
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis of a key series of vinblastines modified at C4 that define the importance and surprising trends in activity†
Shouliang
Yang
,
Kuppusamy
Sankar
,
Colin K.
Skepper
,
Timothy J.
Barker
,
John C.
Lukesh III
,
Daniel M.
Brody
,
Manuela M.
Brütsch
and
Dale L.
Boger
*
Department of Chemistry, The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 N. Torrey Pines Road, La Jolla, California 92037, USA. E-mail: boger@scripps.edu
First published on 3rd November 2016
Abstract
The total synthesis and evaluation of a key systematic series of vinblastines that incorporate the first deep-seated changes to the substituent at C4 are detailed. The synthetic approach features an expanded and redefined scope of a 1,3,4-oxadiazole [4 + 2]/[3 + 2] cycloaddition cascade in which electronically mismatched electron-deficient trisubstituted alkenes and unactivated trisubstituted alkenes were found to productively initiate the cycloaddition cascade with tethered electron-deficient 1,3,4-oxadiazoles. Such cycloaddition cascades were used to directly introduce altered C4 substituents, providing the basis for concise total syntheses of a series of C4 modified vindolines and their subsequent single-step incorporation into the corresponding synthetic vinblastines in routes as short as 8–12 steps. Evaluation of the synthetic vinblastines revealed a surprisingly large impact and role of the C4 substituent on activity even though it was previously not thought to intimately interact with the biological target tubulin. Only the introduction of a C4 methyl ester, a constitutional isomer of vinblastine in which the carbonyl carbon and ester oxygen of the C4 acetate are transposed, provided a synthetic vinblastine that matched the potency of the natural product. In contrast, even introduction of a C4 acetamide or N-methyl carboxamide, which incorporate single heavy atom exchanges (amide NH for ester oxygen) in vinblastine or the C4 methyl ester, provided compounds that were ≥10-fold less active than vinblastine. Other C4 acetate replacements, including a C4 amine, carboxylic acid, hydroxymethyl or acetoxymethyl group, led to even greater reductions in potency. Even replacement of the C4 acetoxy group or its equally active C4 methyl ester with an ethyl or isopropyl ester led to 10-fold or more reductions in activity. These remarkable trends in activity, which correlate with relative tubulin binding affinities, retrospectively may be ascribed to the role the substituent serves as a H-bond acceptor for α-tubulin Lys336 and Asn329 side chains at a site less tolerant of a H-bond donor, placing the methyl group of the C4 acetate or C4 methyl ester in a spatially restricted and well-defined hydrophobic half pocket created by a surrounding well-ordered loop. This remarkable impact of the C4 substituent, its stringency, and even the magnitude of its effect are extraordinary, and indicate that its presence was selected in Nature to enhance the effects of vinblastine and related natural products.
Introduction
The most widely recognized members of the Vinca alkaloids are the antitumor drugs vinblastine (1) and vincristine, originally isolated from Catharanthus roseus (L.) G. Don1,2 (Fig. 1). Because of this intrinsic importance, their complex structures, and their role in the discovery of an important antineoplastic mechanism of action,3 they have attracted extensive synthetic and mechanistic efforts since their discovery.4–7 In our own efforts first targeting the natural products, we reported the discovery of a powerful intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of 1,3,4-oxadiazoles8,9 inspired by the structure of vindoline, the lower subunit of vinblastine. A concise total synthesis of (−)- and ent-(+)-vindoline10 was developed in which this reaction cascade was used to assemble the full pentacyclic skeleton of 2 with incorporation of all necessary functionality and stereochemistry in a single key step. The extension of this methodology to the preparation of a series of related natural products,10–15 its applications in the total syntheses of additional alkaloid natural products,16–19 and its use in the subsequent development of an asymmetric total synthesis20 of vindoline followed shortly thereafter. This work along with the use of a biomimetic Fe(III)-promoted coupling of vindoline with catharanthine21,22 and the development of a subsequent in situ Fe(III)/NaBH4-mediated free radical alkene oxidation for C20′-alcohol introduction22–24 allowed for the single-step incorporation of vindoline and its analogues into total syntheses of vinblastine, related natural products including vincristine, and key analogues in routes as short as 8–12 steps. In these latter efforts, we found that subtle modifications at C4 of vinblastine led to surprisingly significant changes in biological activity (Fig. 1).15,22 The origin of these substantial effects was not clear and must be subtle since substituents at this site were not thought to intimately interact with the biological target tubulin, rather they are believed to form an interface with solvent in the vinblastine bound complex (Fig. 1).25 As a result, we initiated efforts to more clearly define the impact of the C4 substituent by examination of a series of systematic deep-seated modifications at this site complimentary to early semi-synthetic modifications that examined alternative acyl groups (vs. Ac).5 Because hydrolysis of the C4 acetate represents the major in vivo metabolic reaction of the clinical drugs and since the corresponding C4 alcohol is often used as a productive but labile functionalization or bioconjugation site,4c,5 the results of these studies were anticipated to be of special interest.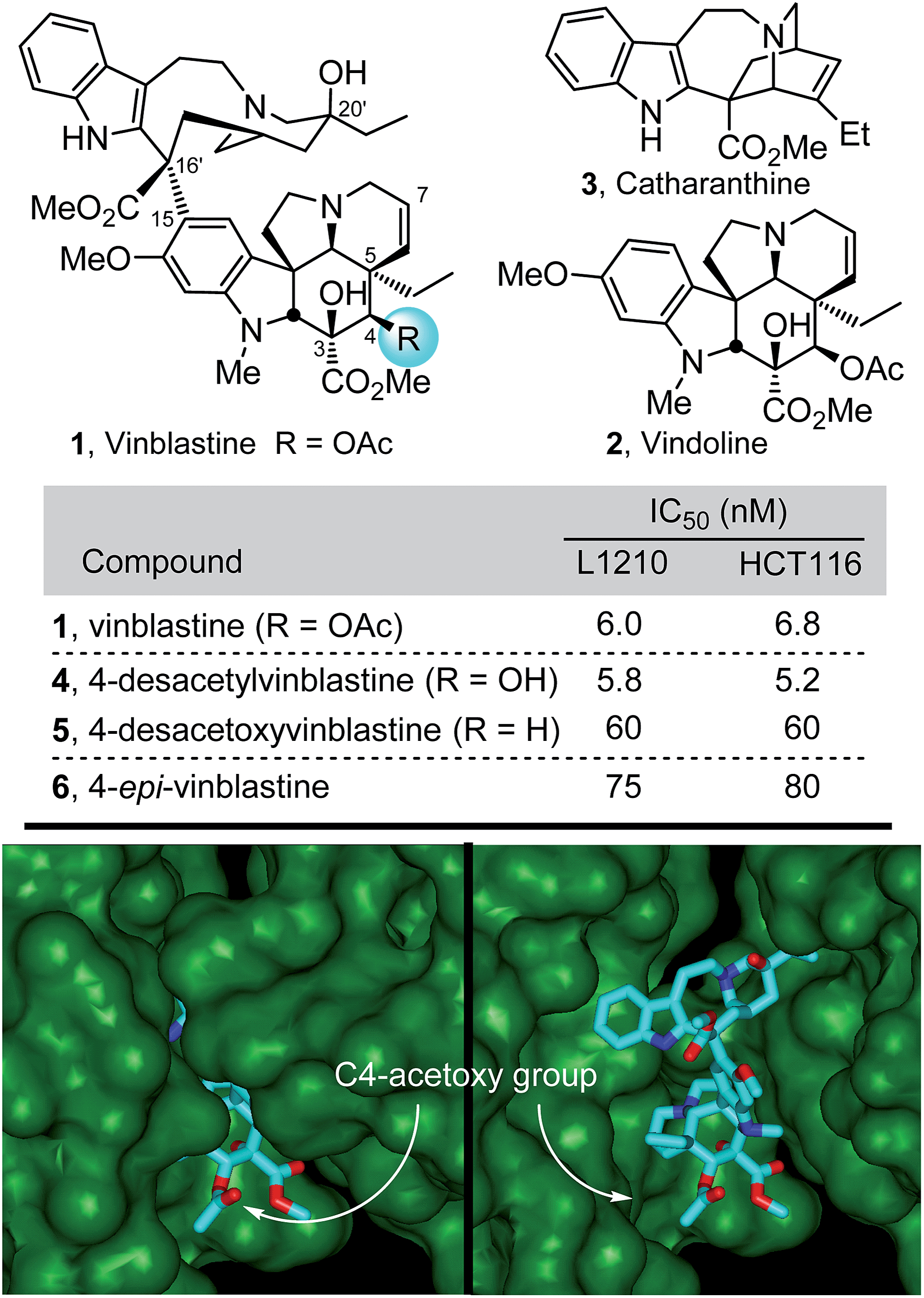 | ||
