage or a silver oxidant. Mono- and the rarely reported bis-arylated phenol derivatives of different oxidation states were prepared in one step. This convenient, one-step synthesis of a series of DG-preserved products could facilitate the continued generation of a library of fluorescent probes. Switching between internal and external oxidation could be a general strategy in other directed C–H functionalization reactions to realize the bis-functionalized products.Acknowledgements
Financial support was provided by the Shenzhen Government (JCYJ20150626110741712, JCYJ20150331100341865 and JCYJ20140627145302109), the Guangdong Government (S20120011226), the National Science Foundation of China (21332005 and 21571098) and the MOST of China (2014AA020512).Notes and references
- Related work on various directing groups assisted C–H activation: (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (d) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed; (e) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (f) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed.
- (a) C. Wang, H. Chen, Z. Wang, J. Chen and Y. Huang, Angew. Chem., Int. Ed., 2012, 51, 7242 CrossRef CAS PubMed; (b) C. Wang and Y. Huang, Synlett, 2013, 24, 0145 CAS; (c) C. Wang, H. Sun, Y. Fang and Y. Huang, Angew. Chem., Int. Ed., 2013, 125, 5907 CrossRef; (d) Y. Chen, D. Wang, P. Duan, R. Ben, L. Dai, X. Shao, M. Hong, J. Zhao and Y. Huang, Nat. Commun., 2014, 5, 4610 CAS; (e) J. Jayakumar, K. Parthasarathy, Y.-H. Chen, T.-H. Lee, S.-C. Chuang and C.-H. Cheng, Angew. Chem., Int. Ed., 2014, 53, 9889 CrossRef CAS PubMed; (f) J. Kim, S.-W. Park, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2015, 137, 13448 CrossRef CAS PubMed.
- (a) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908 CrossRef CAS PubMed; (b) P. C. Too, Y.-F. Wang and S. Chiba, Org. Lett., 2010, 12, 5688 CrossRef CAS PubMed; (c) L. Ackermann and S. Fenner, Org. Lett., 2011, 13, 6548 CrossRef CAS PubMed; (d) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449 CrossRef CAS PubMed; (e) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846 RSC; (f) F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1977 CrossRef CAS PubMed; (g) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350 CrossRef CAS PubMed; (h) X. Zhang, D. Chen, M. Zhao, J. Zhao, A. Jia and X. Li, Adv. Synth. Catal., 2011, 353, 719 CrossRef CAS; (i) S.-C. Chuang, P. Gandeepan and C.-H. Cheng, Org. Lett., 2013, 15, 5750 CrossRef CAS PubMed; (j) J. R. Huckins, E. A. Bercot, O. R. Thiel, T. L. Hwang and M. M. Bio, J. Am. Chem. Soc., 2013, 135, 14492 CrossRef CAS PubMed; (k) B. Liu, C. Song, C. Sun, S. Zhou and J. Zhu, J. Am. Chem. Soc., 2013, 135, 16625 CrossRef CAS PubMed; (l) G. Liu, Y. Shen, Z. Zhou and X. Lu, Angew. Chem., Int. Ed., 2013, 52, 6033 CrossRef CAS PubMed; (m) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2013, 135, 66 CrossRef CAS PubMed; (n) Y. Shen, G. Liu, Z. Zhou and X. Lu, Org. Lett., 2013, 15, 3366 CrossRef CAS PubMed; (o) D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426 CrossRef CAS PubMed; (p) W. Han, G. Zhang, G. Li and H. Huang, Org. Lett., 2014, 16, 3532 CrossRef CAS PubMed; (q) F. Hu, Y. Xia, F. Ye, Z. Liu, C. Ma, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 1364 CrossRef CAS PubMed; (r) Y. Liang, K. Yu, B. Li, S. Xu, H. Song and B. Wang, Chem. Commun., 2014, 50, 6130 RSC; (s) K. Muralirajan and C.-H. Cheng, Adv. Synth. Catal., 2014, 356, 1571 CrossRef CAS; (t) H. Zhang, K. Wang, B. Wang, H. Yi, F. Hu, C. Li and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 13234 CrossRef CAS PubMed; (u) L. Zheng and R. Hua, Chem.–Eur. J., 2014, 20, 2352 CrossRef CAS PubMed; (v) B. Li, J. Lan, D. Wu and J. You, Angew. Chem., Int. Ed., 2015, 54, 14008 CrossRef CAS PubMed; (w) S. Yu, S. Liu, Y. Lan, B. Wan and X. Li, J. Am. Chem. Soc., 2015, 137, 1623 CrossRef CAS PubMed; (x) J. Zhou, J. Shi, X. Liu, J. Jia, H. Song, H. E. Xu and W. Yi, Chem. Commun., 2015, 51, 5868 RSC.