| Fig. 1 Top: Natural product structures and cell growth inhibition data. Bottom: X-ray co-crystal structure of tubulin-bound vinblastine25a (pdb 1Z2B) highlighting the solvent exposed C4 acetoxy group at the tubulin head-to-tail dimer–dimer interface where vinblastine binds (left) and site of binding with top of proteins removed to visualize bound vinblastine (right). | ||
Results and discussion
Like vinblastine itself, the targeted modifications were anticipated to be available by synthesis of vindoline analogues bearing the deep-seated C4 modifications through use of the oxadiazole cycloaddition cascade. In initial studies of the intramolecular 1,3,4-oxadiazole cycloaddition cascade, we demonstrated that the initiating inverse electron demand Diels–Alder reaction proceeds with a faster rate and under milder reaction conditions with electronically matched electron-rich dienophiles, and that increasing substitution progressively slows the reaction.8 Nonetheless, because of the intramolecular nature of the initiating Diels–Alder reaction, mono and disubstituted unactivated dienophiles as well as mono and disubstituted electron-deficient dienophiles were found capable of initiating the reaction cascade with the electron-deficient oxadiazole, albeit at progressively slower rates. However, two dienophile classes that failed to productively participate in the reaction cascade in our initial survey8a were trisubstituted unactivated alkenes and trisubstituted electron-deficient alkenes. Because of our interest in exploring the impact of vinblastine C4 substituents, we reexamined such substrates and herein report conditions under which they may now be employed (Fig. 2).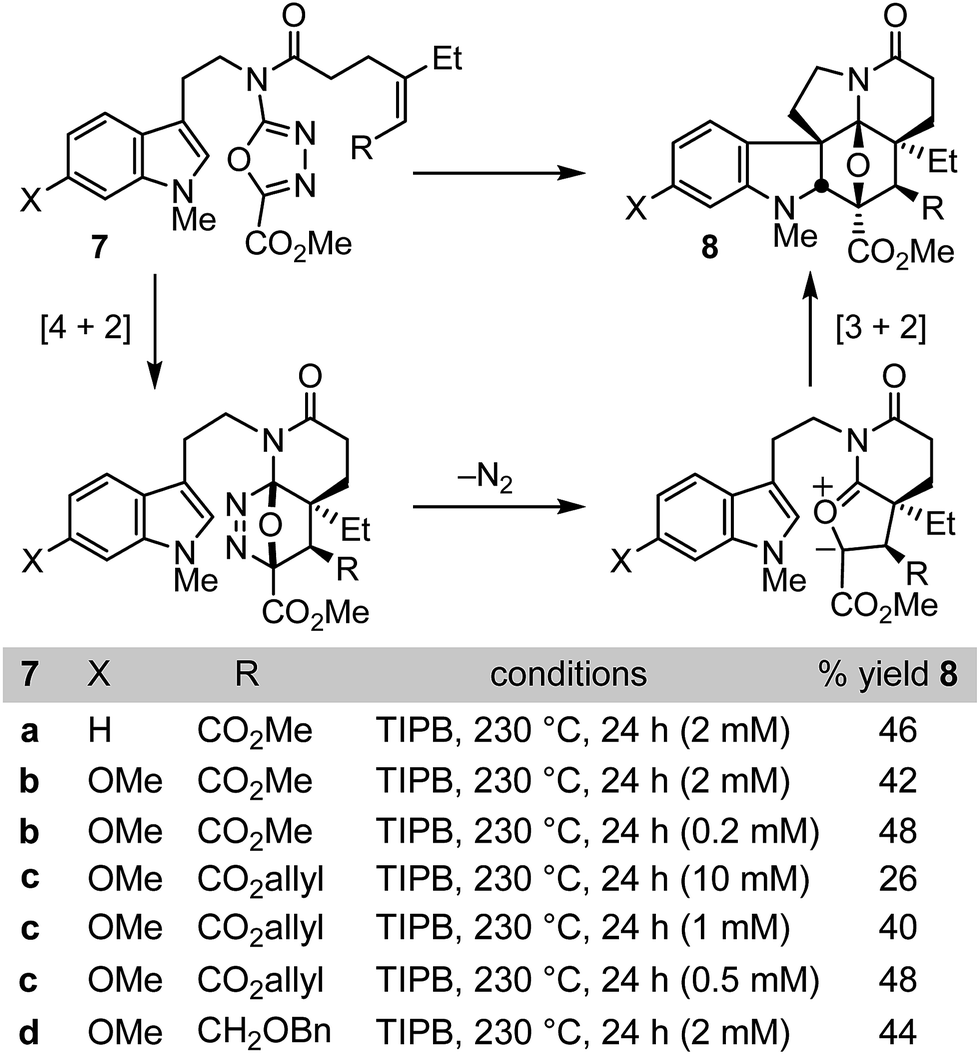 | ||
| Fig. 2 Cycloaddition cascade. | ||
Both electron-deficient trisubstituted alkenes (7a–c) and unactivated trisubstituted alkenes (7d) were found to productively initiate the 1,3,4-oxadiazole [4 + 2]/[3 + 2] cycloaddition cascade when the reaction was conducted in triisopropylbenezene (TIPB, 230 °C, 24 h) under dilute reaction conditions (Fig. 2). The concentration dependence of the reaction, which is highlighted most clearly with 7c, is more dramatic than observed with most substrates. Little product was observed at lower reaction temperatures or at concentrations much greater than 10 mM (e.g. 100 mM) and these combined features are likely the reason their productive participation in the reaction cascade was overlooked in early studies.8 In addition, resubjection of the products 8a–d to the reaction conditions led to complete recovery, indicating they are stable under the reaction conditions, and we observed no byproducts that indicated the final [3 + 2] cycloaddition reactions are reversible under these conditions. A single diastereomer of the reaction product was produced, providing the highly functionalized cycloadducts 8a–d in yields (44–48%) more than sufficient to explore their conversion to synthetic vindolines modified at C4. Four C–C bonds, three rings, all six stereocenters about the newly formed central six-membered ring including four quaternary centers, all the requisite functionality required for preparation of the targeted compounds including the deep-seated C4 modifications, and the full pentacyclic skeleton found in vindoline are formed in a single transformation, offsetting the modest yields of the cycloaddition cascade.
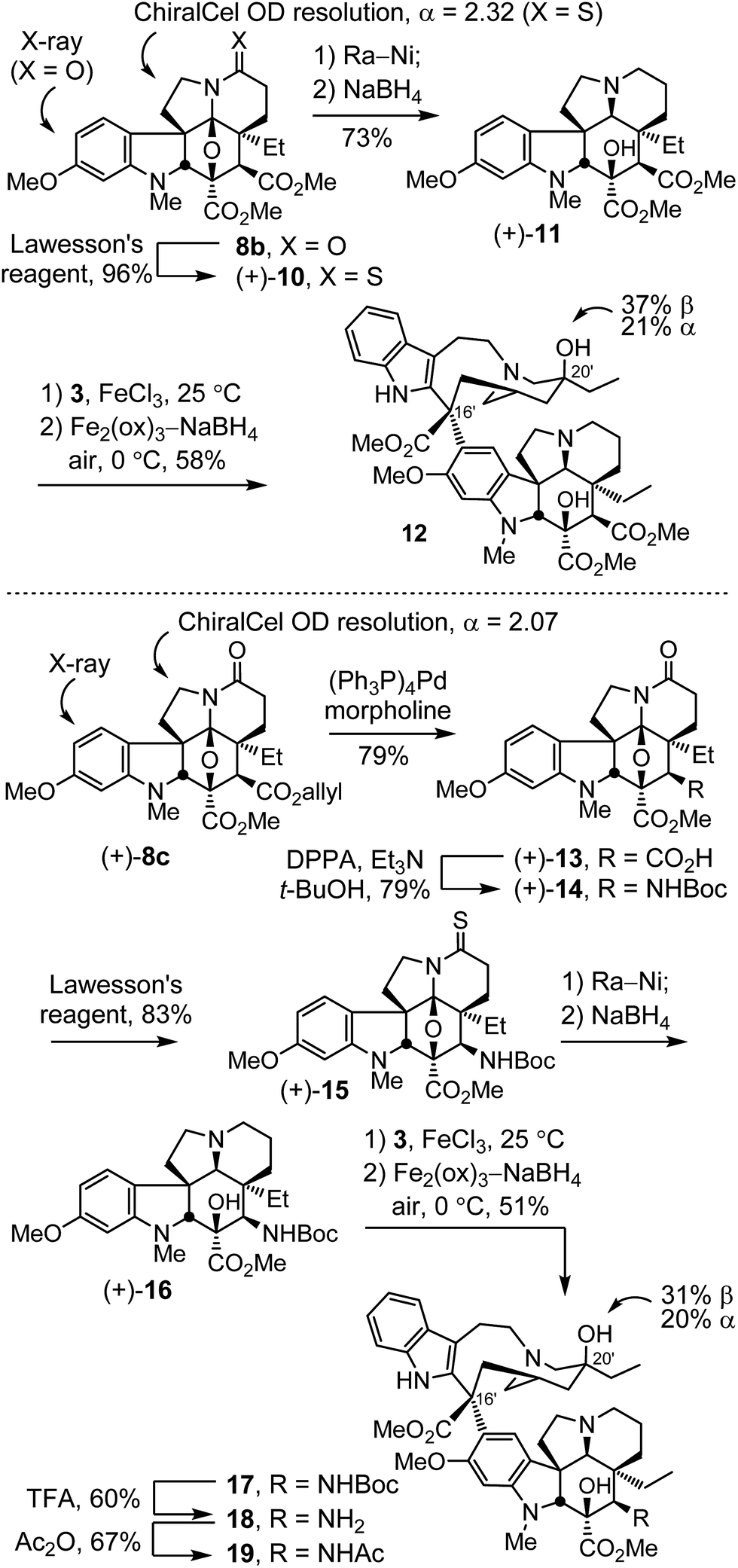
Initial studies conducted with 8b and 8c, targeting C4 analogues of 6,7-dihydrovinblastine (9),22b are summarized in Scheme 1. Although anticipated to be less potent than vinblastine because of the removal of the 6,7-double bond,24e these studies were conducted first to establish whether the targeted C4 modifications would prove important to pursue. Treatment of cycloadduct 8b with Lawesson's reagent (1 equiv., toluene, 110 °C, 1 h) provided the thioamide 10 (96%), which was easily resolved into its enantiomers by chiral phase chromatography (semi-preparative ChiralCel OD, 60% i-PrOH/hexane, α = 2.32). This chromatographic separation proved both remarkable in its resolution (see ESI†) and scalability for enantiomer separation (>500 mg per injection), making it far preferable to existing6,20 or candidate asymmetric synthetic routes.26 Removal of the thioamide of (+)-10 with RANEY® nickel (Ra-Ni, 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 THF/MeOH, 23 °C) followed by diastereoselective reductive ring opening of the oxido bridge (10 equiv. NaBH4, MeOH, 0 °C, 1 h) provided (+)-11 (73%). Without optimization, single-step Fe(III)-promoted coupling of (+)-11 with catharanthine (3) proceeded with complete control of the C16′ stereochemistry (5 equiv. FeCl3, 10% TFE–0.05 N aq. HCl, 25 °C, 2 h), and in situ Fe(III)/NaBH4-mediated introduction of the C20′ alcohol via free radical hydrogen atom transfer to the intermediate trisubstituted alkene (10 equiv. Fe2(ox)3, 20 equiv. NaBH4, 0 °C, 30 min) in the presence of air (O2) proceeded with a 2:1 C20′ diastereoselectivity to afford the 6,7-dihydrovinblastine analogue 12, bearing a C4 methyl ester in place of the acetoxy group. As such, 12 was prepared by total synthesis in only 5 steps from 7b, and 8 steps from 6-methoxy-1-methyltryptamine.
:1 THF/MeOH, 23 °C) followed by diastereoselective reductive ring opening of the oxido bridge (10 equiv. NaBH4, MeOH, 0 °C, 1 h) provided (+)-11 (73%). Without optimization, single-step Fe(III)-promoted coupling of (+)-11 with catharanthine (3) proceeded with complete control of the C16′ stereochemistry (5 equiv. FeCl3, 10% TFE–0.05 N aq. HCl, 25 °C, 2 h), and in situ Fe(III)/NaBH4-mediated introduction of the C20′ alcohol via free radical hydrogen atom transfer to the intermediate trisubstituted alkene (10 equiv. Fe2(ox)3, 20 equiv. NaBH4, 0 °C, 30 min) in the presence of air (O2) proceeded with a 2:1 C20′ diastereoselectivity to afford the 6,7-dihydrovinblastine analogue 12, bearing a C4 methyl ester in place of the acetoxy group. As such, 12 was prepared by total synthesis in only 5 steps from 7b, and 8 steps from 6-methoxy-1-methyltryptamine.
 | ||
| Scheme 1 | ||
Through selective ester deprotection, the allyl ester 8c26 provided the opportunity to introduce additional C4 functionality of which we targeted the acetamide 19 (Scheme 1). This compound incorporates a single heavy atom replacement within the structure with substitution of an amide nitrogen for the acetoxy ester oxygen presumably providing a non-hydrolyzable amide replacement for the metabolically labile C4 acetoxy ester. Resolution of 8c by chiral phase chromatography proceeded with a similarly remarkable and scalable separation of the enantiomers (semi-preparative ChiralCel OD, 60% i-PrOH/hexane, α = 2.07).26 Allyl ester cleavage of (+)-8c (0.1 equiv. (Ph3P)4Pd, 10 equiv. morpholine, 10:1 THF/DMSO, 23 °C, 1 h) cleanly provided the carboxylic acid (+)-13 (79%). Treatment of (+)-13 with diphenylphosphoryl azide (2 equiv. DPPA, 2.2 equiv. Et3N, t-BuOH, 85 °C, 16 h) led to in situ formation of the acyl azide and subsequent Curtius rearrangement to give (+)-14 (79%) with trap of the intermediate isocyanate by t-BuOH. Conversion to the thioamide with Lawesson's reagent (1 equiv., toluene, 110 °C, 1 h) provided (+)-15 (83%). Removal of the thioamide with Ra-Ni (1:1 THF/MeOH, 23 °C) followed by diastereoselective reductive ring opening of the oxido bridge (10 equiv. NaBH4, MeOH, 0 °C, 1 h) provided (+)-16. Without optimization, single-step Fe(III)-promoted coupling with catharanthine (3) and in situ Fe(III)/NaBH4-mediated C20′ oxidation provided 17. Acid-catalyzed Boc removal (TFA–CH2Cl2 1:4, 23 °C, 2 h) afforded 18 (60%) and amine acetylation (1:1 Ac2O/pyridine, 23 °C, 30 min; 10 equiv. K2CO3, MeOH, 23 °C, 1 h) provided the C4 acetamide 19 (67%), incorporating the single heavy atom replacement at C4 of 6,7-dihydrovinblastine (9).
Compounds 12 and 17–19 were assessed alongside 6,7-dihydrovinblastine (9) as a direct comparison in cell growth inhibition assays against both mouse leukemia (L1210) and human colon cancer (HCT116) cell lines that have been used to initially examine vinblastine analogues (Fig. 3). Each of the C4 amine derivatives including the acetamide 19 and free amine 18 matched the biological potency of 9 and mirrored the relative activity observed with 1vs.4 (acetate vs. alcohol). Even more significantly, the C4 methyl ester 12 (IC50 = 60–80 nM) exceeded the potency of 9 by nearly 10-fold and proved to be only 10-fold (vs. 100-fold) less active than vinblastine itself. These results, especially the potent activity of 12, inspired the following efforts to incorporate these and related C4 modifications into synthetic vinblastines in anticipation that they would be well tolerated, potentially provide additional advantages, and reveal insights into the importance and role of the C4 substituent.
 | ||
| Fig. 3 Cell growth inhibition, C4 modifications on 6,7-dihydrovinblastine. | ||
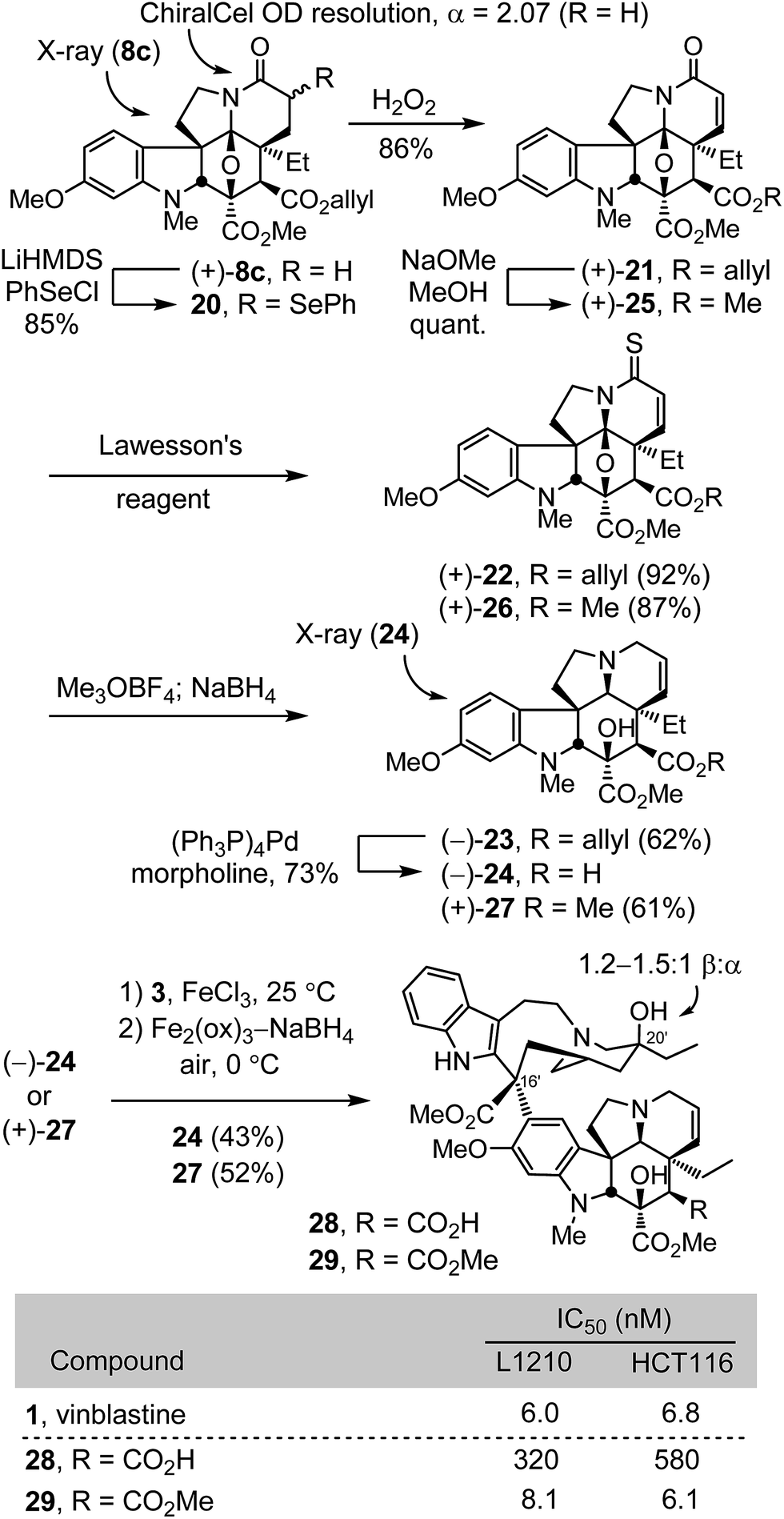
Conversion of (+)-8c to a diastereomeric mixture of α-selenides 20 (85%) was accomplished by treatment of the lactam enolate with phenylselenyl chloride (LiHMDS, THF, 1 h, −78 °C) (Scheme 2). The mixture of selenides 20 was treated with H2O2 (3 equiv.) in THF (0 °C) to provide the α,β-unsaturated lactam (+)-21 in good yield (86%). Treatment of (+)-21 with Lawesson's reagent (1.2 equiv., toluene, 100 °C, 1 h) provided thioamide (+)-22 (92%), which was subjected to methylation with Meerwein's salt (3 equiv., Me3OBF4, CH2Cl2, 23 °C, 1 h) followed by NaBH4 reduction (3 equiv., MeOH, 0 °C) of the S-methyl iminium salt in the same vessel to provide (−)-23 cleanly (62%). Allyl ester cleavage of (−)-23 (0.1 equiv. (Ph3P)4Pd, 10 equiv. morpholine, 10:1 THF/DMSO, 23 °C, 1 h) cleanly provided (−)-2426 (73%), bearing a C4 carboxylic acid in place of the vindoline C4 acetate. In a complementary fashion, the allyl ester (+)-21 was converted to the corresponding methyl ester (+)-25 by transesterification (5 equiv. NaOMe, MeOH, 23 °C, 1 h, quant) and carried through the same sequence of conversion to the thioamide (+)-26 (1.1 equiv. Lawesson's reagent, toluene, 100 °C, 1 h, 87%), S-methylation with Meerwein's salt (3 equiv. Me3OBF4, CH2Cl2, 23 °C, 30 min) and NaBH4 reduction (9 equiv., MeOH, 0 °C) to provide (+)-27 (61%), a synthetic vindoline bearing a C4 methyl ester. Without optimization, single-step Fe(III)-promoted coupling of (−)-24 and (+)-27 with catharanthine (3) and in situ Fe2(ox)3/NaBH4-mediated C20′ oxidation afforded 28 and 29, synthetic vinblastines containing the C4 carboxylic acid and methyl ester, respectively, each prepared by total synthesis in 7 steps from 7c or 10 steps from 6-methoxy-1-methyltryptamine. In part, the conciseness of the approach may be attributed to use of an early stage15versus penultimate10 introduction of the vindoline 6,7-double bond that, in this case, also avoided late stage competitive lactone formation between the C4 ester and a C7 β-alcohol.