- P. Duan, Y. Yang, R. Ben, Y. Yan, L. Dai, M. Hong and J. Zhao, Chem. Sci., 2014, 5, 1574 RSC.
- P. Duan, X. Lan, Y. Chen, S. S. Qian, J. J. Li, L. Lu and J. Zhao, Chem. Commun., 2014, 50, 12135 RSC.
- Related work on cyclometalation intermediates of Iridium: J. Zhou, J. Shi, Z. Qi, X. Li, H. E. Xu and W. Yi, ACS Catal., 2015, 5, 6999 CrossRef CAS.
- For Rhodium-catalyzed C–H activation reviews, see: (a) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (b) J. Wencel-Delord, T. Drçge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (c) S. Chiba, Chem. Lett., 2012, 41, 1554 CrossRef CAS; (d) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (e) F. W. Patureau, J. WencelDelord and F. Glorius, Aldrichimica Acta, 2012, 45, 31 CAS; (f) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443 CrossRef CAS; (g) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007 CrossRef CAS PubMed.
- Related work on Rhodium-catalyzed (hetero)arylation of arenes: (a) N. Kuhl, M. N. Hopkinson and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 8230 CrossRef CAS PubMed; (b) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (c) Y. Shang, X. Jie, H. Zhao, P. Hu and W. Su, Org. Lett., 2014, 16, 416 CrossRef CAS PubMed; (d) D. Qin, J. Wang, X. Qin, C. Wang, G. Gao and J. You, Chem. Commun., 2015, 51, 6190 RSC.
- Related work on the direct C–H bond functionalization of 1,3-azoles: (a) X. Zhao, G. Wu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 3296 CrossRef CAS PubMed; (b) T. Yao, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 775 CrossRef CAS PubMed.
- (a) A. Heller and D. L. Williams, J. Phys. Chem., 1970, 74, 4473 CrossRef CAS; (b) R. Hu, J. Feng, D. Hu, S. Wang, S. Li, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2010, 49, 4915 CrossRef CAS PubMed; (c) M. Santra, B. Roy and K. H. Ahn, Org. Lett., 2011, 13, 3422 CrossRef CAS PubMed; (d) J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483 RSC; (e) J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803 RSC; (f) P. Xu, M. Liu, T. Gao, H. Zhang, Z. Li, X. Huang and W. Zeng, Tetrahedron Lett., 2015, 56, 4007 CrossRef CAS.
- (a) N. Guimond and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 12050 CrossRef CAS PubMed; (b) P. M. Boyer, C. P. Roy, J. M. Bielski and J. S. Merola, Inorg. Chim. Acta, 1996, 245, 7 CrossRef CAS.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- (a) Y. H. Kim, S. G. Roh and D. W. Cho, Photochem. Photobiol. Sci., 2010, 9, 722 RSC; (b) R. Wang, D. Liu, K. Xu and J. Li, J. Photochem. Photobiol., A, 2009, 205, 61 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds. CCDC 1047606, 1426755, 1438599 and 1442845. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc03169b |
| This journal is © The Royal Society of Chemistry 2017 |