 | ||
| Scheme 2 | ||
The compounds 28 and 29 were assessed in cell growth inhibition assays where the methyl ester 29 (IC50 = 6–8 nM) matched the activity of vinblastine, whereas the carboxylic acid 28 was found to be 50–100 fold less potent. Compound 29 is a constitutional isomer of vinblastine in which the ester oxygen and carbonyl of the C4 acetate are simply transposed such that it is now the carbonyl carbon that is directly attached to C4. In addition to this nuanced constitutional isomeric relationship between 1 and 29, a significant ramification of the change is that the C4 methyl ester, which is flanked by two quaternary centers, is not nearly as susceptible to hydrolysis as the natural C4 acetate. In fact, treatment of 29 with LiOH at room temperature led only to recovered 29, and reaction was observed only at elevated temperatures with excess base over extended reaction times (excess LiOH, 3:2:1 THF:MeOH:H2O, 50 °C, 17 h), providing preferential and selective C3 (and not C4) methyl ester hydrolysis. Similar room temperature treatment of vinblastine leads to rapid and selective C4 acetate hydrolysis, suggesting 29 is likely to be metabolically much more stable toward C4 hydrolysis than vinblastine. Finally, the reduced activity of the carboxylic acid 28 represents a direct impact the C4 substituent has on tubulin binding affinity (see Fig. 4), although we cannot rule out whether poor cellular uptake also contributes to the diminished activity.
The extension of the studies to the preparation of synthetic vinblastines bearing a functionalized C4 amine, including an acetamide is summarized in Scheme 3. Allyl ester cleavage conducted on the intermediate thioamide (+)-22 (0.1 equiv. (Ph3P)4Pd, 10 equiv. morpholine, 10:1 THF/DMSO, 23 °C, 1 h) was followed by Curtius rearrangement of the resulting carboxylic acid 30 (2 equiv. DPPA, 3 equiv. Et3N, t-BuOH, 85 °C, 16 h) to provide (+)-31 (64% for two steps). S-Methylation with Meerwein's salt (3 equiv. Me3OBF4, 20 equiv. 2,6-di-t-butylpyridine, CH2Cl2, 23 °C, 30 min) followed by NaBH4 reduction (9 equiv., MeOH, 0 °C) in the same vessel provided (+)-32, a modified vindoline bearing a protected C4 amine. Without optimization, single-step Fe(III)-promoted coupling of (+)-32 with catharanthine (3) and in situ Fe2(ox)3/NaBH4-mediated free radical C20′ oxidation afforded 33. Acid-catalyzed Boc removal (TFA, CH2Cl2, 23 °C, 2 h) afforded amine 34 (74%) and acetylation (Ac2O, DMAP, CH2Cl2, 23 °C, 30 min) provided the C4 acetamide 35 (63%). All three compounds 33–35 were assessed in cell growth inhibition assays where both the Boc protected derivative 33 and the free amine 34 were found to be 50 to 75-fold less potent than vinblastine. In contrast, the acetamide 35 exhibited improved activity (IC50 = 50–60 nM) relative to the corresponding dihydro compound 19 (ca. 10-fold), although it still proved to be roughly 10-fold less potent than vinblastine.
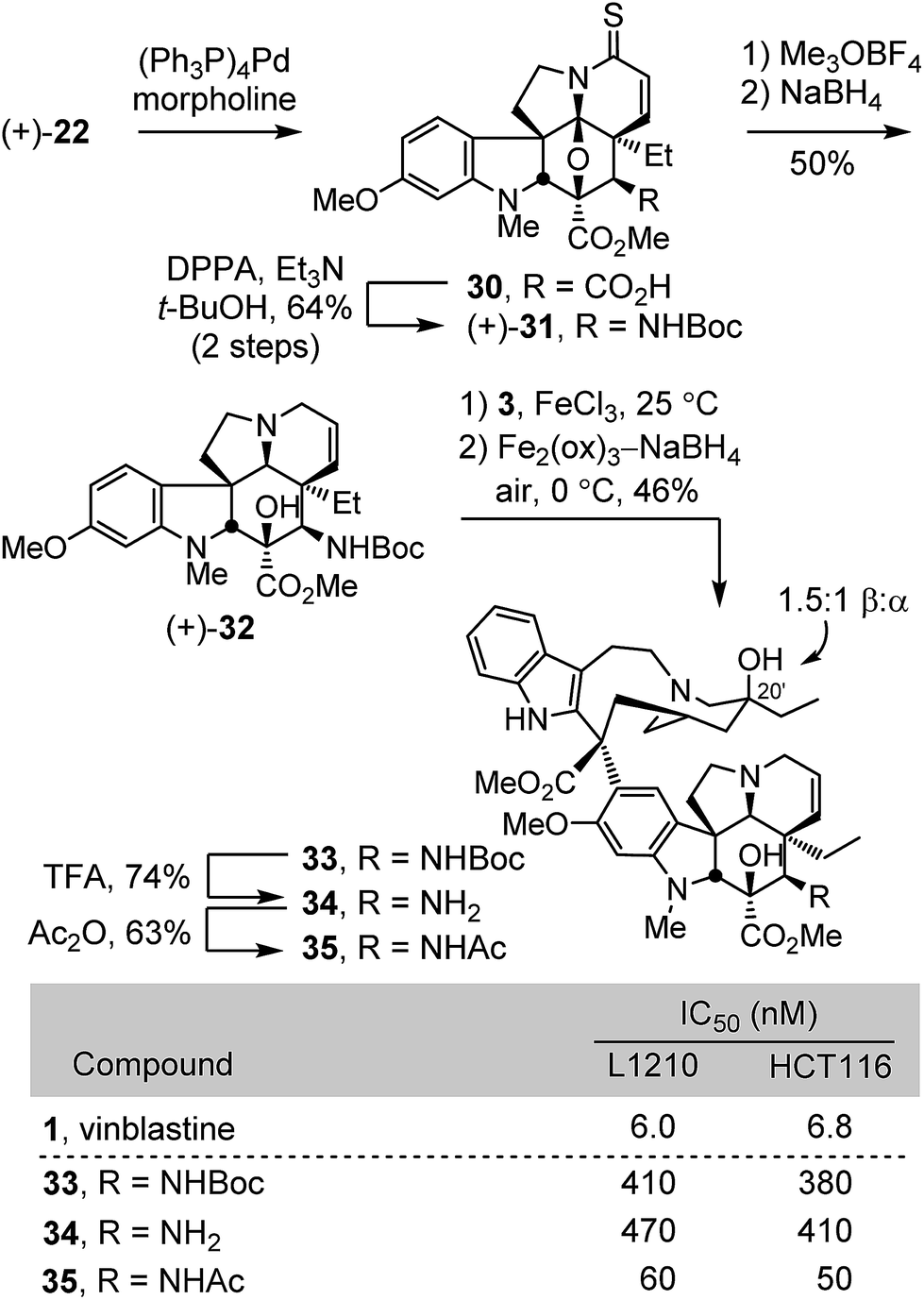 | ||
| Scheme 3 | ||
Concurrent with these studies, we prepared compounds that incorporate a C4 hydroxymethyl or acetoxymethyl substituent from the cycloadduct 8d, introducing an additional carbon between C4 and the polar functional group found in 4-desacetylvinblastine or vinblastine, respectively (Scheme 4). Conversion of 8d to a diastereomeric mixture of α-selenides 36 (63%) was accomplished by treatment of the lactam enolate with phenylselenyl chloride (2 equiv., 3 equiv. LiHMDS, THF, 1 h, −78 °C). The mixture of selenides 36 was subjected to treatment with m-CPBA (1.25 equiv.) in THF (excess pyridine, 0–23 °C, 2 h) to provide the α,β-unsaturated lactam 37 (72%). These latter two reactions were most conveniently conducted without the intermediate purification of the diastereomeric mixture 36, providing 37 directly in further improved overall yield (72%, 2 steps). Resolution of 37 by chiral phase chromatography provided the two enantiomers (semi-preparative ChiralCel OD, 60% i-PrOH/hexane, α = 1.41). Treatment of (+)-37 with Lawesson's reagent (1 equiv., toluene, 80 °C, 30 min) provided thioamide (+)-38 (92%), which was subjected to methylation with Meerwein's salt (2 equiv. Me3OBF4, CH2Cl2, 23 °C, 1 h) followed by in situ NaBH4 reduction (6 equiv., 1:1 MeOH/CF3CH2OH, 0 °C) of the S-methyl iminium ion, provided (−)-39 (71%). A single crystal X-ray structure determination conducted with the natural enantiomer of 39 confirmed its structure, relative stereochemistry, and absolute configuration.26 For comparison purposes and with (−)-39 in hand, the synthetic vinblastines 40–42 were prepared. Single-step Fe(III)-promoted coupling of (−)-39 with catharanthine (3), which proceeded with complete control of the newly formed C16′ quaternary stereocenter, and subsequent in situ Fe2(ox)3/NaBH4-mediated free radical C20′ oxidation afforded 40 and its C20′ isomer. O-Debenzylation (H2, Pd/C, 50:1 MeOH/TFA, 71%) and subsequent O-acetylation of the liberated alcohol 41 (Ac2O, DMAP, 23 °C, CH2Cl2, 54%) provided 42. The latter two compounds constitute analogues of desacetylvinbastine (4) and vinblastine (1) containing a carbon inserted at C4 between the natural product core and the polar substituent. Both 41 and 42 proved to be 20- to 70-fold less potent than the corresponding natural products, indicating that the added change significantly reduces activity. Interestingly, the benzyl ether precursor 40 was found to be more potent than either 41 or 42, but remained ca. 10-fold less active than vinblastine and exhibited activity on par with desacetoxyvinblastine (5) lacking a C4 substituent.
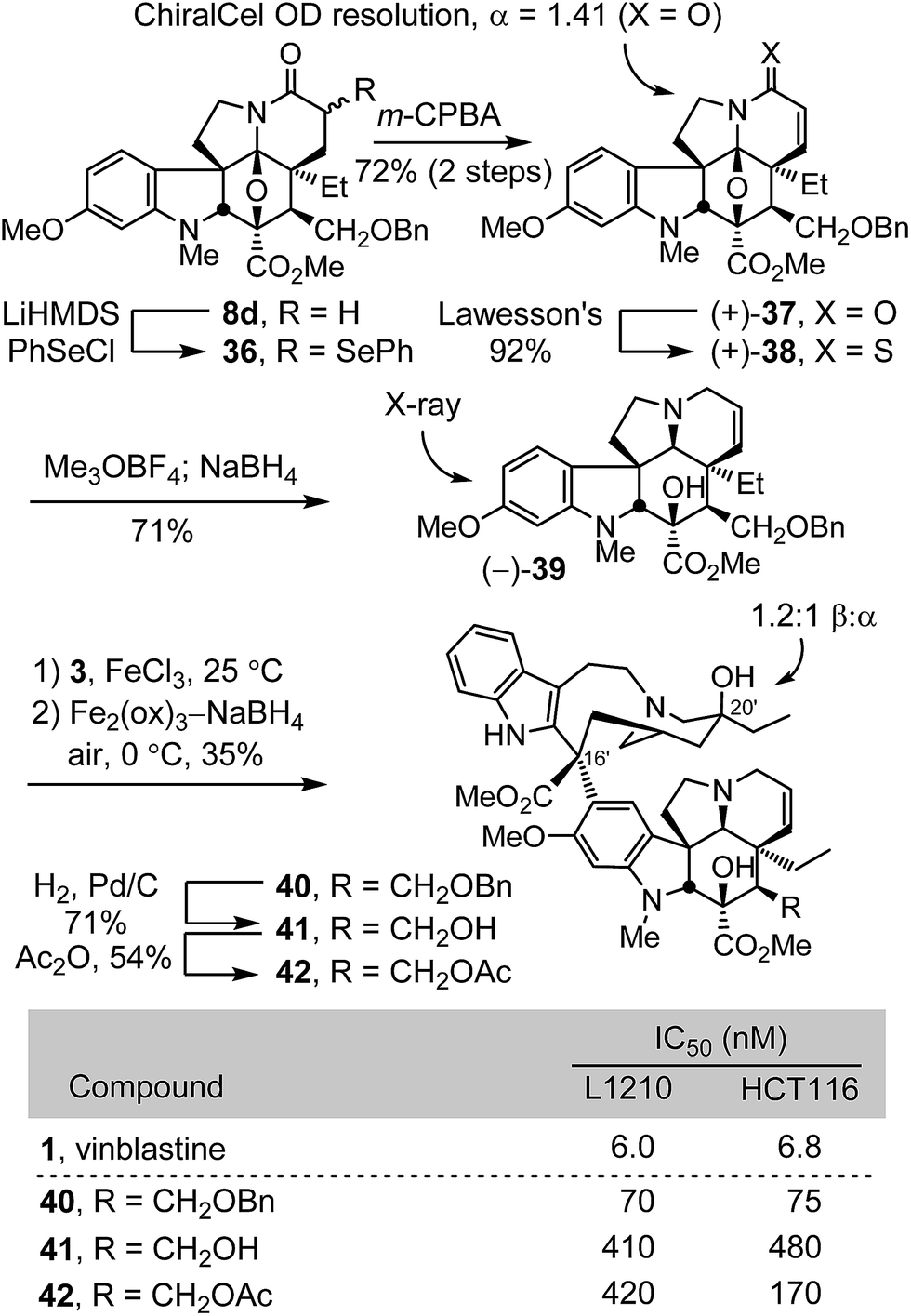 | ||
| Scheme 4 | ||
Given the unique behavior of the methyl ester 29, being the only C4 modified compound that matched the activity of vinblastine, three additional esters were examined to define its sensitivity to modification. Thus, alkylative esterification of carboxylic acid 24, derived from deprotection of the allyl ester (−)-23, with ethyl iodide (2 equiv. CsF, DMF, 25 °C, 1 h, 62%), isopropyl iodide (2 equiv. CsF, DMF, 40 °C, 1 h, 60%), or benzyl bromide (2 equiv. CsF, DMF, 25 °C, 1 h, 80%) provided 43–45 without purification of the intermediate carboxylic acid (Scheme 5). Without optimization, their single-step Fe(III)-promoted coupling with catharanthine (3) and subsequent in situ Fe2(ox)3/NaBH4-mediated free radical C20′ oxidation afforded 46–48. Stunningly, even the apparently benign changes to the ethyl or isopropyl esters 46 and 47 led to 10-fold reductions in cell growth inhibition activity. Only the benzyl ester 48 approached the activity of 29, desacetylvinblastine (4), and vinblastine.
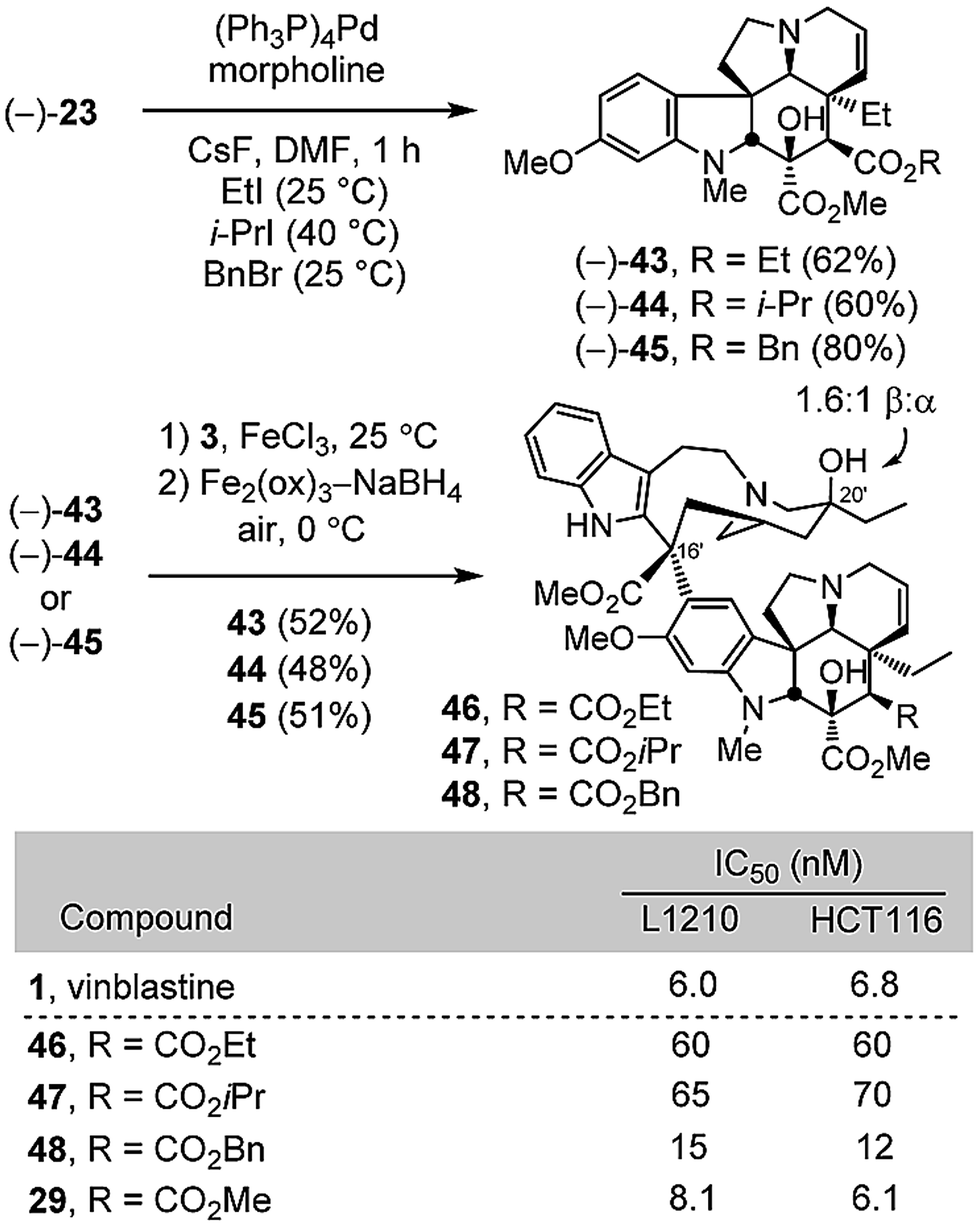 | ||
| Scheme 5 | ||
Finally, the C4 N-methyl carboxamide 52 and N-benzyl carboxamide 53 were prepared for comparison in part to establish whether the substituent polarity may contribute to the functional activity of vinblastine (Scheme 6). The former N-methyl carboxamide serves as a direct amide comparison with the methyl ester 29, replacing the ester oxygen with an amide NH. Their synthesis involved a unique closure of the carboxylic acid 24 first to the characterized but reactive β-lactone 4927 (2 equiv. DMAP, 2 equiv. EDCI, CH2Cl2, 25 °C, 12 h), an intermediate preferentially generated upon activation of the carboxylic acid under a range of conditions (EDCI, HATU, or PyBOP), followed by its subsequent reaction with methylamine or benzylamine (i-PrOH, 80 and 60 °C respectively, 12 h) to provide 50 (61%) and 51 (50%) in good yields from 23 (3 steps) without intermediate purifications. Without optimization, their single-step Fe(III)-promoted coupling with catharanthine (3) and subsequent in situ Fe2(ox)3/NaBH4-mediated free radical C20′ oxidation afforded 52 and 53. Again and remarkably, the N-methyl amide proved to be more than 10-fold less active than vinblastine or the methyl ester 29 and, while more potent, the N-benzyl amide was also 5–10-fold less active.
 | ||
| Scheme 6 | ||
In order to establish whether the substituent effects observed in the cell growth functional assays were derived from on target effects, key members of the initial series of compounds were examined in a competitive tubulin binding assay, measuring their ability to displace tubulin-bound BODIPY-vinblastine (Fig. 4).24h Consistent with their functional activity but contrary to expectations based on their perceived placement in a tubulin-bound X-ray (see Fig. 1), these key derivatives displayed easily distinguishable relative tubulin binding affinities that correlated with their relative cell growth inhibition activity (R: CO2Me > NHAc > CH2OAc > CO2H, NH2). Thus, the vinblastine C4 substituent significantly impacts tubulin binding affinity and the trends observed correlate with the resulting functional cell growth inhibition. Moreover, the site and functionality are remarkably sensitive to seemingly benign structural modification.
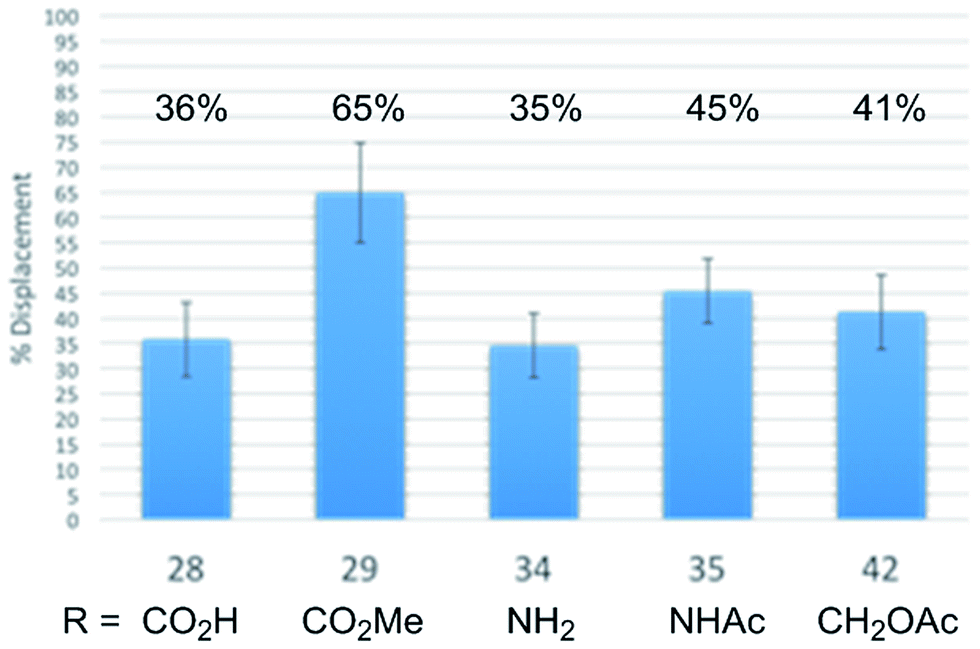 | ||
| Fig. 4 Relative tubulin binding affinities assessed by measuring competitive % displacement of tubulin-bound BODIPY-vinblastine. | ||
Retrospective examination of the original >4 Å tubulin-bound X-ray of vinblastine25a was not helpful in defining additional roles for the C4 substituent beyond serving as a polar interface for the bound complex. However, a higher resolution 2.2 Å structure just published25b proved much more informative. In it, the C4 acetate serves as a stabilizing H-bond acceptor with the protonated amine side chain of Lys336 (ester O) and Asn329 (carbonyl O) in a now well-ordered lip on the tubulin protein surface that interacts with and wraps around the side of the acetate, placing the acetate methyl group in a spatially well-defined small hydrophobic half pocket adjacent to and lined by Ile332 and Ala333 (Fig. 5). These same H-bond acceptor roles may be functionally satisfied by swapping the protein residues interacting with the carbonyl oxygen (H-bond from Lys336) and ester oxygen (H-bond from Asn329) of the C4 methyl ester 29 within this ordered flexible loop, still placing the methyl group in the same hydrophobic half pocket adjacent to the side chains of Ile332 and Ala333. This would not be possible with C4 substituents that incorporate H-bond donors versus acceptors at these sites (C4 acetamide, N-methyl carboxamide), with substituents that displace the H-bond acceptors (the homologated C4 hydroxymethyl or acetoxymethyl groups), or with substituents that incorporate larger acyl or ester substituents. Such substituents do not preclude vinblastine binding, just that the well-ordered interaction of the C4 acetate/methyl ester with the protein loop and the resulting stabilizing interactions would be lost. As a result, it is not surprising that the alternative C4 substituents at best behave analogous to desacetoxyvinblastine lacking a C4 substituent altogether (5vs.35, 46, 47, 52, and 53) or may further destabilize binding (28, 33, 34, 41, and 42). Perhaps still superimposed on this role as a H-bond acceptor for Lys336 and Asn329, the C4 functionality may still serves as part of the polar interface for tubulin bound vinblastine. Additionally, the equipotent activity of the C4 alcohol (4), the surprisingly good activity of the benzyl ester 53 and even the activity of the benzyl ether 40 (vs.41 and 42), which do not conform to this interpretation, suggest there may be additional unrecognized ways in which this flexible lip of tubulin at the solvent interface of the complex may productively interact with selected C4 substituents and stabilize the binding of vinblastine analogues. Finally, it is surprising the C4 carboxylic acid binds so poorly to tubulin given the potential stabilizing electrostatic interaction with tubulin Lys336, perhaps reflecting the impact of necessarily residing alongside the hydrophobic half pocket defined by Ile332 and Ala333.
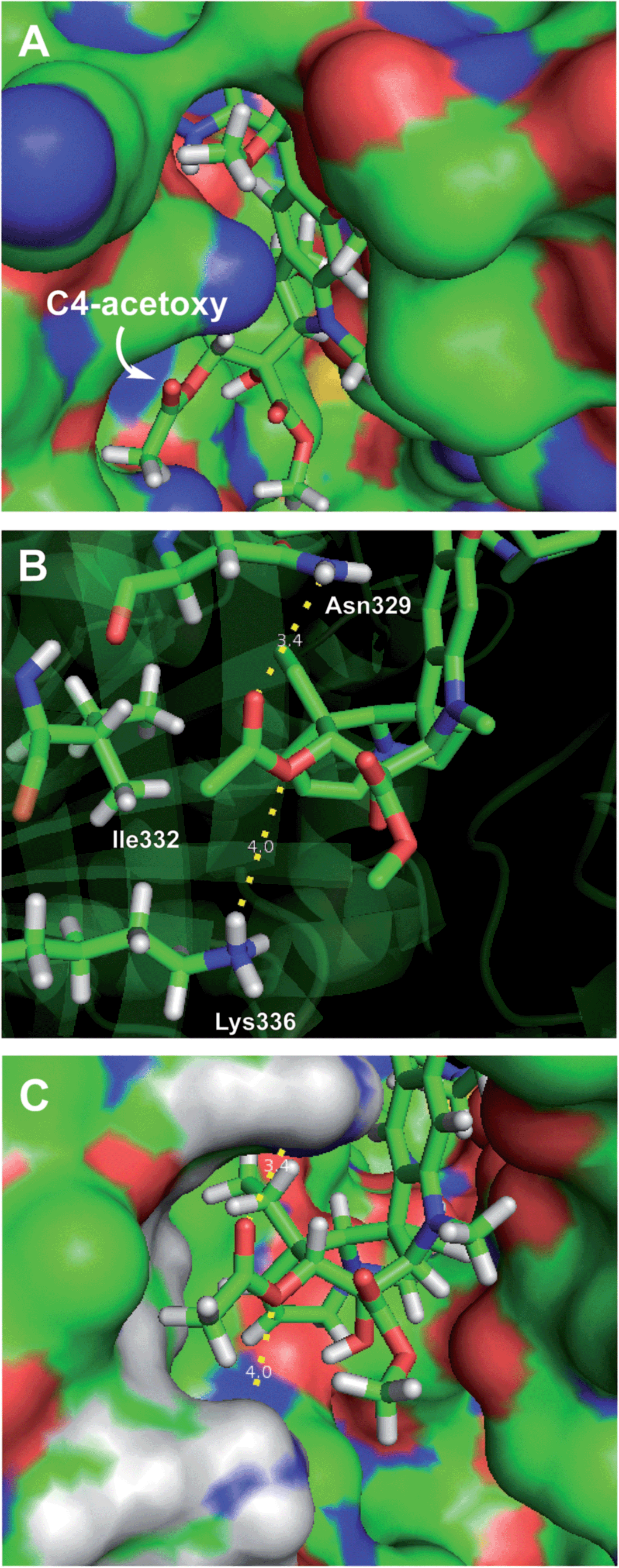 | ||
| Fig. 5 Recent X-ray co-crystal structure of tubulin-bound vinblastine25b (pdb 5J2T) highlighting the (A) solvent exposed C4 acetoxy group. (B) Key residues interacting with the C4 substituent: Lys336 (H-bond to acetoxy ester oxygen), Ile332 (close contact to methyl group), and Asn329 (H-bond to acetoxy carbonyl oxygen). (C) Space filling model of B showing the fit of the acetoxy methyl group in the hydrophobic pocket defined by Ala333 and Ile332 residues. | ||
Conclusions
Herein, we described concise divergent28 total syntheses of a key series of C4 modified vinblastines, containing changes at a site that was not thought to intimately interact with the biological target tubulin yet impacts potency and constitutes a metabolic liability. Central to the approach was the development of an expanded scope of the 1,3,4-oxadiazole [4 + 2]/[3 + 2] cycloaddition cascade that permitted the direct installation of the desired C4 modifications in the vindoline subunit. Although originally reported to be unproductive, conditions where both electron-deficient trisubstituted alkenes and unactivated trisubstituted alkenes productively initiate the cycloaddition cascade were disclosed. The use of the cycloaddition cascade of three such substrates in concise total syntheses of a series of C4 modified vindolines, their single-step incorporation into the total synthesis of a series of 17 synthetic vinblastines, and their use in defining both the importance and surprising trends of the C4 substituent contribution to the biological properties of vinblastine were detailed (Fig. 6). Whereas the C4 acetate or a C4 alcohol contribute to the productive biological properties of vinblastine and desacetylvinblastine (1 and 4vs.5), replacement with a C4 amine, carboxylic acid, hydroxymethyl or acetoxymethyl substituent led to substantial reductions in potency (28, 34, 41 and 42). Introduction of an acetamide or N-methyl carboxamide substituent, which incorporate single heavy atom exchanges (amide NH for ester oxygen), provided compounds that matched the activity of desacetoxyvinblastine (35 and 52vs.5) but were still ca. 10-fold less active than vinblastine. In contrast, only the introduction of a C4 methyl ester (29vs.1), a constitutional isomer of vinblastine in which the carbonyl carbon and ester oxygen are transposed, provided a compound that matched the potency of vinblastine. Even the seemingly benign alteration of this equally active C4 methyl ester to the corresponding ethyl or isopropyl esters, and N-methyl carboxamide led to 10-fold reductions in activity (29vs.46, 47 and 52). Examination of the early series of the C4 modified vinblastines revealed that they bind to tubulin with affinities that correlate with their functional growth inhibition. We suggest that this unanticipated impact on target binding affinity is derived from a previously unrecognized role the C4 substituent plays as a H-bond acceptor for α-tubulin Lys336 and Asn329 side chains at a site incapable of accommodating a substituent H-bond donor, placing the methyl groups of the C4 acetate and C4 methyl ester in a spatially well-defined small hydrophobic half pocket of a well-ordered loop that organizes around the side of bound substituent. This remarkable impact of the C4 substituent, its stringency, and even the magnitude of its effect are extraordinary, yet are analogous to observations made with other peripheral substituents found on vinblastine (e.g., N1–Me, C16-methoxy, C20′-hydroxy,22b C16′,22c and C524a). In instances when the productive properties of a natural product are directly related to its emergence in Nature and has undergone continued optimization by natural selection as is likely the case with vinblastine, it may be that such peripheral functionality is not easily subjected to structural modifications or simplifications.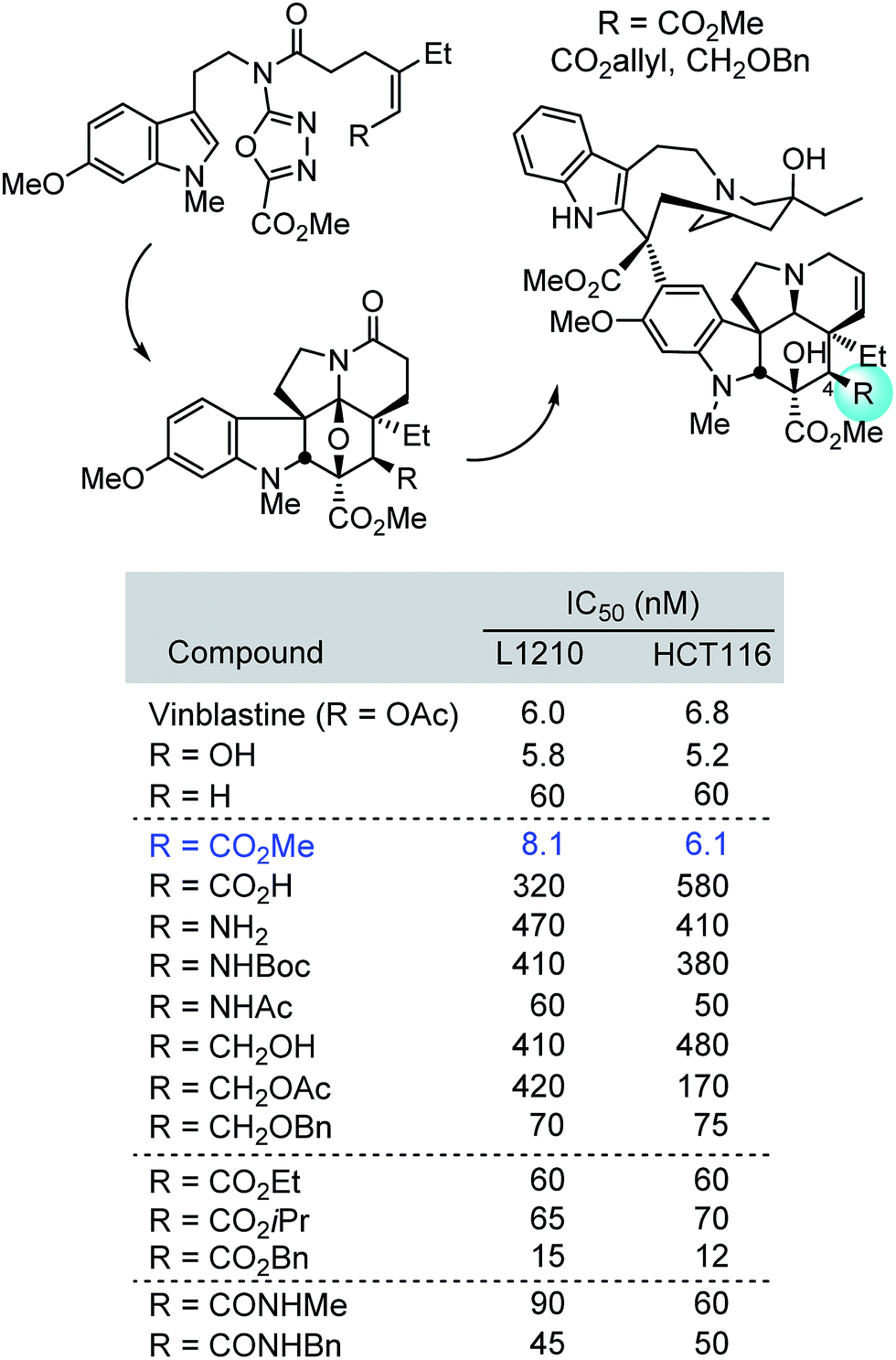 | ||
| Fig. 6 Summary of synthetic approach and cell growth inhibition properties of C4 modified vinblastines. | ||
Significantly, the compounds examined herein represent synthetic vinblastines prepared by total synthesis enabled by a powerful and now expanded oxadiazole cycloaddition cascade inspired by structures once thought too complex for such studies. Moreover, the compounds examined are themselves presently inaccessible by alternative methods, including natural product derivatization, late-stage functionalization, or biosynthetic methods. The examination of the key series of compounds revealed an unanticipated importance and defined the role of the C4 substituent in the expression of the natural product biological properties and provided one key analogue, the C4 methyl ester 29, that matched the activity of the natural product. The important difference being that the C4 methyl ester, which is flanked by two quaternary centers, was found to be not nearly as susceptible to hydrolysis as the natural C4 acetate, suggesting the known metabolically labile site on the natural product might be replaced with a more stable, less accessible C4 methyl ester.
Acknowledgements
We gratefully acknowledge the financial support of the National Institutes of Health (CA115526 and CA042056) and the Skaggs Institute for Chemical Biology. We especially wish to thank Katharine K. Duncan for the biological assessment of early samples, and Professor Arnold L. Rheingold and Dr Curtis E. Moore (UCSD) for the single crystal X-ray structure determinations of 8b, 8c, 24 and 39.References
- (a) R. L. Noble, C. T. Beer and J. H. Cutts, Ann. N. Y. Acad. Sci., 1958, 76, 882 CrossRef CAS PubMed; (b) R. L. Noble, Lloydia, 1964, 27, 280 Search PubMed; (c) Review: R. L. Noble, Biochem. Cell Biol., 1990, 68, 1344 CrossRef CAS PubMed.
- G. H. Svoboda, N. Neuss and M. Gorman, J. Am. Pharm. Assoc., Sci. Ed., 1959, 48, 659 CrossRef CAS.
- Reviews: (a) S. Timasheff, J. Andreu, M. Gorbunoff, F. Medranot and V. Prakash, Cell. Pharmacol., 1993, 1, S27 CAS; (b) M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253 CrossRef CAS PubMed.
- For a comprehensive review of the chemistry, medicinal chemistry, biology, and clinical applications, see: (a) M. E. Kuehne and I. Marko, in The Alkaloids, ed. A. Brossi and M. Suffness, Academic, San Diego, 1990, vol. 37, pp. 77–131 Search PubMed; (b) L. S. Borman and M. E. Kuehne, in The Alkaloids, ed. A. Brossi and M. Suffness, Academic, San Diego, 1990, vol. 37, pp. 133–144 Search PubMed; (c) N. Neuss and M. N. Neuss, in The Alkaloids, ed. A. Brossi and M. Suffness, Academic, San Diego, 1990, vol. 37, pp. 229–240 Search PubMed.
- Review: H. L. Pearce, in The Alkaloids, ed. A. Brossi and M. Suffness, Academic, San Diego, 1990, vol. 37, pp. 145–204 Search PubMed.
- (a) N. Langlois, F. Gueritte, Y. Langlois and P. Potier, J. Am. Chem. Soc., 1976, 98, 7017 CrossRef CAS PubMedP. Mangeney, R. Z. Andriamialisoa, N. Langlois, Y. Langlois and P. Potier, J. Am. Chem. Soc., 1979, 101, 2243 CrossRef CAS; (b) J. P. Kutney, T. Hibino, E. Jahngen, T. Okutani, A. H. Ratcliffe, A. M. Treasurywala and S. Wunderly, Helv. Chim. Acta, 1976, 59, 2858 CrossRef CAS PubMedJ. P. Kutney, L. S. L. Choi, J. Nakano, H. Tsukamoto, M. McHugh and C. A. Boulet, Heterocycles, 1988, 27, 1845 CrossRef; (c) M. E. Kuehne, P. A. Matson and W. G. Bornmann, J. Org. Chem., 1991, 56, 513 CrossRef CASW. G. Bornmann and M. E. Kuehne, J. Org. Chem., 1992, 57, 1752 Search PubMed; M. E. Kuehne, T. C. Zebovitz, W. G. Bornmann and I. Marko, J. Org. Chem., 1987, 52, 4340 Search PubMed; (d) P. Magnus, A. Stamford and M. Ladlow, J. Am. Chem. Soc., 1990, 112, 8210 Search PubMedP. Magnus, J. S. Mendoza, A. Stamford, M. Ladlow and P. Willis, J. Am. Chem. Soc., 1992, 114, 10232 Search PubMed; (e) S. Yokoshima, T. Ueda, S. Kobayashi, A. Sato, T. Kuboyama, H. Tokuyama and T. Fukuyama, J. Am. Chem. Soc., 2002, 124, 2137 Search PubMed.
- Reviews: (a) P. Potier, J. Nat. Prod., 1980, 43, 72 CrossRef CAS; (b) J. P. Kutney, Nat. Prod. Rep., 1990, 7, 85 RSC; (c) J. Sears and D. L. Boger, Acc. Chem. Res., 2015, 48, 653 CrossRef CAS PubMed.
- (a) G. D. Wilkie, G. I. Elliott, B. S. J. Blagg, S. E. Wolkenberg, D. R. Soenen, M. M. Miller, S. Pollack and D. L. Boger, J. Am. Chem. Soc., 2002, 124, 11292 CrossRef CAS PubMed; (b) G. I. Elliott, J. R. Fuchs, B. S. J. Blagg, H. Ishikawa, H. Tao, Z. Q. Yuan and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 10589 CrossRef CAS PubMed.
- Reviews: (a) J. E. Sears and D. L. Boger, Acc. Chem. Res., 2016, 49, 241 CrossRef CAS PubMed; (b) D. Margetic, P. Troselj and M. R. Johnston, Mini-Rev. Org. Chem., 2011, 8, 49 CrossRef CAS; (c) D. L. Boger, Tetrahedron, 1983, 34, 2869 CrossRef; (d) D. L. Boger, Chem. Rev., 1986, 86, 781 CrossRef CAS; (e) D. L. Boger and S. M. Weinreb, Hetero Diels-Alder Methodology in Organic Synthesis, Academic, San Diego, 1987 Search PubMed.
- (a) H. Ishikawa, G. I. Elliott, J. Velcicky, Y. Choi and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 10596 CrossRef CAS PubMed; (b) Y. Choi, H. Ishikawa, J. Velcicky, G. I. Elliott, M. M. Miller and D. L. Boger, Org. Lett., 2005, 7, 4539 CrossRef CAS PubMed.
- G. I. Elliott, J. Velcicky, H. Ishikawa, Y. Li and D. L. Boger, Angew. Chem., Int. Ed., 2006, 45, 620 CrossRef CAS PubMed.
- Z. Q. Yuan, H. Ishikawa and D. L. Boger, Org. Lett., 2005, 7, 741 CrossRef CAS PubMed.
- H. Ishikawa and D. L. Boger, Heterocycles, 2007, 72, 95 CrossRef CAS.
- S. E. Wolkenberg and D. L. Boger, J. Org. Chem., 2002, 67, 7361 CrossRef CAS PubMed.
- J. E. Sears, T. J. Barker and D. L. Boger, Org. Lett., 2015, 17, 5460 CrossRef CAS PubMed.
- J. P. Lajiness, W. Jiang and D. L. Boger, Org. Lett., 2012, 14, 2078 CrossRef CAS PubMed.
- E. L. Campbell, A. M. Zuhl, C. M. Lui and D. L. Boger, J. Am. Chem. Soc., 2010, 132, 3009 CrossRef CAS PubMed.
- (a) J. Xie, A. L. Wolfe and D. L. Boger, Org. Lett., 2013, 15, 868 CrossRef CAS PubMed; (b) K. Lee and D. L. Boger, Tetrahedron, 2015, 71, 3741 CrossRef CAS PubMed.
- K. Lee and D. L. Boger, J. Am. Chem. Soc., 2014, 136, 3312 CrossRef CAS PubMed.
- (a) D. Kato, Y. Sasaki and D. L. Boger, J. Am. Chem. Soc., 2010, 132, 3685 CrossRef CAS PubMed; (b) Y. Sasaki, D. Kato and D. L. Boger, J. Am. Chem. Soc., 2010, 132, 13533 CrossRef CAS PubMed.
- (a) J. Vukovic, A. E. Goodbody, J. P. Kutney and M. Misawa, Tetrahedron, 1988, 44, 325 CrossRef CAS; (b) C. Szantay Jr, J. Balazs, J. Bolcskei and C. Szantay, Tetrahedron, 1991, 47, 1265 CrossRef; (c) R. J. Sundberg, J. Hong, S. Q. Smith, M. Sabato and I. Tabakovic, Tetrahedron, 1998, 54, 6259 CrossRef CAS.
- (a) H. Ishikawa, D. A. Colby and D. L. Boger, J. Am. Chem. Soc., 2008, 130, 420 CrossRef CAS PubMed; (b) H. Ishikawa, D. A. Colby, S. Seto, P. Va, A. Tam, H. Kakei, T. J. Rayl, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2009, 131, 4904 CrossRef CAS PubMed; (c) A. Tam, H. Gotoh, W. M. Robertson and D. L. Boger, Bioorg. Med. Chem. Lett., 2010, 20, 6408 CrossRef CAS PubMed; (d) H. Gotoh, J. E. Sears, A. Eschenmoser and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 13240 CrossRef CAS PubMed.
- E. K. Leggans, T. J. Barker, K. K. Duncan and D. L. Boger, Org. Lett., 2012, 14, 1428 CrossRef CAS PubMed.
- For additional applications, see: (a) P. Va, E. L. Campbell, W. M. Robertson and D. L. Boger, J. Am. Chem. Soc., 2010, 132, 8489 CrossRef CAS PubMed; (b) H. Gotoh, K. K. Duncan, W. M. Robertson and D. L. Boger, ACS Med. Chem. Lett., 2011, 2, 948 CrossRef CAS PubMed; (c) T. J. Barker and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 13588 CrossRef CAS PubMed; (d) T. J. Barker, K. K. Duncan, K. Otrubova and D. L. Boger, ACS Med. Chem. Lett., 2013, 4, 985 CrossRef CAS PubMed; (e) K. D. Schleicher, Y. Sasaki, A. Tam, D. Kato, K. K. Duncan and D. L. Boger, J. Med. Chem., 2013, 56, 483 CrossRef CAS PubMed; (f) E. K. Leggans, K. K. Duncan, T. J. Barker, K. D. Schleicher and D. L. Boger, J. Med. Chem., 2013, 56, 628 CrossRef CAS PubMed; (g) O. Allemann, M. Brutsch, J. C. Lukesh III, D. M. Brody and D. L. Boger, J. Am. Chem. Soc., 2016, 138, 8376 CrossRef CAS PubMed; (h) D. W. Carney, J. C. Lukesh III, D. M. Brody, M. M. Brutsch and D. L. Boger, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9691 CrossRef CAS PubMed.
- (a) B. Gigant, C. Wang, R. B. G. Ravelli, F. Roussi, M. O. Steinmetz, P. A. Curmi, A. Sobel and M. Knossow, Nature, 2005, 435, 519 CrossRef CAS PubMed; (b) A. B. Waight, K. Bargsten, S. Doronina, M. O. Steinmetz, D. Sussman and A. E. Prota, PLoS One, 2016, 11, e0160890 Search PubMed.
- Single-crystal X-ray structure determinations provided the structure, relative stereochemistry, and absolute configuration of (−)-8b (unnatural enantiomer, CCDC 1475228), (+)-24 (unnatural enantiomer, CCDC 1475226), and (−)-39 (natural enantiomer, CCDC 1475227). The structure and relative stereochemistry of 8c (CCDC 1475225) were confirmed by X-ray and the absolute stereochemistry of (−)-8c (unnatural enantiomer) was assigned by correlation with (−)-8b (NaOMe, MeOH, 60 °C, 24 h; TMSCHN2, MeOH–toluene, 23 °C, 30 min, 98%).
- For 49: 1H NMR (600 MHz, CDCl3) δ 7.00 (d, J = 8.2 Hz, 1H), 6.38 (dd, J = 8.2, 2.3 Hz, 1H), 6.12 (d, J = 2.3 Hz, 1H), 5.92 (ddd, J = 9.8, 4.4, 1.9 Hz, 1H), 5.42 (dt, J = 9.8, 2.3 Hz, 1H), 3.89 (s, 3H), 3.77 (s, 3H), 3.77 (s, 1H), 3.70 (s, 1H), 3.43 (ddd, J = 16.6, 4.4, 1.9 Hz, 1H), 3.32–3.24 (m, 1H), 2.81 (dd, J = 16.6, 2.3 Hz, 1H), 2.69 (s, 3H), 2.49 (s, 1H), 2.43 (ddd, J = 10.8, 8.9, 7.5 Hz, 1H), 2.34 (ddd, J = 13.0, 7.5, 1.6 Hz, 1H), 2.23 (ddd, J = 13.0, 10.6, 7.8 Hz, 1H), 1.64 (dt, J = 14.3, 7.3 Hz, 1H), 1.24 (dq, J = 14.7, 7.6 Hz, 1H), 0.41 (t, J = 7.4 Hz, 3H); ESI-TOF HRMS m/z 425.2069 (M+H+, C24H28N2O5 requires 425.2071).
- (a) D. L. Boger and C. E. Brotherton, J. Org. Chem., 1984, 49, 4050 CrossRef CAS; (b) M. D. Mullican and D. L. Boger, J. Org. Chem., 1984, 49, 4033 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and copies of 1H and 13C NMR spectra are provided. CCDC 1475225–1475228. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04146a |
| This journal is © The Royal Society of Chemistry 2017 |